

Abstract
Hypoxia and doxorubicin can cause cardiotoxicity and loss of myocardial function. These effects are due, in part, to an induction of apoptosis. Herein we identify the apoptotic pathways activated in H9c2 cells in response to hypoxia (O2/N2/CO2, 0.5:94.5:5) and doxorubicin (0.5 μM). Although the apoptosis induced was accompanied by induction of Fas and Fas ligand, the death receptor pathway was not critical for caspase activation by either stimulus. Hypoxia induced the expression of endoplasmic reticulum (ER) stress mediators and processed ER-resident pro-caspase-12 whereas doxorubicin did not induce an ER stress response. Most importantly, both stimuli converged on mitochondria to promote apoptosis. Accumulation of cytochrome c in the cytosol coincided with the processing of pro-caspase-9 and -3. Increasing the expression of the anti-apoptotic protein Bcl-xL, either by dexamethasone or adenovirus-mediated transduction, protected H9c2 cells from doxorubicin- and hypoxia-induced apoptosis. Bcl-xL attenuated mitochondrial cytochrome crelease and reduced downstream pro-caspase processing and apoptosis. These data demonstrate that two distinct cardiomyocyte-damaging stimuli converge on mitochondria thus presenting this organelle as a potentially important therapeutic target for anti-apoptotic strategies for cardiovascular diseases.
Keywords: apoptosis, gene transfer, hypoxia/anoxia, mitochondria, SR (function)
Introduction
Cardiomyocyte apoptosis has important pathophysiological consequences contributing to functional abnormalities in the myocardium. It has been reported in a variety of cardiovascular diseases, including myocardial infarction, end-stage heart failure, arrhythmogenic right ventricular dysplasia and doxorubicin-induced cardiomyopathy [1–3]. The diverse stresses and conditions that trigger apoptosis do so by activating one or more signal transduction pathways, which converge to activate a conserved family of aspartic acid-specific cysteine proteases, referred to as caspases [4]. Caspases are constitutively expressed as inactive zymogens and are activated in response to apoptotic stimuli by dimerization or specific proteolytic cleavage. Activated caspases cleave and activate other family members as well as several key proteins, which when cleaved dismantle the cell [4]. Activation of caspases is central to apoptosis and can be initiated by either of three distinct mechanisms: ligation of death receptors, release of cytochrome c from mitochondria and stress to the ER [1, 5].
The death receptor or extrinsic pathway is activated when an extracellular ligand, like Fas ligand (FasL) binds to its specific cell surface death receptor (Fas). Binding of the ligand results in receptor trimerization, recruitment of multiple pro-caspase-8 molecules to the receptor and activation of caspase-8 at this death inducing signalling complex (DISC) [6]. The mitochondrial or intrinsic pathway is triggered by intracellular stresses which cause mitochondrial membrane depolarization, cytochrome c release, apoptosome formation and pro-caspase-9 activation [7, 8]. When active, caspase-8 and -9 cleave and activate downstream effector caspases, such as pro-caspase-3. Finally, stresses to the ER cause loss of Ca2+ homeostasis and protein aggregation, events sensed by three ER transmembrane sensors; ATF6, Ire1 and PERK [9]. Activation of these sensors launches a coordinated response, called the Unfolded Protein Response (UPR) which attempts to restore ER homeostasis. However, failure of the UPR to restore normal ER function, results in the triggering of apoptosis and cleavage of ER-localized pro-caspase-12 [10].
Members of the Bcl-2 family of proteins are major regulators of mitochondrial cytochrome c release and downstream caspase activation and as such play an important role in the regulation of cardiomyocyte apoptosis [11–13]. The family includes pro-apoptotic (e.g. Bax and Bid) and anti-apoptotic (e.g. Bcl-2 and Bcl-xL) members. Bcl-2 and Bcl-xL localize to the intracellular membranes of the mitochondria, ER and nuclear envelope. Overexpression of anti-apoptotic members of Bcl-2 family of proteins have previously been shown to protect cardiomyocytes from doxorubicin and hypoxia-induced death [11–13].
Apoptosis is a tightly controlled process offering targets for therapeutic intervention against pathophysiological conditions, including cardiovascular diseases [1]. Pathophysiological stresses induce multiple apoptotic pathways with the exact contribution of each to cellular demise unknown. In an attempt to dissect the mechanisms of caspase activation in cardiomyocytes, we examined the effects of two cardiomyocyte-damaging insults, hypoxia and doxorubicin (a chemotherapeutic drug with cardiotoxic effect) on the rat neonatal cardiomyocyte cell line H9c2. We examined the effect of Bcl-xL by adenovirus transduction. We found that both stimuli activated components of the death receptor and the mitochondrial death pathways, with hypoxia also inducing ER stress. These results indicate that despite the multiple death pathways activated by cardiomyocyte-damaging insults, prevention of mitochondrial cytochrome c release is sufficient to protect cardiomyocytes from apoptosis.
Materials and methods
Cell culture and treatments
The embryonic rat heart derived cell line H9c2(2-1) was obtained from ATCC. Cells were maintained in DMEM medium supplemented with 10% foetal bovine serum, 2 mM glutamine, 50 U/ml penicillin and 50 mg/ml streptomycin at 37°C, 5% CO2 in a humidified incubator. H9c2 cells were seeded at 100,000 cells/ml for all experiments. To induce apoptosis, cells were treated with either 0.5 μM doxorubicin or were subjected to hypoxia (O2/N2/CO2, 0.5:94.5:5), using an In Vivo2 200 hypoxic chamber (Ruskinn Technology) for the time periods indicated. The dose of doxorubicin was selected based on its physiological significance [14] and its IC50 as determined by cell viability assays (data not shown). Jurkat cells (ATCC) were cultured in RPMI medium supplemented with 10% foetal bovine serum, 2 mM glutamine, 1 mM sodium pyruvate, 50 U/ml penicillin and 50 mg/ml streptomycin. Jurkat cells were seeded at 500,000 cells/ml 24 hrs prior to treatment. To examine receptor-mediated apoptosis, 2 μg/ml recombinant FasL (Upstate Biotechnology) or 2 μg/ml recombinant Fas:Fc with 1 μg/ml enhancer (Alexis Biochemicals) were administered alone or in combination with hypoxia or doxorubicin. To determine the effect of dexamethasone, cells were pre-treated with 1 μM dexamethasone for 24 hrs prior to exposure to hypoxia or doxorubicin. The glucocorticoid receptor antagonist, RU486 (4 ìM), was added 30 min prior to administration of dexamethasone. All reagents were from Sigma-Aldrich unless otherwise stated.
Microscopy
Cells were seeded in 24-well plates. After treatment, cells were fixed in methanol for 5 min at room temperature, stained with Eosin Y for 4 min, rinsed in tap water and stained in Harris haematoxylin solution for 5 min. Gross morphological features were observed by light microscopy at 400x overall magnification. For Hoechst staining, cells were grown on coverslips. After treatment cells were fixed in 3.7% formaldehyde for 5 min and rinsed in PBS. Hoechst 33342 (100 μg/ml) in 80% glycerol was added to the coverslips which were inverted and sealed onto microscope slides. Hoechst stained nuclei were visualized using fluorescent microscopy at 200x overall magnification.
Western blot analysis
Cells were lysed in buffer containing 20 mM HEPES, pH 7.5, 350 mM NaCl, 1 mM MgCl2, 0.5 mM EDTA, 0.1 mM EGTA, 1% Nonidet P-40, 0.5 mM DTT, 100 μM PMSF, 2 μg/ml pepstatin A, 25 μM ALLN, 2.5 μg/ml aprotinin and 10 μM leupeptin. The protein concentration of the lysates was determined by the Bradford method, using BSA as standard. Cellular proteins were separated by electrophoresis on 8–12% SDS-PAGE and transferred onto nitrocellulose membranes. After blocking in 5% non-fat milk and 0.05% Tween-20 in PBS, blots were incubated with rabbit polyclonal antibodies to caspase-3 (1:1000; Cell Signaling Technologies), caspase-9 (1:1000; Cell Signaling Technologies), Grp78 (1:1000; Stressgen), CHOP (1:1000; Santa Cruz Biotechnology), actin (1:1000; Sigma-Aldrich), mouse monoclonal antibodies to cytochrome c(1:1000; BD Pharmingen, 7H8.2C12), Bcl-xL (1:200; Santa Cruz Biotechnology, H-5) and rat monoclonal antibody to caspase-12 (1:1000; Sigma-Aldrich, 14F7). For detection the appropriate horseradish peroxidase (HRP) conjugated goat secondary antibodies (Pierce) were used at a 1:5000 dilution. Protein bands were visualized with Super Signal Ultra Chemiluminescent Substrate (Pierce) on X-ray film (Agfa).
Caspase activity assay
Cells were harvested and pelleted by centrifugation at 350 x g. After washing in PBS, cell pellets were re-suspended in 50 μl of PBS and 25 μl was transferred to duplicate wells of a microtitre plate and snap-frozen by floating on liquid nitrogen. To initiate the reaction, 50 μM of the caspase substrate DEVD-AMC (Peptide Institute Inc.) in assay buffer (100 mM HEPES, pH 7.5, 10% sucrose, 0.1% CHAPS, 5 mM DTT and 0.0001% Igepal-630, pH 7.25) was added to cell lysates. Liberated free AMC was measured by a Wallac Victor 1420 Multilabel counter (Perkin Elmer Life Sciences) using 355 nm excitation and 460 nm emission wavelengths at 37 °C at 60 sec intervals for 25 cycles. The data was analysed by linear regression and enzyme activity was expressed as nmoles AMC released/min/mg total cellular protein.
Preparation of cytosolic- and mitochondrial-rich fractions
To separate the cytosolic and mitochondrial-rich fractions, cells were washed once in ice cold PBS and lysed in 100 μl of buffer (250 mM Sucrose, 70 mM KCl, 0.5 mM DTT, 100 μM PMSF, 2 μg/ml pepstatin A, 25 μM ALLN, 2.5 μg/ml aprotinin and 10 μM leupeptin in PBS) containing 200 μg/ml digitonin (Calbiochem) on ice for 5 min. The cell suspension was centrifuged at 20,000 x g for 5 min. The supernatant was kept as the cytosolic-rich fraction whilst the pellet was resuspended in whole cell lysis buffer to form the mitochondrial-rich fraction.
RNA extraction and RT-PCR
Total RNA was isolated using Tri reagent (Molecular Research Centre Inc.) according to the manufacturer's instructions. Reverse transcription (RT) was carried out with 2 μg RNA and Oligo dT (12–18) (Invitrogen) using 20 U AMV Reverse Transcriptase (Sigma-Aldrich). The cDNA product was subjected to 25-35 cycles of PCR using primers specific for FasL (forward: CTGCACTACTGGCCAGA; reverse: GCCTCTGCATTGCCACA), Fas receptor (forward: CGAATGCAAGGGACTGA; reverse: AAGTCCACACGAGGTGCA), cFLIP (forward: TTCGCTGCCTTAGCTGA; reverse: TCTTGCTTCTTGGCTGGA), XBP-1 unspliced (forward: CAGACTACGTGCGCCTCTGC; reverse: CTTCTGGGTAGACCTCTGGG), XBP-1 spliced (forward: TCTGCTGAGTCCGCAGCAGG; reverse: CTCTAAGACTAGAGGCTTGG), GADD34 (forward: TTTCTAGGCCAGACACATGG; reverse: TGTTCCTTTTTCCTCCGTGG) and ATF4 (forward: CCGAGATGAGCTTCCTGA; reverse: CTCCTTGCCGGTGTCTGA). GAPDH (forward: ACCACAGTCCATGCCATC; reverse: TCCACCACCTGTTGCTG) was used as an endogenous standard unchanged between treatments.
Annexin V assay
Externalization of phosphatidylserine (PS) to the outer leaflet of the plasma membrane of apoptotic cells was assessed with annexin V-FITC (IQ Corporation) as described earlier [15]. Briefly, cells were collected by centrifugation at 350 x g, washed once in ice-cold calcium buffer (10 mM HEPES/NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) and incubated with annexin V-FITC for 15 min on ice. A wash step in calcium buffer was carried out prior to acquisition on a FACSCalibur flow cytometer (Becton Dickinson).
Adenoviral constructs and transduction
The Bcl-xL construct was generated and the virus was propagated and purified as described before [16–18]. AdNull was used as an empty vector control. H9c2 cells were transduced with adenovirus diluted in DMEM at a multiplicity of infection (MOI) of 1000, for 1 hr. After 1 hr the virus solution was removed and the cells were cultured for an additional 24 hrs prior to exposure to doxorubicin or hypoxia. At this MOI, no cytotoxic effects were observed.
Statistical analysis
Differences in annexin V staining and DEVDase activity between the treatment groups were assessed using oneway ANOVA with pairwise comparisons performed using Tukey's post hoc test with a significance of P < 0.05. All statistics were carried out using SPSS 14.0 for Windows.
Results
Hypoxia and doxorubicin induce apoptosis in H9c2 cells
To confirm treatment with doxorubicin or exposure to hypoxia triggers H9c2 cell apoptosis, caspase activation and cellular morphology were examined. Cells were subjected to hypoxia (0.5% O2, 5% CO2 and 94.5% N2) or treated with 0.5 μM doxorubicin following which cellular morphology was examined. Nuclear condensation and fragmentation, features typical of apoptosis, were evident following both treatments (Fig. 1A). These morphological changes were accompanied by processing of pro-caspase-3 as demonstrated by Western blot analysis (Fig. 1B). At 36 hrs hypoxia, cleaved caspase-3 was readily detected, while doxorubicin-induced cleavage of procaspase-3 was distinguished as early as 12 hrs. These data confirm that exposure of H9c2 cells to hypoxia or doxorubicin induces classical features of apoptosis.
1.
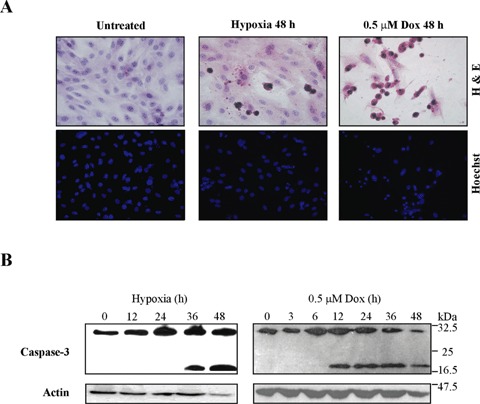
Hypoxia and doxorubicin induce H9c2 cell apoptosis. H9c2 cells were exposed to hypoxia or treated with 0.5 μM doxorubicin for the times indicated. (A) Gross morphological features were assessed by light microscopy of H&E-stained cells whilst nuclear alterations were examined by fluorescence microscopy of Hoechst-stained nuclei. (B) Caspase-3 processing in response to hypoxia and doxorubicin in whole cell lysates (30 μg), analysed by Western blotting.
Fas/FasL pathway is not critical for hypoxia- or doxorubicin-induced H9c2 apoptosis
To investigate whether hypoxia- or doxorubicin-induced apoptosis involved the Fas/FasL pathway, H9c2 cells were subjected to hypoxia or doxorubicin for up to 24 hrs and the mRNA levels of FasL, Fas and cFLIP monitored (Fig. 2A). H9c2 cells subjected to hypoxia and doxorubicin exhibited increased expression of FasL and Fas mRNA while the expression of cFLIP, the endogenous inhibitor of the pathway, remained unaltered. Induced expression of Fas occurred as early as 3 hrs for both treatments, whereas FasL expression was detectable after 3 and 24 hrs for hypoxia and doxorubicin, respectively. To determine if the induced mRNA expression of Fas and FasL could augment H9c2 cell susceptibility to Fas-mediated apoptosis, recombinant FasL was added to cells 3 hrs into hypoxia and doxorubicin treatments and cell death was assessed by measuring externalization of phosphatidylserine (PS) (Fig. 2B). Addition of FasL to doxorubicin treated cells did not alter the extent of PS externalization. However, addition of exogenous FasL to hypoxic cells did increase PS externalization and thus cell death. To determine whether Fas-mediated apoptosis was required for hypoxia- and doxorubicin-induced apoptosis, recombinant Fas:Fc was added to cells 3 hrs into treatments to neutralize endogenously present FasL. Fas:Fc did not decrease cell death in response to either hypoxia or doxorubicin as measured by Annexin V assay. In contrast, FasL-induced cell death in the Fas-sensitive Jurkat cells was completely blocked by Fas:Fc (P< 0.05) (Fig. 2C). These data demonstrate that hypoxia but not doxorubicin increases H9c2 cell sensitivity to FasL but that neither doxorubicin- nor hypoxia-induced apoptosis is dependent on Fas/FasL signalling events.
2.
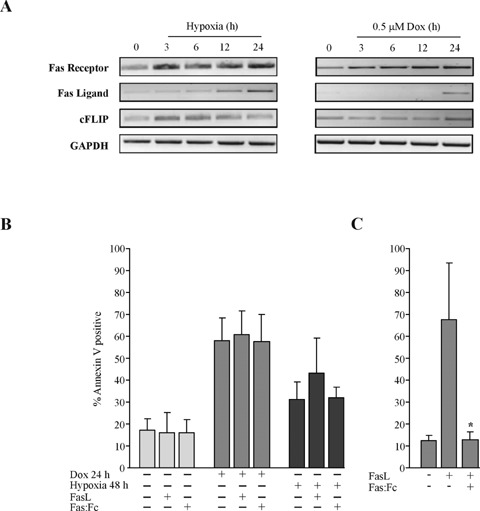
Expression and activity of death receptor pathway mediators in response to hypoxia and doxorubicin. (A) Expression of Fas, Fas ligand (FasL) and cFLIP mRNA during exposure to hypoxia or doxorubicin. Total RNA was isolated at the time-points indicated and examined for levels by RT-PCR. GAPDH was used as an endogenous loading control as it remained unaltered by treatment. (B) Activity and requirement of the Fas death receptor pathway for hypoxia and doxorubicin-induced cardiomyocyte death. H9c2 cells were exposed to hypoxia or 0.5 μM doxorubicin for 24 hrs, after 3 hrs of treatment, FasL (2 μg/ml) and/or Fas:Fc (2 μg/ml) plus enhancer (1 μg/ml) were added. Cell death was measured by annexin V staining and analysed by flow cytometry. (C) Fas sensitive Jurkat cells were used as positive control to test the functionality of recombinant FasL and neutralizing Fas:Fc. Jurkat cells were treated with 2 μg/ml recombinant FasL with or without 2 μg/ml recombinant Fas:Fc with 1 μg/ml Fas:Fc enhancer for 24 hrs. The values displayed are the means ± S.D. of three independent experiments, *P < 0.05.
Hypoxia, but not doxorubicin, initiates the ER stress response in H9c2 cells
To assess the activation of the ER stress response during hypoxia and doxorubicin treatment, we examined the activation of specific targets of the three ER stress sensors, ATF6, Ire1 and PERK. Activation of PERK was monitored by the induction of two of its targets, GADD34 and ATF4. Activation of ATF6 was assessed by examining the induction of its specific target XBP-1, while activation of the RNase and kinase Ire1 was followed by examining the splicing of XBP-1 [19]. RT-PCR analysis revealed that while doxorubicin had no effect on the transcription of UPR-mediators, hypoxia induced the mRNA expression of XBP-1 (unspliced and spliced), GADD34 and ATF4 (Fig. 3A). Examination of ER stress markers, Grp78 and CHOP, by Western blot analysis confirmed an upregulation in Grp78 expression before 12 hrs hypoxia with CHOP induction a much later event occurring at 36 hrs hypoxia (Fig. 3B). ER resident pro-caspase-12 was processed following 24 hrs hypoxia. Doxorubicin treatment failed to alter the expression of Grp78 or CHOP and did not lead to processing of pro-caspase-12. These data may suggest that while hypoxia compromises ER function and activates an ER stress response, doxorubicin does not interfere with ER events and does not exploit the ER stress-signalling pathway to activate H9c2 cell apoptosis.
3.
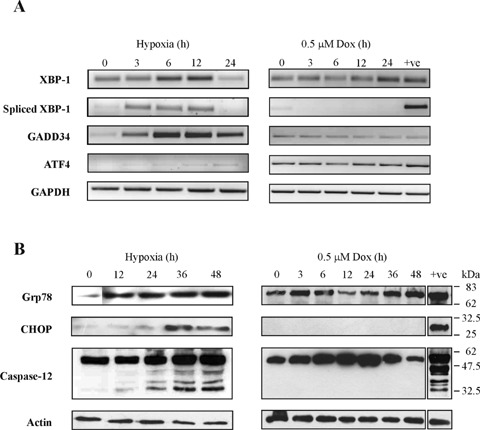
Hypoxia, but not doxorubicin, initiates the ER stress response in H9c2 cells. (A) mRNA expression levels of the unfolded protein response (UPR) mediators XBP-1, GADD34 and ATF4 assessed by RT-PCR of total RNA samples isolated from cells exposed to hypoxia or 0.5 μM doxorubicin for the times indicated. (B) Alterations in the expression of ER stress associated proteins analysed by Western blotting using protein harvested after exposure to hypoxia or 0.5 μM doxorubicin for up to 48 hrs. Whole cell lysates were analysed for levels of Grp78 (10 μg) and CHOP (30 μg) and processing of pro-caspase-12 (30 μg). ER stress inducer thapsigargin (2 μM; 4 hrs for mRNA, 36 hrs for protein) was used as a positive control (+ve). displayed are the means ± S.D. of three independent experiments, *P < 0.05.
Hypoxia and doxorubicin initiate mitochondria-mediated apoptosis
The effect of hypoxia and doxorubicin on mitochondrial integrity was studied by examining release of cytochrome cinto the cytosol. Western blot analysis demonstrated a time-dependent accumulation of cytochrome c in the cytosolic-rich fractions (Fig. 4A). The processing of pro-caspase-9 was detectable between 24 and 36 hrs hypoxia (Fig. 4B) which correlates with the accumulation of cytochrome c at 24 hrs. Accumulation of cytochrome c together with the processing of pro-caspase-9 following doxorubicin treatment was detectable between 6 and 12 hrs. These data indicate that both hypoxia and doxorubicin utilize mitochondrial-mediated signalling to trigger apoptosis in cardiomyocytes.
4.
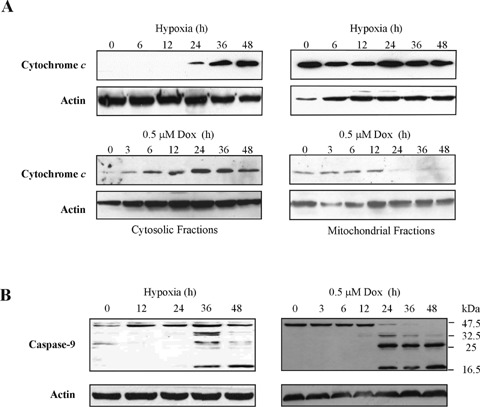
Hypoxia and doxorubicin induce mitochondrial cytochrome c release and pro-caspase-9 processing. (A) Cytosolic accumulation of cytochrome c during hypoxia and doxorubicin treatment. After treatment, cells were harvested at the time-points indicated and separated into cytosolic- and mitochondrial-rich fractions by centrifugation. The cytosolic-rich (left-hand panel) and mitochondrial-rich (righthand panel) fractions (10 μg) were analysed by Western blotting using anti-cytochrome c antibody. (B) Pro-caspase-9 processing during hypoxia (left-hand panel) and doxorubicin (righthand panel) treatment. Whole cell lysates (30 μg) were analysed by Western blotting for cleavage of pro-caspase-9 using anti-caspase-9 antibody. Actin levels in samples were analysed to serve as loading control.
Bcl-xL reduces hypoxia- and doxorubicin-induced H9c2 cell apoptosis
To test whether the mitochondrial death pathway is an essential component of hypoxia- and doxorubicin-induced cardiomyocyte death, the effect of antiapoptotic Bcl-xL was studied. Since corticosteroids are cytoprotective in a number of cell models through induction of Bcl-xL[20,21], we tested the effect of dexamethasone on hypoxia and doxorubicin-induced apoptosis in H9c2 cells. Pre-treatment of cells with 1 μM dexamethasone increased Bcl-xL protein expression, an effect reversed by glucocorticoid receptor antagonist RU486 and partially reversed by doxorubicin treatment (Fig. 5A and B). Dexamethasone pre-treatment reduced hypoxia and doxorubicininduced caspase-3 activity significantly (P< 0.05) (Fig. 5C). To achieve higher Bcl-xL protein expression and determine the protective effect of such increased expression, a Bcl-xL-expressing adenovirus construct was generated (AdBcl-xL). Cells transduced with AdBcl-xL (Fig. 5A) were exposed to hypoxia or doxorubicin and cytochrome crelease was analysed in cytosolic-rich fractions. The subcellular fractionation revealed that Bcl-xL reduced release of cytochrome c induced by both hypoxia and doxorubicin (Fig. 6B). Bcl-xL overexpressing cells also demonstrated reduced pro-caspase-3 processing (Fig. 6B). Furthermore, AdBcl-xL transduced cultures displayed less pyknotic nuclei then untreated or AdNull transduced cells after exposure to 48 hrs hypoxia or 24 hrs 0.5 μM doxorubicin (Fig. 6C). These data show that hypoxia and doxorubicin require and depend on mitochondrial events to induce cardiomyocyte apoptosis and that Bcl-xL can maintain cellular viability against both stimuli.
5.
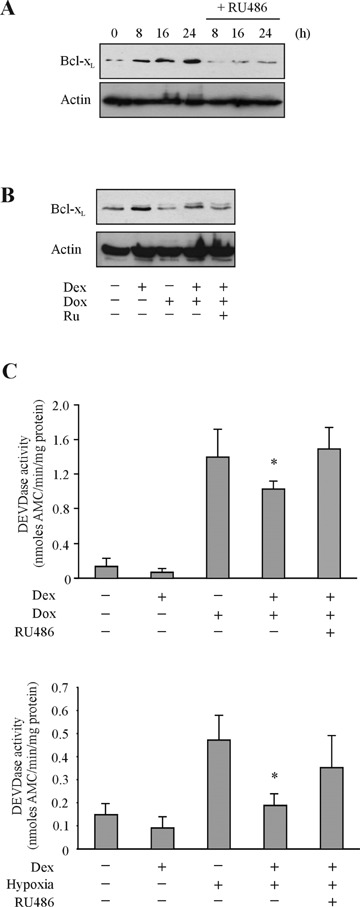
Dexamethasone increases Bcl-xL protein expression and reduces hypoxia- and doxorubicin-induced caspase-3-like activity. (A) Bcl-xL protein expression was examined by Western blotting following 1 μM dexamethasone treatment (0–24 hrs) with and without RU486 pre-treatment. (B) Bcl-xL protein expression was also examined following treatment with dexamethasone (1 μM; 24 hrs) or doxorubicin (0.5 μM; 24 hrs) separately and in combination with RU486. (C) H9c2 cells were exposed to doxorubicin (upper graph) or hypoxia (lower graph) with or without pre-treatment with dexamethasone (1 μM;for 24 hrs) and RU486 (4 μM; 30 min prior to dexamethasone pre-treatment). Caspase-3-like activity was detected by monitoring the cleavage of synthetic substrate DEVD-AMC by active caspases. Results are expressed as nmoles AMC released in 1 min by 1 mg total cellular protein and plotted as mean values ± S.D. of three independent experiments, *P< 0.05.
6.
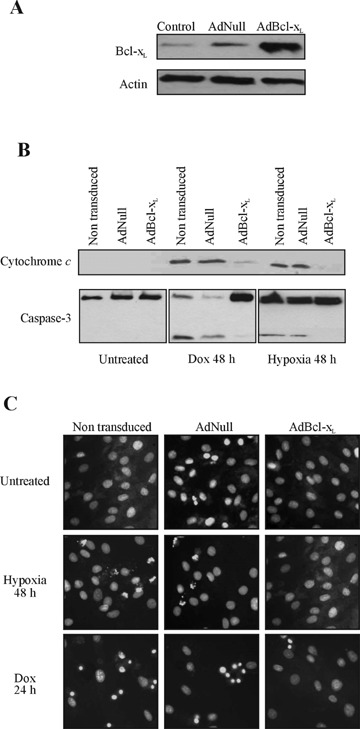
Adenoviral-mediated overexpression of Bcl-xL reduces hypoxiaand doxorubicin-induced H9c2 cell apoptosis. Cells were transduced with AdBcl-xL or AdNull at an MOI of 1000 for 1 hr and cultured for 24 hrs before treatment with hypoxia and doxorubicin (0.5 μM) for 48 hrs. (A) Overexpression of Bcl-xL confirmed by Western blotting in samples 24 hrs after transduction. (B) Hypoxia- and doxorubicin-induced accumulation of cytochrome c in the cytosol is prevented by overexpression of Bcl-xL. Subcellular fractionation was performed by differential centrifugation of cell lysates exposed to hypoxia or doxorubicin with the cytosolic-rich fractions analysed for presence of cytochrome c by Western blotting. Bcl-xL prevents pro-caspase-3 processing induced by hypoxia and doxorubicin. Whole cell lysates of nontransduced, AdNull- and AdBcl-xLtransduced cells, untreated and treated, were analysed for pro-caspase-3 cleavage by Western blotting. (C) Non-transduced, AdNull and AdBcl-xL infected cells were exposed to hypoxia for 48 hrs or doxorubicin for 24 hrs and stained with Hoechst to analyse nuclear fragmentation. Overexpression of Bcl-xL protects cells from hypoxia- and doxorubicin-induced apoptosis.
Discussion
In the developed world, cardiovascular disease is a most prominent killer. Apoptosis of terminally differentiated cardiomyocytes has been recognized as essential to the pathogenesis of cardiovascular diseases which include myocardial infarction, coronary heart disease, cardiomyopathy and heart failure. Although apoptosis is one of the main contributors to myocardium loss and dysfunction, due to its highly regulated and sequential activation processes it offers targets for therapeutic interventions.
Studies have shown that hypoxia and doxorubicin induce cardiomyocyte apoptosis [22–25]. In this study, these pathologically relevant insults were selected to determine the role of the different caspase activation pathways to the cardiomyocyte apoptosis induced. We, like others, report that both hypoxia and doxorubicin activate caspases to induce H9c2 cell apoptosis. Despite the numerous reports on the induction of Fas and/or FasL in a wide range of myocardial pathologies, the relevance of death receptor-mediated apoptosis in cardiomyocytes is still unclear. In agreement with other studies we found that doxorubicin induced expression of Fas and FasL mRNA. However, the induction of Fas and FasL expression was insufficient to activate the pathway as the doxorubicin-treated cells remained unresponsive to recombinant FasL. Similarly, hypoxic cells also demonstrated induced expression of Fas and FasL [26,27]. In contrast to doxorubicin, the addition of recombinant FasL enhanced hypoxia-induced apoptosis, although this increase was marginal and statistically insignificant. In addition to induction of Fas and/or FasL, decreased cFLIP expression has also been implicated in sensitization of cardiomyocytes to toxic stimuli [27–29]. However, in our hands neither hypoxia nor doxorubicin treatments reduced cFLIP mRNA levels which may account for the lack of sensitivity to externally added FasL. Furthermore, the Fas/FasL pathway was not critical for either hypoxia- or doxorubicin-induced H9c2 cell apoptosis, as neutralization of endogenous FasL by recombinant Fas:Fc failed to reduce hypoxia- or doxorubicin-induced cell death.
Hypoxia also activated ER stress-induced apoptosis. Ischaemia has been shown to trigger an ER stress response and if prolonged activates the ER stress death pathway [24]. Our results indicate that one component of the ischaemic insult, the lack of oxygen or hypoxia, is sufficient to activate ER stress. Although the apoptotic signaling cascade initiated by ER stress is not fully understood, studies demonstrate a possible role for caspase-12 and mitochondrial cytochrome c released [30]. Therefore, hypoxia-induced ER stress may rely on mitochondria-mediated signaling to execute apoptosis. Doxorubicin did not activate an ER stress response and this coupled with the early cytochrome crelease observed suggests doxorubicin targets mitochondria differently from hypoxia, potentially through excessive reactive oxygen species generated [31]. Though dissimilar insults, we illustrate that ultimately, both hypoxia and doxorubicin converge on mitochondria and use this organelle to provoke cardiomyocyte apoptosis. Therefore, maintaining outer mitochondrial membrane integrity is important to preserve cellular viability.
Mitochondria-mediated apoptosis is modulated by the Bcl-2 family of proteins. We and others have shown that corticosteroids such as dexamethasone inhibit apoptosis through the increased expression of Bcl-xL[20, 21]. Dexamethasone pre-treatment attenuated hypoxia and doxorubicin induced caspase-3 processing. However, as dexamethasone has other downstream targets, AdBcl-xL was utilized to examine whether increased expression of Bcl-xL was sufficient to protect H9c2 cells. Cells transduced with AdBcl-xL demonstrated reduced apoptosis following exposure to hypoxia or doxorubicin confirming the importance of mitochondrial homeostasis in cardiomyocyte vitality and demise. These results point out that despite the multiple death pathways activated by cardiomyocyte-damaging insults, prevention of mitochondrial cytochrome crelease is sufficient to protect cardiomyocytes. Examination of these events in primary cardiomyocytes and assessment of their contractile function post-rescue warrants study as this would deduce the feasibility of such cardioprotective strategies in vivo.
Acknowledgments
This work was supported in part by grants from the Millennium Research Fund of NUI Galway, the Higher Education Authority of Ireland and SFI grant to REMEDI. Janice Reeve is supported by an EMBARK scholarship from IRCSET. Afshin Samali is an SFI Principal Investigator.
References
- 1.Reeve JL, Duffy AM, O’Brien T, Samali A. Don't lose heart–therapeutic value of apoptosis prevention in the treatment of cardiovascular disease. J Cell Mol Med. 2005;9:609–22. doi: 10.1111/j.1582-4934.2005.tb00492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gill C, Mestril R, Samali A. Losing heart: the role of apoptosis in heart disease–a novel therapeutic target? FASEB J. 2002;16:135–46. doi: 10.1096/fj.01-0629com. [DOI] [PubMed] [Google Scholar]
- 3.Logue SE, Gustafsson AB, Samali A, Gottlieb RA. Ischaemia/reperfusion injury at the intersection with cell death. J Mol Cell Cardiol. 2005;38:21–33. doi: 10.1016/j.yjmcc.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 4.Fischer U, Janicke RU, Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 2003;10:76–100. doi: 10.1038/sj.cdd.4401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Samali A, Zhivotovsky B, Jones D, Nagata S, Orrenius S. Apoptosis: cell death defined by caspase activation. Cell Death Differ. 1999;6:495–6. doi: 10.1038/sj.cdd.4400520. [DOI] [PubMed] [Google Scholar]
- 6.Muzio M, Stockwell BR, Stennicke HR, Salvesen GS, Dixit VM. An induced proximity model for caspase- 8 activation. J Biol Chem. 1998;273:2926–30. doi: 10.1074/jbc.273.5.2926. [DOI] [PubMed] [Google Scholar]
- 7.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 8.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–13. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 9.Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–33. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 10.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 11.Sugioka R, Shimizu S, Funatsu T, Tamagawa H, Sawa Y, Kawakami T, Tsujimoto Y. BH4-domain peptide from Bcl-xL exerts anti-apoptotic activity in vivo. Oncogene. 2003;22:8432–40. doi: 10.1038/sj.onc.1207180. [DOI] [PubMed] [Google Scholar]
- 12.Kunisada K, Tone E, Negoro S, Nakaoka Y, Oshima Y, Osugi T, Funamoto M, Izumi M, Fujio Y, Hirota H, Yamauchi-Takihara K. Bcl-xl reduces doxorubicininduced myocardial damage but fails to control cardiac gene downregulation. Cardiovasc Res. 2002;53:936–43. doi: 10.1016/s0008-6363(01)00506-5. [DOI] [PubMed] [Google Scholar]
- 13.Chen Z, Chua CC, Ho YS, Hamdy RC, Chua BH. Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am J Physiol Heart Circ Physiol. 2001;280:H2313–20. doi: 10.1152/ajpheart.2001.280.5.H2313. [DOI] [PubMed] [Google Scholar]
- 14.Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol. 1999;57:727–41. doi: 10.1016/s0006-2952(98)00307-4. [DOI] [PubMed] [Google Scholar]
- 15.Concannon CG, FitzGerald U, Holmberg CI, Szegezdi E, Sistonen L, Samali A. CD95-mediated alteration in Hsp70 levels is dependent on protein stabilization. Cell Stress Chaperones. 2005;10:59–65. doi: 10.1379/CSC-69R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ritter T, Vogt K, Rieck P, Schilling-Schon A, Kolls J, Hartmann C, Volk HD, Pleyer U. Adenovirus-mediated gene transfer of interleukin-4 to corneal endothelial cells and organ cultured corneas leads to high IL-4 expression. Exp Eye Res. 1999;69:563–8. doi: 10.1006/exer.1999.0731. [DOI] [PubMed] [Google Scholar]
- 17.Fallaux FJ, Kranenburg O, Cramer SJ, Houweling A, Van Ormondt H, Hoeben RC, Van Der Eb AJ. Characterization of 911: a new helper cell line for the titration and propagation of early region 1-deleted adenoviral vectors. Hum Gene Ther. 1996;7:215–22. doi: 10.1089/hum.1996.7.2-215. [DOI] [PubMed] [Google Scholar]
- 18.Graham FL, Prevec L. Manipulation of adenovirus vectors. In: Murray EJ, Walker JM, editors. Methods in molecular biology. Gene transfer and expression protocols. Clifton, NJ: Humana Press; 1991. pp. 109–27. [DOI] [PubMed] [Google Scholar]
- 19.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–5. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen QM, Alexander D, Sun H, Xie L, Lin Y, Terrand J, Morrissy S, Purdom S. Corticosteroids inhibit cell death induced by doxorubicin in cardiomyocytes: induction of antiapoptosis, antioxidant, and detoxification genes. Mol Pharmacol. 2005;67:1861–73. doi: 10.1124/mol.104.003814. [DOI] [PubMed] [Google Scholar]
- 21.Ni Chonghaile T, Concannon CG, Szegezdi E, Gorman AM, Samali A. Dexamethasone inhibits apoptosis in C6 glioma cells through increased expression of Bcl-X(L) Apoptosis. 2006;11:1247–55. doi: 10.1007/s10495-006-7233-1. [DOI] [PubMed] [Google Scholar]
- 22.Arola OJ, Saraste A, Pulkki K, Kallajoki M, Parvinen M, Voipio-Pulkki LM. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. 2000;60:1789–92. [PubMed] [Google Scholar]
- 23.O’Mahoney ME, Logue S, Szegezdi E, Stenson-Cox C, Fitzgerald U, Samali A. Hypoxia and ischaemia induce nuclear condensation and caspase activation in cardiomyocytes. Ann N Y Acad Sci. 2003;1010:728–32. doi: 10.1196/annals.1299.131. [DOI] [PubMed] [Google Scholar]
- 24.Szegezdi E, Duffy A, O’Mahoney ME, Logue SE, Mylotte LA, O’Brien T, Samali A. ER stress contributes to ischaemia-induced cardiomyocyte apoptosis. Biochem Biophys Res Commun. 2006;349:1406–11. doi: 10.1016/j.bbrc.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 25.Wu S, Ko YS, Teng MS, Ko YL, Hsu LA, Hsueh C, Chou YY, Liew CC, Lee YS. Adriamycin-induced cardiomyocyte and endothelial cell apoptosis: in vitro and in vivo studies. J Mol Cell Cardiol. 2002;34:1595–607. doi: 10.1006/jmcc.2002.2110. [DOI] [PubMed] [Google Scholar]
- 26.Tanaka M, Ito H, Adachi S, Akimoto H, Nishikawa T, Kasajima T, Marumo F, Hiroe M. Hypoxia induces apoptosis with enhanced expression of Fas antigen messenger RNA in cultured neonatal rat cardiomyocytes. Circ Res. 1994;75:426–33. doi: 10.1161/01.res.75.3.426. [DOI] [PubMed] [Google Scholar]
- 27.Yaniv G, Shilkrut M, Lotan R, Berke G, Larisch S, Binah O. Hypoxia predisposes neonatal rat ventricular myocytes to apoptosis induced by activation of the Fas (CD95/Apo-1) receptor: Fas activation and apoptosis in hypoxic myocytes. Cardiovasc Res. 2002;54:611–23. doi: 10.1016/s0008-6363(02)00264-x. [DOI] [PubMed] [Google Scholar]
- 28.Nitobe J, Yamaguchi S, Okuyama M, Nozaki N, Sata M, Miyamoto T, Takeishi Y, Kubota I, Tomoike H. Reactive oxygen species regulate FLICE inhibitory protein (FLIP) and susceptibility to Fas-mediated apoptosis in cardiac myocytes. Cardiovasc Res. 2003;57:119–28. doi: 10.1016/s0008-6363(02)00646-6. [DOI] [PubMed] [Google Scholar]
- 29.Yamaoka M, Yamaguchi S, Suzuki T, Okuyama M, Nitobe J, Nakamura N, Mitsui Y, Tomoike H. Apoptosis in rat cardiac myocytes induced by Fas ligand: priming for Fas-mediated apoptosis with doxorubicin. J Mol Cell Cardiol. 2000;32:881–9. doi: 10.1006/jmcc.2000.1132. [DOI] [PubMed] [Google Scholar]
- 30.Di Sano F, Ferraro E, Tufi R, Achsel T, Piacentini M, Cecconi F. Endoplasmic reticulum stress induces apoptosis by an apoptosome-dependent but caspase 12-independent mechanism. J Biol Chem. 2006;281:2693–700. doi: 10.1074/jbc.M509110200. [DOI] [PubMed] [Google Scholar]
- 31.Spallarossa P, Garibaldi S, Altieri P, Fabbi P, Manca V, Nasti S, Rossettin P, Ghigliotti G, Ballestrero A, Patrone F, Barsotti A, Brunelli C. Carvedilol prevents doxorubicin-induced free radical release and apoptosis in cardiomyocytes in vitro. J Mol Cell Cardiol. 2004;37:837–46. doi: 10.1016/j.yjmcc.2004.05.024. [DOI] [PubMed] [Google Scholar]