

Abstract
Clinical trials have shown life-prolonging effects of antithrombotics in cancer patients, but the molecular mechanisms remain unknown due to the multitude of their effects. We investigated in a mouse model whether one of the targets of antithrombotic therapy, fibrin deposition, stimulates tumour development. Fibrin may provide either protection of cancer cells in the circulation against mechanical stress and the immune system, or form a matrix for tumours and/or angiogenesis in tumours to develop. Mice homozygous for Factor V Leiden (FVL), a mutation in one of the coagulation factors that facilitates fibrin formation, were used to investigate whether hypercoagulability affects tumour development in an experimental metastasis model. Liver metastases of colon cancer were induced in mice with the FVL mutation and wild-type littermates. At day 21, number and size of tumours at the liver surface, fibrin/fibrinogen distribution, vessel density and the presence of newly formed vessels in tumours were analysed. Number and size of tumours did not differ between mice with and without the FVL mutation. Fibrin/fibrinogen was found in the cytoplasm of hepatocytes and cancer cells, in blood vessels in liver and tumour tissue and diffusely distributed outside vessels in tumours, indicating leaky vessels. Vessel density and angiogenesis varied widely between tumours, but a pre-dominance for vessel-rich or vessel-poor tumours or vessel formation could not be found in either genotype. In conclusion, the FVL mutation has no effect on the development of secondary tumours of colon cancer in livers of mice. Fibrin deposition and thus inhibition of fibrin formation by anticoagulants do not seem to affect tumour development in this model.
Keywords: mouse model colon carcinoma metastasis Factor V Leiden fibrin liver
Introduction
Cancer patients are at an increased risk of developing venous thromboembolism (VTE) as compared with the general population [1,2]. Cancer itself can induce a hypercoagulable state by pressure on veins, damage of vessel walls and by direct or indirect activation of the coagulation system [3]. Evidence is accumulating that this hypercoagulable state may help cancer progression: fibrin and platelets may aggregate on cancer cells and protect them against mechanical stress and the host immune system in the circulation, and/or provide a matrix for tumour development and tumour-associated angiogenesis and/or facilitate adhesion to endothelium [4–7]. The coagulation system may also activate platelets and endothelial cells to release growth factors that facilitate cancer cell proliferation [3, 6, 8].
Clinical trials have shown life-prolonging effects of anticoagulants in cancer patients [9–13], but the molecular mechanisms are not understood yet [5, 7]. Anticoagulants have many effects besides inhibition of fibrin formation in biological systems [5, 7]. Therefore, we decided to study the effects of fibrin formation per se on the formation of secondary tumours. A mouse model of hypercoagulability on the basis of the factor V Leiden (FVL) mutation [14] was used to determine effects of elevated fibrin deposition on the development of metastases of colon cancer in the liver [15]. Factor V is an important cofactor in the coagulation cascade and the Leiden mutation makes it resistant to degradation by activated protein C, thereby inhibiting an important negative feedback loop in the system [16,17]. Mice that have been ‘knocked-in’ with the same mutation show an increased tendency to develop thrombosis [14]. Moreover, increased fibrin deposition in tissues including the liver has been demonstrated in these mice [14].
Number and size of tumours were determined on the surface of livers of mice homozygous for the FVL mutation and their wild-type littermates at day 21 after inoculation of syngenic colon cancer cells. Furthermore, fibrin/fibrinogen distribution was analysed in liver and tumour tissue and density of blood vessels and angiogenesis were studied in tumours as well because in vitro experiments have indicated that angiogenesis is affected by anticogulants and particularly low-molecular-weight heparin [18].
Materials and methods
Animals
FVL mice carrying a single amino acid mutation (arginine to glutamine mutation at position 504) have been described previously by Cui and coworkers [14]. FVL mice were back-crossed to C57Bl/6J mice for four generations (N4). Inter-crossing of N4 FVL heterozygous mice resulted in the homozygous and wild-type mice used in the current study. Since the difference in genetic background between these mice and C57Bl/6J mice may affect the results, wild-type littermates were used as controls. All mice were bred and maintained at the animal care facility at the Academic Medical Center, with free access to food and water. The data of two separate experiments were combined. In total, 18 homozygous FVL mice and 17 wild-type littermates were used. Three homozygous FVL mice and four wild-type littermates were used as sentinels in the first experiment (see below). The experiments were performed in agreement with the Animal Ethics Committee of the Academic Medical Center.
Cancer cells
An established syngenic murine colon adenocasein cell line, MCA-38, was kindly provided by Dr. E. Nelson, National Cancer Institute, Frederick MD, USA. Cells were cultured at 37°C as monolayers in RPMI 1640 Dutch modification without L-glutamine (GIBCO/BRL, Grand Island NY, USA) supplemented with 10% (v/v) fetal calf serum, 2 mM L-glutamine, 1 mM sodium pyruvate, 10 mM HEPES, 0.1 mM non-essential amino acids for minimum essential medium Eagle, 0.001% (v/v) β-mercaptoenthanol, 100 IU penicillin/ml and 100 mg streptomycin/ml. Cells were washed with phosphate-buffered saline (PBS) and, after detachment with trypsin (0.05% w/v) and EDTA (0.02% w/v) in PBS followed by centrifugation (250 g, room temp, 10 min), single cell suspensions were obtained with a viability of at least 95% as checked with Evans blue staining.
Surgery
An established liver metastasis model was used, previously described by Griffini et al. [15]. Briefly, a small midline incision was made in the abdominal wall of the mice under anaesthesia with FFM mix (1ml Hypnorm, 1 ml Midazolam, and 2 ml water (FFM), 0.07 ml/10 g body weight, intraperitoneally administered). A suspension containing the cancer cells (3 × 105 in the first experiment and 2.5 × 105 in the second experiment) in 100 μl PBS was injected into the portal vein using a 30-gauge needle. In order to prevent peritoneal seeding, the portal vein was closed by applying pressure with a piece of Spongostan (Medical Workshop, Groningen, The Netherlands) until bleeding had stopped. The abdomen was closed using stitches (Perma-hand woven non-absorbable sutures 5/0; Ethicon, Johnson & Johnson, New York NY, USA).
Monitoring of the animals
In order to prevent overgrowth of the experimentally-induced metastases in the liver, seven sentinel mice were used in the first experiment, three homozygous FVL mice and four wild-type littermates. These animals were injected with cancer cells at the same time as the other mice, but their abdomen was opened on day 14, 16 and 17 of the experiment to inspect the liver for overgrowth. It allowed for earlier termination of the experiment when the animals would show signs of excessive tumour growth. However, this was not the case in any of the animals. Therefore, the experimental mice were kept for 21 days after administration of the cancer cells. The sentinel mice were excluded from further analysis.
Preparation of liver tissue and evaluation of tumour growth
The animals were sacrificed at day 21 with an overdose of FFM mix (0.1 ml/10 g body weight). The livers were removed immediately and the surfaces of the liver lobes were macroscopically examined for the presence of tumours.
The size of all tumour nodules was measured using callipers [19]. The livers were weighed and subsequently frozen in liquid nitrogen and stored at –80°C until used. Serial sections (6 μm thick) were cut at a cabinet temperature of –25°C using a motor-driven cryostat (Bright, Huntingdon, UK), collected on clean glass slides (Star Frost; Knittel Gläser, Braunschweig, Germany) and stored at –20°C until further use.
Statistics
Because of the small numbers of animals, data of groups of animals were pooled over the two experiments. Variables that were normally distributed within the groups were compared using a Student's t-test and those that were not normally distributed were compared using a non-parametric Mann–Whitney test. Values of < 0.05 were considered to indicate significant differences between animal groups.
Histochemistry
Immunohistochemical staining of serial cryostat sections was performed as described previously [15] with the use of both the goat polyclonal antibody against fibrin/fibrinogen (Accurate, Westbury NY, USA) in a dilution of 1:500 and the rat monoclonal antibody against mouse CD105 or endoglin (Clone MJ7/18; BD Biosciences, Erembodegem, Belgium) in a dilution of 1:10 [20] as primary antibodies and secondary rabbit anti-goat or rabbit anti-rat antibodies coupled with horseradish peroxidase (Dako, Glostrup, Denmark), in a dilution of 1:100 or 1:200, respectively. Visualization was performed by using the chromogen diaminobenzidine (Fluka, Buchs, Switzerland).
The anti-fibrin/fibrinogen antibody stains fibrin and fibrinogen equally well and the anti-CD105/endoglin antibody stains newly-formed vessels [21, 22]. Sections were counterstained with haematoxylin for 5 sec and mounted in glycerin–gelatin. Control incubations were performed in the absence of the primary antibody in the incubation medium.
Furthermore, we performed staining of alkaline phosphatase activity [23], that is expressed by endothelial cells in general in various types of tumours [24]. After incubation, sections were rinsed and mounted in glycerin–gelatin. Control incubations were performed in the absence of substrate.
Staining of the tumours was performed semi-quantitatively using the following grading system: 0, absence of staining; 1, moderate staining in patches in the tumour area; 2, moderate staining all over the tumour area; 3, strong staining all over the tumour area.
Results
In the second experiment, two FVL mice and one wild-type mouse died during or directly after surgery, leaving a total of 13 homozygous FVL mice and 12 wild-type littermates for analysis.
Tumour load
Twenty-one days after cancer cell inoculation, tumours were macroscopically visible on the livers of all animals. Numbers and sizes of tumours in each animal varied strongly irrespective of the genotype (Fig. 1 and Table 1). Differences between mice with and without FVL were not statistically significant. The mean values of numbers and sizes of tumours were similar (Fig. 1). The median values (Table 1) differed considerably (4.5 tumour per FVL mouse and one per wild-type mouse, whereas 48.3% of the tumours were larger than 1 mm in diameter in the FVL mice and 28.6% in the wild-type mice). However, these differences were not significant either.
1.
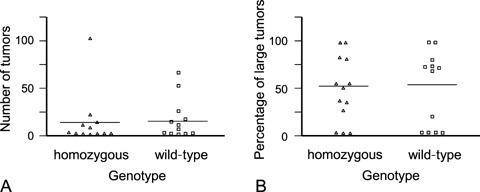
Numbers (A) and sizes (expressed as percentage of tumours larger than 1 mm in diameter; (B) of experimentally-induced colon cancer metastases in homozygous FVL mice and wild-type mice at 21 days after cancer cell inoculation. Horizontal bars represent the mean values.
1.
Quantitative analysis of colon cancer metastases in livers of mice with a homozygous FVL mutation and wild-type littermates
| Parameter* | Homozygous FVL mutation (n = 13) | Wild-type (n = 12) |
|---|---|---|
| Liver weight | 1.5 ± 0.5 | 1.6 ± 0.9 |
| Number of tumours | 4.5 (1–104) | 11 (1–67) |
| Percentage of large tumours | 48.3 (0–100) | 28.6 (0–100) |
Liver weight is given in g as mean ± standard deviation, numbers of tumours are given as median value and range in between brackets, and percentage of large tumours as median value and range in between brackets.
Fibrin/fibrinogen localisation
Since the antibody against fibrin cross-reacted with fibrinogen, both forms were demonstrated.
Weak staining was detected in the cytoplasm of both liver parenchymal cells and cancer cells (Fig. 2A and B and Fig. 3A–D). This was expected for the former cell type as it produces fibrinogen. Staining of the cytoplasm of cancer cells indicated production of fib-rinogen by these cells as well. In addition, strong staining was present in the lumen of vessels both in liver tissue and tumours (Fig. 2C and D), probably representing clots formed during postmortem stasis of blood in these vessels. Endothelial cells of vessels both in liver parenchyma and tumours were also positive for fibrinogen/fibrin (Fig. 2C and D). Staining was also diffusely localized around vessels in tumours (Fig. 2D and 3), which would be in line with earlier observations of fibrinogen leakage from intra-tumoural vessels with subsequent conversion into fibrin in cancer stroma [25–27]. We expected to find increased fibrin deposition in the homozygous FVL mice, but differences in distribution patterns of fibrin/fibrinogen between tumours of these mice and their wild-type littermates could not be detected by semi-quantitative analysis (Fig. 3).
2.
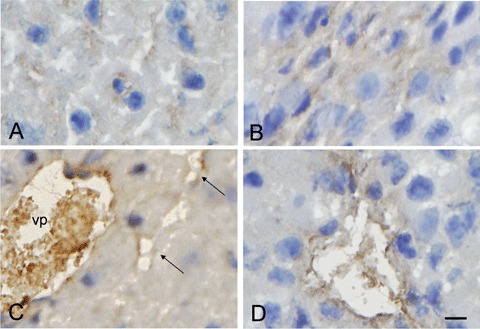
Immunolocalization of fibrin/fibrinogen in cytoplasm of liver parenchymal cells (A) and colon cancer cells (B) and in the lumen of a branch of the vena portae (vp) and sinusoids (arrows) in liver parenchyma (C) and in a vessel in colon cancer liver metastasis (D) of a homozygous FVL mouse. Bar = 10 μm.
3.
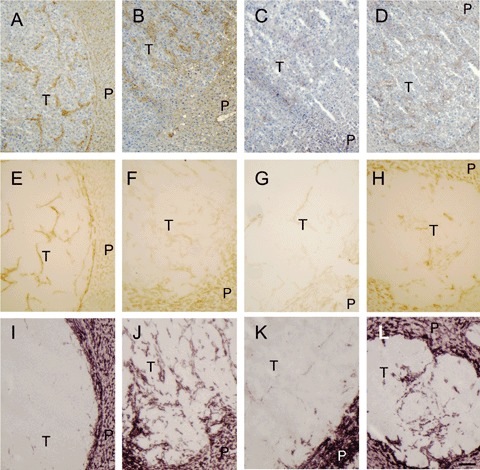
Staining of fibrin/fibrinogen and endothelial cells in colon cancer liver metastases in mice varied strongly irrespec-tive of the FVL genotype. Serial sections of livers of both mice with FVL mutation and wild-type littermates with tumours were stained for fibrin/fibrinogen (A–D), CD105/endoglin (E–H) and alkaline phosphatase activity (I–L). (A, E, I) Micrographs of a tumour (T) and liver parenchyma (P) of a mouse homozygous for the FVL mutation. In this tumour, CD105/endoglin, but not alkaline phosphatase activity co-localized with fibrin/fibrinogen depositions indicating that virtually all vessels were newly formed. (B, F, J) Micrographs of a tumour and liver parenchyma of a mouse with FVL mutation. In this tumour alkaline phosphatase activity, but not CD105/endoglin co-localized with fibrin/fibrinogen depositions. (C, G, K) Micrographs of a vessel-poor tumour in a wild-type littermate. (D, H, L) Micrographs of a tumour with vessels that were positive for all three markers in a wild-type littermate. Bar = 100 μm.
Vascularization of tumours
Since fibrin provides a scaffold for angiogenesis [4, 5, 8, 18], we investigated whether there were differences in vascularization of the tumours in animals with and without the FVL mutation. In tumours, strand-like structures that co-localized with fibrin/fib-rinogen were positive for either CD105/endoglin, the marker for newly-formed endothelial cells or alkaline phosphatase activity, the marker for endothelial cells in general or both (Fig. 3E–I).
Semi-quantitative analysis revealed that the vessel content of tumours varied largely in individual mice and intra-individually. Vascularization appeared to be independent of tumour size since vessels were present in both small and larger tumours. Vessels were not distributed homogeneously in tumours. Some areas were rich in vessels, whereas other areas in the same tumour were without any staining for endoglin or alkaline phosphatase activity. These large individual variations in vascularization and angiogenesis in tumours were independent of the FVL genotype of the mice.
Discussion
In our search for molecular mechanisms of the life-prolonging effects of anticoagulants in cancer patients [9–13], we have studied metastasis of colon cancer in the liver in a mouse model of hypercoagu-lability. The FVL mutation is a risk factor for thrombosis because of a defective negative feedback loop in the formation of fibrin by the coagulation system. Cancer patients with the FVL mutation have a higher risk to develop thrombosis than patients without the mutation [28]. On the other hand, there are no clear indications that the FVL mutation is a risk factor for developing cancer [29, 30]. However, an effect of the FVL mutation on tumour development in our animal model was expected for at least two reasons. Firstly, fibrin around cancer cells may protect the cells from the host immune system. Secondly, fibrin can act as a scaffold for growth of tumours or the development of angiogenesis. Since FVL has been found to affect the amount of fibrin in tissues including the liver [14], indirect effects on both tumour development and angiogenesis in tumours were expected. However, statistically significant differences were not found in the present study in numbers of tumours or size of the tumours between homozygous FVL mice and wild-type littermates.
We realize that inter-individual variation in number and size of tumours is large and therefore, subtle differences are not detectable. This is inherent to the cancer model, but biologically-significant effects can be detected [15]. The effects of anticoagulants in cancer patients were rather dramatic. For example, Klerk et al. [11] found that survival was prolonged by 6 months after treatment with low-molecular-weight heparin for 6 weeks. Therefore, we like to conclude that the FVL mutation does not affect tumour development in this model, indicating that the life-prolonging effects of antithrombotics in cancer patients are not simply due to inhibition of fibrin formation.
Antithrombotics do not appear to exert their effects on primary tumours, but rather on metastases [7]. On the basis of the findings in the present study, we like to suggest that the effects of antithrombotics in cancer patients are involving specific cancer cell-platelet and/or cancer cell-endothelial cell interactions rather than inhibition of thrombus formation and blood coagulation. This is in agreement with the fact that chemically-modified heparins with no or limited antithrombotic activity also have shown anti-metastatic properties [7].
References
- 1.Alikhan R, Cohen AT, Combe S, Samama MM, Desjardins L, Eldor A, Janbon C, Leizorovicz A, Olsson CG, Turpie AG MEDENOX Study. Risk factors for venous thromboembolism in hospitalized patients with acute medical illness: analysis of the MEDENOX Study. Arch Intern Med. 2004;164:963–8. doi: 10.1001/archinte.164.9.963. [DOI] [PubMed] [Google Scholar]
- 2.Smorenburg SM, Hutten BA, Prins MH. Should patients with venous thromboembolism and cancer be treated differently? Haemostasis. 1999;29:91–7. doi: 10.1159/000054122. [DOI] [PubMed] [Google Scholar]
- 3.De Cicco M. The prothrombotic state in cancer: pathogenic mechanisms. Crit Rev Oncol Hematol. 2004;50:187–96. doi: 10.1016/j.critrevonc.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Dvorak HF, Senger DR, Dvorak AM. Fibrin as a component of the tumor stroma: origins and biological significance. Cancer Metastasis Rev. 1983;2:41–73. doi: 10.1007/BF00046905. [DOI] [PubMed] [Google Scholar]
- 5.Smorenburg SM, Van Noorden CJF. The complex effects of heparins on cancer progression and metas-tasis in experimental studies. Pharmacol Rev. 2001;53:93–105. [PubMed] [Google Scholar]
- 6.Nash GF, Turner LF, Scully MF, Kakkar AK. Platelets and cancer. Lancet Oncol. 2002;3:425–30. doi: 10.1016/s1470-2045(02)00789-1. [DOI] [PubMed] [Google Scholar]
- 7.Niers TMH, Klerk CPW, DiNisio M, Van Noorden CJF, Bueller HR, Reitsma PH, Richel DJ. Mechanisms of heparin induced anti-cancer activity in experimental cancer models. Crit Rev Oncol Hematol. 2007;61:195–207. doi: 10.1016/j.critrevonc.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 8.Morganti M, Carpi A, Nicolini A, Gorini I, Glaviano B, Fini M, Giavaresi G, Mittermayer Ch, Giardino R. Atherosclerosis and cancer: common pathways on the vascular endothelium. Biomed Pharmacother. 2002;56:317–24. doi: 10.1016/s0753-3322(02)00242-1. [DOI] [PubMed] [Google Scholar]
- 9.Altinbas M, Coskun HS, Er O, Ozkan M, Eser B, Unal A, Cetin M, Soyuer S. A randomized clinical trial of combination chemotherapy with and without low molecular weight heparin in small cell lung cancer. J Thromb Haemost. 2004;2:1266–71. doi: 10.1111/j.1538-7836.2004.00871.x. [DOI] [PubMed] [Google Scholar]
- 10.Kakkar AK, Levine MN, Kadziola Z, Lemoine NR, Low V, Patel HK, Rustin G, Thomas M, Quigley M, Williamson RCN. Low molecular weight heparin, therapy with dalteparin, and survival in advanced cancer: the fragmin advanced malignancy outcome study (FAMOUS) J Clin Oncol. 2004;22:1944–8. doi: 10.1200/JCO.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 11.Klerk CPW, Smorenburg SM, Otten HM, Lensing AWA, Prins MH, Piovella F, Prandoni P, Bos MMEM, Richel DJ, Van Tienhoven G, Büller HR. The effect of low molecular weight heparin on survival in patients with advanced malignancy. J Clin Oncol. 2005;23:2130–5. doi: 10.1200/JCO.2005.03.134. [DOI] [PubMed] [Google Scholar]
- 12.Lee AYY, Rickles FR, Julian JA, Gent M, Baker RI, Bowden C, Kakkar AK, Prins M, Levine MN. Randomized comparison of low molecular weight heparin and coumarin derivatives on the survival of patients with cancer and venous thromboembolism. J Clin Oncol. 2005;23:2123–9. doi: 10.1200/JCO.2005.03.133. [DOI] [PubMed] [Google Scholar]
- 13.Falanga A, Piccioli A. Effect of anticoagulant drugs in cancer. Curr Opin Pulm Med. 2005;11:403–7. doi: 10.1097/01.mcp.0000174247.23009.06. [DOI] [PubMed] [Google Scholar]
- 14.Cui J, Eitzman DT, Westrick RJ, Christie PD, Xu ZJ, Yang AY, Purkayastha AA, Yang TL, Metz AL, Gallagher KP, Tyson JA, Rosenberg RD, Ginsburg D. Spontaneous thrombosis in mice carrying the factor V Leiden mutation. Blood. 2000;96:4222–6. [PubMed] [Google Scholar]
- 15.Griffini P, Fehres O, Klieverik L, Vogels IMC, Tigchelaar W, Smorenburg SM, Van Noorden CJF. Dietary omega-3 polyunsaturated fatty acids promote colon carcinoma metastasis in rat liver. Cancer Res. 1998;58:3312–9. [PubMed] [Google Scholar]
- 16.Bertina RM, Koeleman BP, Koster T, Rosendaal FR, Dirven RJ, De Ronde H, Van Der Velden PA, Reitsma PH. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994;389:64–7. doi: 10.1038/369064a0. [DOI] [PubMed] [Google Scholar]
- 17.Middeldorp S, Meinardi JR, Koopman MM, Van Pampus EC, Hamulyak K, Van Der Meer J, Prins MH, Büller HR. A prospective study of asymptomatic carriers of the factor V Leiden mutation to determine the incidence of venous thromboembolism. Ann Intern Med. 2001;135:322–7. doi: 10.7326/0003-4819-135-5-200109040-00008. [DOI] [PubMed] [Google Scholar]
- 18.Collen A, Smorenburg SM, Peters E, Lupu F, Koolwijk P, Van Noorden CJF, Van Hinsbergh VWM. Unfractionated and low molecular weight heparin affect fibrin structure and angiogenesis in vitro. Cancer Res. 2000;60:6196–200. [PubMed] [Google Scholar]
- 19.Van Noorden CJF, Jonges GN, Meade-Tollin LC, Smith RE, Koehler A. In vivo inhibition of cysteine proteinases delays the onset of growth of human pan-creatic cancer explants. Br J Cancer. 2000;82:931–6. doi: 10.1054/bjoc.1999.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ge AZ, Butcher EC. Cloning and expression of a cDNA encoding mouse endoglin, an endothelial cell TGF-beta ligand. Gene. 1994;138:201–6. doi: 10.1016/0378-1119(94)90808-7. [DOI] [PubMed] [Google Scholar]
- 21.Fonsatti E, Maio M. Highlights on endoglin (CD105): from basic findings towards clinical applications in human cancer. J Transl Med. 2004;2:18. doi: 10.1186/1479-5876-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fonsatti E, Sigalotti L, Arslan P, Altomonte M, Maio M. Emerging role of endoglin (CD105) as a marker of angiogenesis with clinical potential in human malignancies. Curr Cancer Drug Targets. 2003;3:427–32. doi: 10.2174/1568009033481741. [DOI] [PubMed] [Google Scholar]
- 23.Van Noorden CJF, Frederiks WM. Enzyme histo-chemistry. A laboratory manual of current methods. New York: Oxford Univ Press; 1992. [Google Scholar]
- 24.Murray JC, Smith KA, Lauk S. Vascular markers for murine tumours. Radiother Oncol. 1989;16:221–34. doi: 10.1016/0167-8140(89)90022-4. [DOI] [PubMed] [Google Scholar]
- 25.Nagy JA, Brown LF, Senger DR, Lanir N, Van de Water L, Dvorak AM, Dvorak HF. Pathogenesis of tumour stroma generation: a critical role for leaky blood vessels and fibrin deposition. Biochim Biophys Acta. 1989;948:305–26. doi: 10.1016/0304-419x(89)90004-8. [DOI] [PubMed] [Google Scholar]
- 26.Brown LF, Dvorak AM, Dvorak HF. Leaky vessels, fibrin deposition, and fibrosis: a sequence of events common to solid tumours and to many other types of disease. Am Rev Respir Dis. 1989;140:1104–7. doi: 10.1164/ajrccm/140.4.1104. [DOI] [PubMed] [Google Scholar]
- 27.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumour stroma generation and wound healing. N Engl J Med. 1986;315:1650–9. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 28.Blom JW, Doggen CJM, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA. 2005;293:715–22. doi: 10.1001/jama.293.6.715. [DOI] [PubMed] [Google Scholar]
- 29.Pihush R, Danzl G, Scholz M, Harich D, Pihusch M, Lohse P, Hiller E. Impact of thrombophilic gene mutations on thrombosis risk in patients with gastrointestinal carcinoma. Cancer. 2002;94:3120–6. doi: 10.1002/cncr.10590. [DOI] [PubMed] [Google Scholar]
- 30.Paspatis GA, Sfyridaki A, Papanikolaou N, Triantafyllou K, Livadiotaki A, Kapsoritakis A, Lydataki N. Resistance to activated protein C, factor V Leiden and the prothrombin G20210A variant in patients with colorectal cancer. Pathophysiol Haemost Thromb. 2002;32:2–7. doi: 10.1159/000057282. [DOI] [PubMed] [Google Scholar]