

Abstract
Gene transfer into human CD34+ haematopoietic progenitor cells (HPC) and multi-potent mesenchymal stromal cells (MSC) is an essential tool for numerous in vitro and in vivo applications including therapeutic strategies, such as tissue engineering and gene therapy. Virus based methods may be efficient, but bear risks like tumorigenesis and activation of immune responses. A safer alternative is non-viral gene transfer, which is considered to be less efficient and accomplished with high cell toxicity. The truncated low affinity nerve growth factor receptor (ÄLNGFR) is a marker gene approved for human in vivo application. Human CD34+ HPC and human MSC were transfected with in vitro-transcribed mRNA for ΔLNGFR using the method of nucleofection. Transfection efficiency and cell viability were compared to plasmid-based nucleofection. Protein expression was assessed using flow cytometry over a time period of 10 days. Nucleofection of CD34+ HPC and MSC with mRNA resulted in significantly higher transfection efficiencies compared to plasmid transfection. Cell differentiation assays were performed after selecting ΔLNGFR positive cells using a fluorescent activating cell sorter. Neither cell differentiation of MSC into chondrocytes, adipocytes and osteoblasts, nor differentiation of HPC into burst forming unit erythroid (BFU-E) colony forming unit-granulocyte, erythrocyte, macrophage and megakaryocyte (CFU-GEMM), and CFU-granulocyte-macrophage (GM) was reduced. mRNA based nucleofection is a powerful, highly efficient and non-toxic approach for transient labelling of human progenitor cells or, via transfection of selective proteins, for transient manipulation of stem cell function. It may be useful to transiently manipulate stem cell characteristics and thus combine principles of gene therapy and tissue engineering.
Keywords: adult stem cells, mRNA transfection, in vivo application, ΔLNGFR
Introduction
Adult progenitor cell therapy for organ repair is a promising area of biomedical research. Gene therapy is also considered as an important avenue towards the development of novel therapies for human disease. Using gene therapy approaches in order to improve the regenerative capacity of adult progenitor cells is conceivable and, possibly, absolutely necessary to achieve successful cellular therapy.
Multi-potent mesenchymal stromal cells (MSC) obviously have a multi-lineage potential and might gain an increasing impact in cellular therapy [1]. For haematopoietic progenitor cells (HPC), the proposed plasticity remains a controversial issue [2–3]. Gene delivery into human progenitor cells may aim at labelling to track cells in vivo or at transient manipulation of stem cell characteristics to improve their regenerative capacity. Conventional gene delivery into human progenitor cells, however, is associated with high risk and a number of ethical concerns. Namely, virus based gene delivery bears the risk of oncogenesis and mutagenesis as well as elicitation of immune responses, whereas non-viral gene delivery is considered to be inefficient [4]. With the emerging role of HPC and MSC as potential gene and cell therapy vehicles in clinical approaches, there is an increasing need for safe, effective, non-viral gene delivery.
In this paper, we continue a series of reports on the development of a highly efficient, non-toxic methodology of gene transfer into human progenitor cells. Very recently, we have described a rapid, specific and efficient transient genetic labelling of HPC with a plasmid coding for the truncated low affinity nerve growth factor receptor (ΔLNGFR) using nucleofection [5]. Importantly, this method is ready for in vivo application in humans now and is currently being tested in a clinical trial. This trial might establish nucleofection as being feasible and safe. If so, mRNA transfection as described herein is based on a method already applicable in humans and may thus be useful in a wide range of applications.
Materials and methods
Mesenchymal stromal cells
Spongiosa from femur or tibia was obtained from three donors during orthopaedic surgery after receiving informed consent. MSC were isolated from trabecular meshwork after adhesion to a positively charged plastic surface (NUNC, Wiesbaden, Germany) for 24 hrs in complete·αMEM (Cambrex, Verviers, Belgium), supplemented with 20% heat inactivated FBS (Invitrogen, Heidelberg, Germany) Early passages (passage 2–4) were used for experiments. Cells were collected after 10–14 days using trypsin (Invitrogen) and plated at densities of 100–300 cells/cm2.
Medium was changed twice a week. To determine viability, the supernatant of cell culture, containing the dead cells, was withdrawn and together with trypsinised MSC centrifugated at 300 g for 5 min. Viability was determined by trypan-blue exclusion and flow cytometry by scatter exclusion. Antibodies used for characterization of MSC included: CD14 (M5E2), CD29 (HUTS-21), CD34 (581), CD44 (G44-26), CD45 (HI30), CD166 (3A6) (all BD Pharmingen, Heidelberg, Germany), CD105 (SN6) (Biozol-Serotec, Eching, Germany), and CD271 (ME20.4) (Santa Cruz Biotechnology, Heidelberg, Germany). 106 cells were incubated with primary antibodies and relative fluorescence intensity of cells was acquired using a BD FACSDiva Software 4.1.2 on a BD FACSAria (BD Immunocytometry Systems, Heidelberg, Germany).
Differentiation assays
For differentiation assays of MSC, 2.75 × 103–104 cells/cm2 were seeded and differentiation was induced. Adipogenic differentiation medium was purchased from Cambrex BioScience, Walkersville, MD, USA. Chondrogenic and osteogenic differentiation media were purchased from Miltenyi, Bergisch-Gladbach, Germany. Cells were fixed in 4% paraformaldehyde and (i) osteoblasts were stained for alkaline phosphatase activity, (ii) adipogenic differentiation was monitored by staining with a saturated Oil RedO solution/ Meyer's Hematoxylin and (iii) chondrogenic differentiation was performed by Alcian Blue staining. Senescent cells were detected by increase of lysosomal β-galactosidase using a senescent cells histochemical staining kit. MSC were seeded at a density of 0.7–3.5 × 103/cm2 at least one day before staining. (All materials from SIGMA, Taufkirchen, Germany, except Alcian Blue/Nuclear Fast Red staining kit (Dako, Hamburg, Germany).
To evaluate differentiation capacity of HPC, methylcellulose assays were performed as previously described [5]. Briefly, assays using non-, mock- and ΔLNGFR –transfected CD34+ HPC were examined in burst forming unit (BFU) and colony forming unit (CFU). For evaluation of BFU and CFU - granulocyte, erythrocyte, macrophage and megakaryocyte (GEMM) assays, 0.05–0.2 × 105/ml viable cells were plated in methylcellulose medium and 1000 mU/ml rh-Epo, 20 ng/ml rh IL-3 and 100 ng/ml rh-SCF were added, while CFUGM were evaluated using 2 ng/ml GM-colony stimulating factor (CSF), 20 ng/ml rh IL-3 and 100 ng/ml rh-SCF. Colonies were assessed 14 days after plating.
Vector construction
The ΔLNGFR vector was generated by cloning the extracellular and transmembrane domain of the cDNA of the human LNGFR gene (ΔLNGFR) into the eucaryotic pVAX1 expression vector (Invitrogen, Carlsbad, CA, USA). Briefly, the ΔLNGFR 834bp fragment was amplified by polymerase chain reaction (template: cDNA human brain obtained from human brain tissue; Clontech, Palo Alto, CA, USA) with the forward primer 5′-gcgatgggggcaggtgccac-3′ and the reverse primer 5′-ctgtcagcagctgttccacctc-3′, containing an artificially introduced stop codon behind the transmembrane domain of ΔLNGFR.
The pGEM4Z-EGFP-A64 vector was kindly provided by Dr Eli Gilboa, Duke University Medical Center, Durham, NC, USA [6].
In vitro transcription
SpeI-linearized EGFP plasmid and XhoI-linearized ΔLNGFR plasmid were purified using the nucleotid removal kit (Qiagen, Hilden, Germany) and were used as DNA templates for the in vitro transcription reaction. Transcription was conducted using the T7 Opti mRNA transcription kit (Cure Vac GmbH, Tuebingen, Germany). Purification of mRNA was performed by DNaseI digestion. To add a poly-A tail to the mRNA, the Poly(A) Tailing Kit (Ambion, Huntingdon, UK) was used. The mRNAs were finally precipitated by LiCl. The mRNA concentration was assayed by spectrophotometric analysis at OD260.
Nucleofection
CD34+ HPC and MSC were pelleted and re-suspended in human CD34 Cell Nucleofector™ Solution (Amaxa GmbH, Köln, Germany) at 2–3 × 106 or 5 × 105 cells per 100 μl, respectively. Cells were nucleofected with 5 μg mRNA or 2 μg plasmid DNA using program U-08 (for HPC) or C-17 (for MSC) of the nucleofector device (Amaxa GmbH). Mock transfected cells, nucleofected without DNA or mRNA, were used as negative control. After nucleofection, cells were immediately mixed with 500 μl pre-warmed culture medium and transferred into well plates containing pre-warmed medium. Cells were incubated at 37°C over a time period of 10 days. Three independent experiments using samples of three different donors were performed.
Fluorescence activated cell sorting
Non-transfected and mock-transfected cells, as well as cells transfected with ΔLNGFR mRNA were stained with mouse monoclonal anti-NGFR-antibody (Me20.4, Santa Cruz, USA). Transfected living cells (passage 4) were enriched to a purity of 99.4% by fluorescence activated cell sorting using a BD FACSAria (100 μm nozzle, sheet pressure 20 psi).
Untransfected and unstained cells were used as controls (sort gate was set as live gate in the forward scatter versus sideward scatter dot blot). Differentiation assays were performed for all populations as described above, except initial cell count was 7.2 × 104–1.6 × 105 for adipogenic differentiation assays and the corresponding controls.
Results
Human CD34+ HPC were isolated from peripheral blood from donors stimulated with granulocytecolony stimulating factor (G-CSF) [5] after informed consent. Human MSC were characterized by immunophenotyping. They lack haematopoietic stem cell marker CD34 as well as markers for peripheral blood cells CD14, CD45 and CD271 (LNGFR), but do express CD29, CD44, CD105, and CD166 [7] (Fig. 1). Osteogenic, adipogenic and chondrogenic differentiation could be induced (Fig. 2). MSC were not senescent (data not shown).
1.
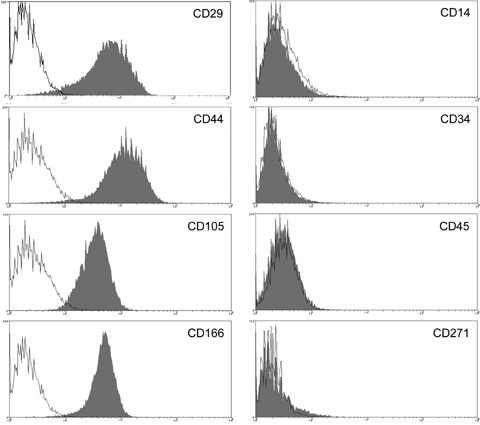
Characterization of undifferentiated MSC by flow cytometry. MSC express CD29, CD44, CD105 (endoglin) and CD166, but are negative for CD14, CD34, CD45, and CD271 (LNGFR).
2.
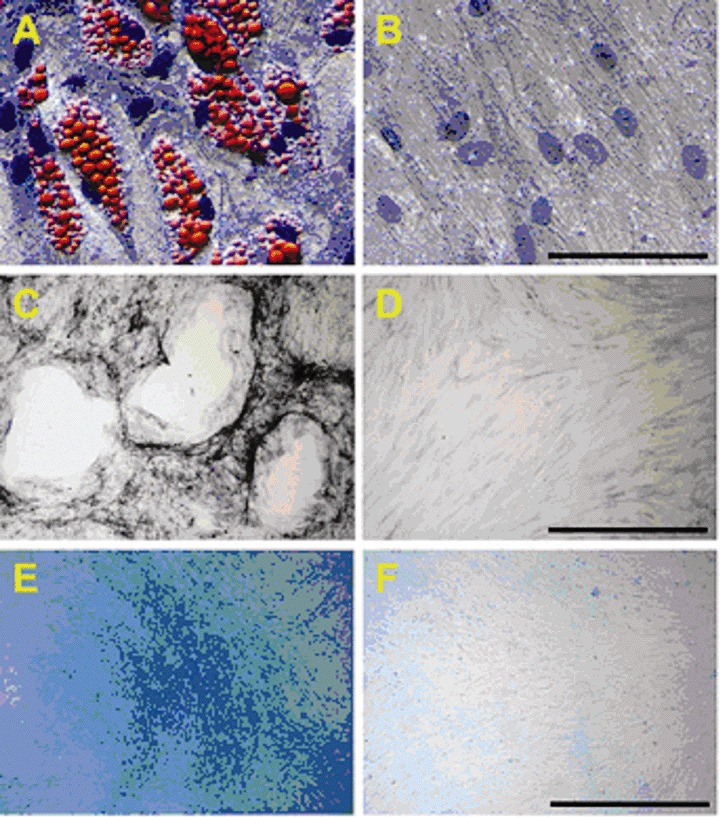
MSC differentiation. Adipogenic (A), osteogenic (C) and chondrogenic (E) differentiation was induced as described above. Negative controls (B, D, F) were set up in ·MEM, supplemented with 20% FBS. Successful differentiation was monitored by histological staining. Initial cell count was 200,000 for adipogenic differentiation assays and 45,000 for osteogenic and adipogenic differentiation assays. Differentiation of one representative MSC preparation is shown. Black bar indicates 1000 μm for adipogenic and 100 μm for chondrogenic and osteogenic differentiation.
Both, MSC and CD34+ HPC were transfected with ΔLNGFR and EGFP using mRNA and plasmid nucleofection. After 24 hrs, mRNA transfection with EGFP resulted in high transfection efficiencies with 89 ± 3% (HPC) or 83 ± 4% (MSC) positive cells, whereas plasmid transfection was markedly less effective (67 ± 11% (HPC), or 42 ± 4 (MSC)). After 3 days, mRNA transfected cells still showed high (81 ± 11% (HPC) or 87 ± 8% (MSC)) marker expression. From day six on, marker expression revealed a reverse pattern with significantly higher expression after plasmid transfection. Antigen expression attenuated intensively with time resulting in less than 30% positive cells after 10 days (Fig. 3A). The high transfection efficiency of mRNA transfection was—in contrast to plasmid transfection—associated with only marginal viability loss (Fig. 3B). Figure 3C shows a representative example of Median Fluorescence Intensity (MFI) of mRNA and plasmid transfected cells to determine transgene expression differences in positive cells 24 hrs after transfection.
3.
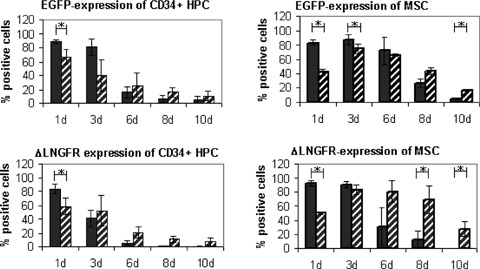
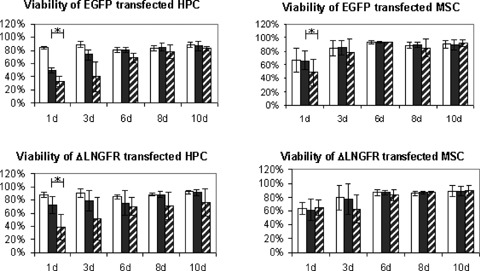
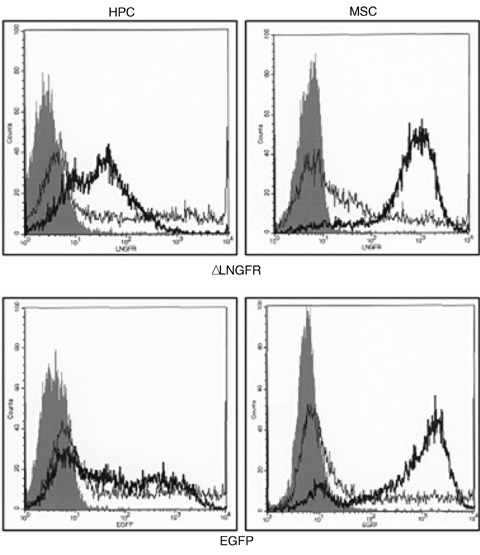
(A) Efficiency of mRNA transfection into human CD34+ HPC and MSC. Cells were transfected with mRNA or plasmid DNA using nucleofection. FACS analysis was performed after 1–10 days. Three independent experiments were performed with cells of three different donors. Upper panel: CD34+ HPC or MSC transfected with EGFP. Lower panel: CD34+ HPC or MSC transfected with ΔLNGFR. 24 hrs transfection efficiency was extremely high. Data are reported ± standard derivation. * significantly different (t-test; P < 0.05). Black bar: mRNA transfected cells, hatched bar: plasmid transfected cells. (B) Viability of human CD34+ HPC and MSC after mRNA transfection. Viability was determined by trypan-blue and flow cytometry (scatter exclusion) 1 to 10 days after nucleofection. Transfection with mRNA is significantly less toxic than with plasmid DNA (*, significantly different; t-test; P < 0.05). Mock transfected cells (nucleofected without DNA or mRNA) were used as negative control. Data are reported ± standard deviation. White bar: control, black bar: mRNA transfected cells, hatched bar: plasmid transfected cells. (C) Representative example of Median Fluorescence Intensity (MFI) of mRNA and plasmid transfected HPC and MSC to determine transgene expression differences in positive cells 24 hrs after transfection. Upper panel: ΔLNGFR; Lower panel: EGFP. Filled grey: control, thick line: mRNA, thin line: plasmid.
To assess whether nucleofection of HPC or MSC altered their capacity to differentiate, mock- and ΔLNGFR mRNA-transfected cells were stained for ΔLNGFR. ΔLNGFR-negative or -positive population was enriched by fluorescence activated cell sorting. Non-transfected, non-stained and stained cells were investigated. Results depicted in Figure 4 show that the differentiation potential is neither influenced by cell sorting nor by nucleofection because, in all assays, MSC differentiate into chondrocytes, osteoblasts and adipocytes (Fig. 4A), whereas HPC differentiate into BFUs, CFU-GEMMs and CFU-GMs (Fig. 4B).
4.
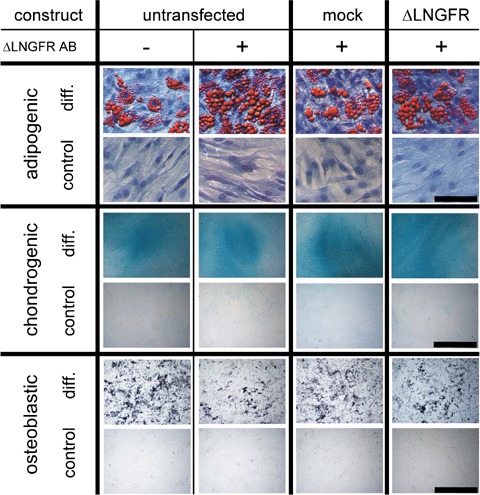
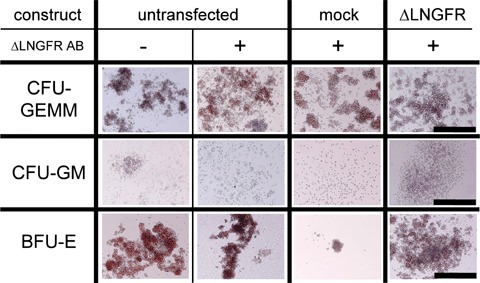
(A) MSC Differentiation potency is not altered by nucleofection. Adipogenic, chondrogenic and osteoblastic differentiation was induced after fluorescence-activated cell sorting of untransfected, mock-transfected and with mRNA of ΔLNGFR-transfected MSC. Control assays were performed in αMEM supplemented with 20% FBS. Black bar indicates 1000 μm for adipogenic and 100 μm for chondrogenic and osteogenic differentiation. (B) Hematopoietic cell differentiation of human CD34+ HPC. Methylcellulose assays of untreated, mock-, and with mRNA of ΔLNGFR-transfected human CD34+ HPC formed all colonies (BFU-E, CFU-GEMM, CFU-GM). BFU-E: burst forming units; CFU-GEMM: colony forming units with granulocytes, erythrocytes, megakaryocytes and macrophages; CFU-GM: colony forming units with granulocytes and macrophages.
Discussion
We demonstrate here that mRNA transfection with ΔLNGFR and EGFP is highly efficient for human HPC and MSC showing > 80–90% positive cells after 24 hrs. A previously published report that used mRNA-based conventional electroporation demonstrated a similar transfection efficiency of 89% in MSC, whereas in HPC only 35% cells where positive for the transfected gene [8]. Marker expression attenuates quickly after 3 days and almost disappears after 10 days. The kinetics of mRNA versus plasmid transfection is easily explainable by the fact that mRNA has a short cellular half-life and thus, expression of the gene of interest does not last for a prolonged period of time. In addition to transfecting marker genes, mRNA transfection may also be useful to either transfect cell surface receptors, like integrins or chemokine receptors in order to improve stem cell homing or transcription factors in order to improve stem cell transdifferentiation. The fact that very high expression levels last for 3 days only and then quickly disappear may be advantageous for any kind of application that requires transient and intense, but no continuous change of stem cell properties. Thus, although short-term gene expression may be seen as a limitation of non-viral gene delivery, it may also be considered as being superior with regards to in vivo application, because it provides a safety advantage in the clinical setting by avoiding uncontrolled gene expression. mRNA has no potential to integrate into the host genome thereby bypassing safety concerns, as for example insertional mutagenesis.The latter is an important advantage of this methodology for potential use in clinical trials [9–10].
A critical parameter in determining quality and efficiency of non-viral gene delivery is the viability of cells after the transfection process. Our results demonstrate that cell viability following mRNA transfection is significantly better compared to that following plasmid transfection. This finding is in accordance with previous reports showing that viability after mRNA transfer by electroporation was markedly improved compared to DNA transfection [11–13]. mRNA is a normal constituent of the cell and thus less toxic than plasmid DNA. Also, only one membrane (i.e. the cell membrane, and not the nuclear membrane), needs to be crossed to create a biologically active cell constituent. Since entry into the nucleus as well as transcriptional regulation associated with plasmids is avoided, mRNA transfection is a valid alternative for non-viral gene delivery [9, 14–15].
In summary, mRNA based nucleofection is a powerful, highly efficient and non-toxic approach for transient labelling of human progenitor cells or, via transfection of selective proteins, for transient manipulation of stem cell function. It is easy to adopt to GMP protocols with the aim to combine principles of gene therapy and tissue engineering.
Acknowledgments
We thank G. Baur and T. Becker, Institute for Transfusion Medicine, University Ulm, for their help in stem cell culture. We thank M. Bienek-Ziolkowski, Internal Medicine II, University of Ulm, for her technical assistance.
References
- 1.Miyahara Y, Nagaya N, Kataoka M, Yanagawa B, Tanaka K, Hao H, Ishino K, Ishida H, Shimizu T, Kangawa K, Sano S, Okano T, Kitamura S, Mori H. Monolayered mesenchymal stem cells repair scarred myocardium after myocardial infarction. Nat Med. 2006;12:459–65. doi: 10.1038/nm1391. [DOI] [PubMed] [Google Scholar]
- 2.Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, Pickel J, McKay R, Nadal- Ginard B, Bodine DM, Leri A, Anversa P. Bone marrow cells regenerate infarcted myocardium. Nature. 2001;410:701–5. doi: 10.1038/35070587. [DOI] [PubMed] [Google Scholar]
- 3.Murry CE, Soonpaa MH, Reinecke H, Nakajima H, Nakajima HO, Rubart M, Pasumarthi KB, Virag JI, Bartelmez SH, Poppa V, Bradford G, Dowell JD, Williams DA, Field LJ. Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature. 2004;428:664–8. doi: 10.1038/nature02446. [DOI] [PubMed] [Google Scholar]
- 4.Liebowitz DN, Smith SL, Williams SF, Dolan ME. Efficient expression of foreign genes in human CD34(+) hematopoietic precursor cells using electroporation. Gene Ther. 2001;8:384–90. doi: 10.1038/sj.gt.3301393. [DOI] [PubMed] [Google Scholar]
- 5.Wiehe JM, Niesler C, Torzewski J, Zimmermann O, Wiesneth M, Schmitt M, Schwarz K, Döhner H, Hombach V, Greiner J. Efficient transient genetic labeling of human CD34+ progenitor cells for in vivo application. Regenerative Med. 2006;1:223–34. doi: 10.2217/17460751.1.2.223. [DOI] [PubMed] [Google Scholar]
- 6.Nair SK, Boczkowski D, Morse M, Cumming RI, Lyerly HK, Gilboa E. Induction of primary carcinoembryonic antigen (CEA)-specific cytotoxic T lymphocytes in vitro using human dendritic cells transfected with RNA. Nat Biotechnol. 1998;16:364–9. doi: 10.1038/nbt0498-364. [DOI] [PubMed] [Google Scholar]
- 7.Jones EA, Kinsey SE, English A, Jones RA, Straszynski L, Meredith DM, Markham AF, Jack A, Emery P, McGonagle D. Isolation and characterization of bone marrow multipotential mesenchymal progenitor cells. Arthritis Rheum. 2002;46:3349–60. doi: 10.1002/art.10696. [DOI] [PubMed] [Google Scholar]
- 8.Smits E, Ponsaerts P, Lenjou M, Nijs G, Van Bockstaele DR, Berneman ZN, Van Tendeloo VF. RNA-based gene transfer for adult stem cells and T cells. Leukemia. 2004;18:1898–902. doi: 10.1038/sj.leu.2403463. [DOI] [PubMed] [Google Scholar]
- 9.Lu D, Benjamin R, Kim M, Conry RM, Curiel DT. Optimization of methods to achieve mRNA-mediated transfection of tumor cells in vitro and in vivo employing cationic liposome vectors. Cancer Gene Ther. 1994;1:245–52. [PubMed] [Google Scholar]
- 10.Ying H, Zaks TZ, Wang RF, Irvine KR, Kammula US, Marincola FM, Leitner WW, Restifo NP. Cancer therapy using a self-replicating RNA vaccine. Nat Med. 1999;5:823–7. doi: 10.1038/10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Tendeloo VF, Ponsaerts P, Lardon F, Nijs G, Lenjou M, Van Broeckhoven C, Van Bockstaele DR, Berneman ZN. Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood. 2001;98:49–56. doi: 10.1182/blood.v98.1.49. [DOI] [PubMed] [Google Scholar]
- 12.Martinet W, Schrijvers DM, Kockx MM. Nucleofection as an efficient non-viral transfection method for human monocytic cells. Biotechnol Lett. 2003;25:1025–9. doi: 10.1023/a:1024157508492. [DOI] [PubMed] [Google Scholar]
- 13.Van De Parre TJ, Martinet W, Schrijvers DM, Herman AG, De Meyer GR. mRNA but not plasmid DNA is efficiently transfected in murine J774A. 1 macrophages. Biochem Biophys Res Commun. 2005;327:356–60. doi: 10.1016/j.bbrc.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 14.Kariko K, Kuo A, Barnathan ES, Langer DJ. Phosphate-enhanced transfection of cationic lipidcomplexed mRNA and plasmid DNA. Biochim Biophys Acta. 1998;1369:320–34. doi: 10.1016/s0005-2736(97)00238-1. [DOI] [PubMed] [Google Scholar]
- 15.Sawai K, Ohno K, Iijima Y, Levin B, Meruelo D. A novel method of cell-specific mRNA transfection. Mol Genet Metab. 1998;64:44–51. doi: 10.1006/mgme.1998.2692. [DOI] [PubMed] [Google Scholar]