

Abstract
Inflammation is a central element of atherogenesis. Innate pathways contribute to vascular inflammation. However, the initial molecular process(es) starting atherogenesis remain elusive. The various risk factors, represented by particular compounds (activators), may cause altered cellular functions in the endothelium (e.g. vascular endothelial cell activation or -dysfunction), in invading cells (e.g. inflammatory mediator production) or in local vessel wall cells (e.g. inflammatory mediators, migration), thereby triggering the innate inflammatory process. The cellular components of innate immunology include granulocytes, natural killer cells and monocytes. Among the molecular innate constituents are innate molecules, such as the toll-like receptors or innate cytokines. Interleukin-1 (IL-1) and IL-6 are among the innate cytokines. Cytokines are potent activators of a great number of cellular functions relevant to maintain or commove homeostasis of the vessel wall. Within the vessel wall, vascular smooth muscle cells (SMCs) can significantly contribute to the cytokine-dependent inflammatory network by: (i) production of cytokines, (ii) response to cytokines and (iii) cytokine-mediated interaction with invading leucocytes. The cytokines IL-1 and IL-6 are involved in SMC-leucocyte interaction. The IL-6 effects are proposed to be mediated by trans-signalling. Dysregulated cellular functions resulting from dysregulated cytokine production may be the cause of cell accumulation, subsequent low-density lipoprotein accumulation and deposition of extracellular matrix (ECM). The deposition of ECM, increased accumulation of leucocytes and altered levels of inflammatory mediators may constitute an ‘innate-immunovascular-memory’ resulting in an ever-growing response to anew invasion. Thus, SMC-fostered inflammation, promoted by invading innate cells, may be a potent component for development and acceleration of atherosclerosis.
Keywords: atherosclerosis, cytokine, ECM accumulation, immunovascular memory, inflammasome, inflammation, innate pathways, interleukin-1, interleukin-6, SMC-monocyte-interaction, vascular smooth muscle cells
Introduction
In the last two decades the suggestion that inflammatory mechanisms are importantly involved in atherogenesis has been substantiated by many investigations. Thus, it appears reasonable to conclude that innate immune/inflammatory mechanisms are involved in the early steps of atherogenesis, and therefore may contribute potently to the initiation and acceleration of atherosclerosis. On the other hand, the facets of the adaptive immune system appear to be important in later periods of atherosclerosis.
Cytokines are central components of inflammatory pathways. Their contribution to cardiovascular malfunction has been substantiated by investigating plasma cytokine levels in cardiovascular patients, tissue mRNA expression, the generation of cytokine-deficient animals and in vitro cell culture investigations. These data showed that functions of vascular endothelial and smooth muscle cells (ECs and SMCs) are modified by exogenous activators (e.g. infectious agents or components thereof) or endogenous triggers (e.g. cytokines or autoantigens), representing the various risk factors (compare Fig. 1– blue boxes and blue arrow). Among the exogenous activation pathways are innate mechanisms, such as toll-like-receptors (TLRs), including the endotoxin receptors TLR-2 and TLR-4. On the other hand (endogenous) innate cytokines, such as interleukin (IL)-1, IL-6 or tumour necrosis factor (TNF), or even autoimmune triggers can activate the cells.
Fig 1.
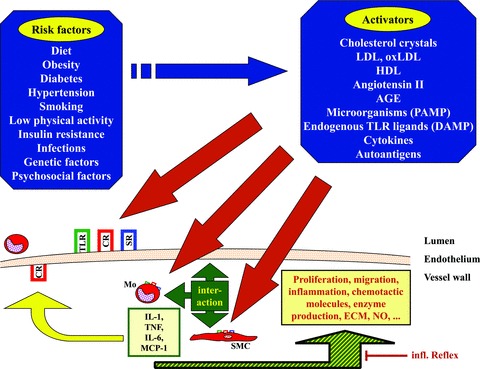
Contribution of cytokines and innate molecules to atherosclerosis by initiation of inflammation and cell interaction related to atherosclerosis (schematic overview). Blue arrow and blue fields: The different classical risk factors may be represented by one or more of the listed activators. Red arrows: These activators trigger cells in the lumen or the vessel wall via innate and cytokine receptors (TLR, SR, CR) expressed on the various cells (on SMCs and Mo the receptor symbols are downscaled). The activators may initiate atherogenesis by either causing endothelial activation/dysfunction, by activation of invading leucocytes, causing enhanced inflammation and/or by reaching the vessel wall tissue and activating an inflammatory response in the local vessel wall cells. Triple-headed green arrow and box: Cytokines will be produced and their production results in cytokine-mediated interaction of the cells inside the vessel wall, followed by (green-yellow hatched arrow) enhanced expression of cytokines, chemokines, enzymes, proliferation, cell death and other functions. Some relevant cytokines are mentioned in the pale yellow box framed in green. Dark yellow arrow: Cytokines and chemokines, produced in the vessel wall upon interaction of monocytes and vessel wall cells, also may cause expression of adhesion molecules and recruitment of more leucocytes. Slim red Tee-arrow: Regulation of inflammation in the vessel wall may be influenced by the ‘inflammatory reflex’ proposed earlier [136]. AGE: advanced glycation end product; CR: cytokine receptor; DAMP: damage-associated molecular pattern; ECM: extracellular matrix; HDL: high-density lipoprotein; LDL: low-density lipoprotein; Mo: monocyte; NO: nitric oxide; oxLDL: oxidized LDL; PAMP: pathogen-associated molecular pattern; SMC: vascular smooth muscle cell; SR: scavenger receptor; TLR: toll-like receptor.
Cytokines are potent regulators of cardiovascular cell functions, and therefore can potently influence homeostasis within the vessel wall. Consequently, SMCs can actively contribute to the inflammatory status in the vessel wall by producing these potent mediators or by responding to them. The cell interaction of vessel wall cells (i.e. ECs or SMCs) with invading cells (monocytes, T cells, mast cells) by cytokines may contribute to vascular inflammation. Cytokines may cause increased accumulation of monocytes, low-density lipoprotein (LDL) and extracellular matrix (ECM) in the vessel wall. If not brought back to physiological levels, cell and ECM accumulation provides an ‘innate-immunovascular-memory’ resulting in an ever-growing response to anew invasion. In this review, we discuss the role of selected inflammatory and innate pathways in atherogenesis, and point to the importance of cytokine-mediated interaction of vascular and invading cells.
Atherosclerosis – overview
General remarks
Atherosclerosis is a multifactorial disease, the pathogenesis of which is still not completely understood. Among the classical risk factors are diet, obesity, metabolic syndrome, diabetes, hypercholesteremia, smoking, hypertension and shortcomings in physical activity. It is now well accepted that inflammatory pathways are involved in the development and progression of atherosclerosis. Thus, formerly regarded as a lipid disease, more recently inflammation is supposed to be an important factor in atherogenesis [1, 2].
Early atherosclerosis
Changes in the vessel wall in early atherosclerosis may start with altered endothelial function (EC dysfunction), occasional endothelial denudation, vasoconstriction, enhanced procoagulation, increased leucocyte adhesion or enhanced plasma protein leakage (compare Fig. 1– red arrows) [3]. On the other hand, ‘patrolling’[4, 5] monocytes of the resident type may enter the vessel wall and initiate the inflammatory response. This subtype of monocytes has been characterized to be GR-1− Ly6C− in mice, and thus corresponds to CD14low CD16hicells in human beings [6]. Further characterization of the patrolling subtype is provided by the chemokine receptor expression: CC-type-chemokine receptor 2 (CCR2) is lacking, whereas the fractalkine receptor is expressed (CCR2− CX3CR1hi[CX3C-type-chemokine receptor]). This subtype may be of importance in early atherogenesis, although the Ly6C+ subtype (CD14hi CD16lo) accumulated more preferentially in the advanced plaque [7]. Based on the ‘response to injury’ proposal by Russel Ross [1] vascular activation may be initiated by one or more of various pathways. These activation pathways may include bacterial membrane components, such as endotoxin (LPS [lipopolysaccharide]), as well as endogenous inflammatory signals like cytokines. Activation results in leucocyte invasion, as well as migration and proliferation of local SMCs. Cellular processes are influenced by variations in cytokine production and reactivity, by altered responses to blood flow or by the ‘inflammatory burden’ caused by infectious or non-infectious triggers.
Exogenous and/or infectious activators
Among the exogenous and/or infectious activators (viral or bacterial), Chlamydiae, herpes simplex virus (HSV), cytomegalovirus (CMV) or epstein barr virus (EBV) are candidates [8]. This suggestion is supported by findings showing that influenza vaccination decreases cardiovascular events in coronary artery disease patients, and that high endotoxin levels predicted an enhanced risk for atherosclerosis in smokers [9]. Animal experiments further supported a role of infection in atherogenesis. Interestingly, the LPS hyporesponsive mouse strain C3H/HeJ belongs to a group of animals least susceptible to atherosclerosis. Although the animal experiments show that infections or infectious agents, such as surface molecules of bacteria or microbial nucleic acids, can have a role in development of atherosclerosis, they indicate that bacterial infections are not essential for the initiation of atherogenesis. Thus, besides infectious pathways, endogenous pathways may exist, resulting in similar downstream mechanisms relevant for development of atherosclerosis.
Endogenous activators
Among the endogenous activators possibly involved in atherogenesis [10] are trauma, disturbed blood flow, modified lipoproteins, crystallized cholesterol [11], autoimmunity and cytokines. Autoimmune pathways are considered to be involved in atherogenesis, because a variety of autoimmune diseases, such as systemic lupus erythematodes (SLE), rheumatoid arthritis or anti-phospholipid syndrome are paralleled by enhanced cardiovascular morbidity caused by enhanced atherosclerosis [12]. Heat shock protein 60, modified LDL, β2-glycoprotein-I or lymphoid protein tyrosine phosphatase PTPN22 (LYP) are possible auto-antigens involved in atherogenesis.
Taken together, various situations or components, including infections, molecules of infectious organisms or endogenous molecules, appear to be initiators of early atherosclerosis. However, it should be considered that not one particular trigger alone, but rather several – parallel or in sequence – cause changes in vessel wall homeostasis that can eventually no longer be controlled or repaired. Under these conditions the increasing cell number and deposition of compounds, such as LDL and ECM, may lead to an “innate-immunovascular-memory”, resulting in ever-growing responses to subsequent invasion.
Later phases of atherosclerosis
In the later phases of atherogenesis the ECM and LDL accumulation, as well as the cell accumulation may reach a time-point where the process becomes irreversible and vessel wall architecture is destroyed. This phase of plaque development is characterized by accumulation of various cell types in the vessel wall. During the establishment of the atherosclerotic plaque distinct monocyte subtypes preferentially invade the vessel wall. In mice the Ly-6Chi monocytes, a subset corresponding to the human CD14+CD16− monocytes, have been shown to invade the vessel wall in high quantities, whereas the Ly-6Clo monocytes invade the vessel wall to a lower degree. Ly-6Clo are proposed to be the source of plaque dendritic cells [7, 13]. However, it is still a matter of debate, which monocyte subset is functionally more important [14–16], also, it is still unclear whether foam cells are derived from Ly-6Chi or Ly-6Clo cells [6]. The ratio of migrating and emigrating cells appears to be important for progression of plaque development [17]. In the wall, monocytes eventually become foam cells, which form a core region in the plaque. However, besides foam cells, which represent the numerical largest part of the invading cells, T cells and other cells have been detected in the plaque [2]. It is now suspected that different subclasses of T cells may have different effects on atherogenesis, some promote atherogenesis, whereas others mute it. Among the cells also involved in atherogenesis are mast cells. They are present in atherosclerotic tissues, and experiments with mast cell-deficient mice showed that mast cell-derived IL-6 and/or interferon-γ (IFN-γ) may contribute to atherosclerosis [18]. Mast cells, as well as other cell types, may contribute to plaque vulnerability in late atherosclerosis by production of cytokines and, subsequently, by production of enzymes, such as matrix-metallo-proteases, cathepsins, chymases or other pro-atherogenic proteases. The vulnerability of the plaque in the late phase of atherosclerosis is a very critical element in the fatal outcome of atherosclerosis in many cases. Inflammation and the resulting activation of proteolytic enzymes has been suggested to be an important determinant of plaque stability and, accordingly, an establishment of treatments directed to stabilize vulnerability of the plaque (and patients) has been recommended.
Innate and inflammatory pathways in atherogenesis
Innate and inflammatory pathways are involved in atherogenesis. These pathways may contribute at various time-points, which have been outlined earlier, to atherogenesis: The steps in the atherogenic processes may start with an increased expression of adhesion molecules on the endothelium, caused by one or more of various activators (compare Fig. 1– blue boxes and blue arrow) in or outside the vessel wall. Adhesion and penetration of monocytes and other cells into the intima is the consequence. This causes accumulation and further attraction of cells by chemokines, such as monocyte chemoattractant protein-1 (MCP-1). Enhanced production of ECM and increased LDL uptake then takes place, followed by transformation of monocytes to foam cells in response to cytokines, such as monocyte-colony stimulating factor. Finally, alteration of plaque stability by cytokine-mediated enzyme production, the resulting modification of the fibrous cap, and, in the worst case, rupture of the plaque and thrombosis, results in stroke or myocardial infarction. The initial activation step of these processes is still not finally resolved (compare below: ‘Future perspectives’). However, in many cases activation of host cells by innate mechanisms may be the origin of the inflammatory processes driving atherogenesis.
Until recently, inflammation was not directly linked to vascular diseases. The cardinal manifestations of inflammation, which are heat, pain, redness and swelling, at first glance, are not apparent in atherosclerosis. However, we know now that inflammatory manifestations are caused by substances produced and released from cells. These mediators include complement factors, prostaglandins, leukotrienes, bradykinins, histamins or cytokines. Cytokines are mediators, produced by leucocytes and tissue cells (for a brief summary see [19]). They are very potent regulators of cell functions and many of them have been described to be involved in atherogenesis [20]. Originally they have been described as monocyte or lymphocyte products (monokines or lymphokines, respectively); however, we know now that many tissue cells also produce cytokines. The term cytokine is derived from the Greek 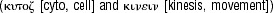 meaning as much as ‘working on and between the cells’. The terms ‘cytokine’ and ‘interleukin’ were coined in the 1970s. A variety of cytokine families are known, they include growth factors, interferons, interleukins, chemokines, tumour necrosis factors, colony stimulating factors and virokines.
meaning as much as ‘working on and between the cells’. The terms ‘cytokine’ and ‘interleukin’ were coined in the 1970s. A variety of cytokine families are known, they include growth factors, interferons, interleukins, chemokines, tumour necrosis factors, colony stimulating factors and virokines.
Under the conditions described in this chapter invading leucocytes may interact in manifold ways with local vessel wall cells by means of cytokines. The activation and cell interaction (compare Fig. 1– triple-headed green arrow and box) may result in further local responses, such as production of various chemokines and cytokines, ECM, enzymes or cell migration (compare Fig. 1– yellow/green crosshatched arrow). Thus, in the following we focus on the role of innate receptors, innate cytokines and cytokine-mediated cell interaction in cardiovascular diseases.
Innate receptors
Infectious pathways may contribute to initiation of atherosclerosis. Infectious ‘agents’, such as bacteria or viruses, are recognized already by the first line of defence of the body, the innate immune system (Table 1). The innate system has been considered to be ‘unspecific’ previously; however, it is obvious now that it recognizes certain particular types of activators, although it has a much smaller repertoire than the adaptive immune system. In addition to these pattern-recognition receptors (PRR; Table 2), natural antibodies and the complement system belong to the innate defence system, but these topics are not addressed here.
Table 1.
Innate and adaptive immune pathways
| Innate pathway | Adaptive pathway |
|---|---|
| Present in most forms of life, including the vertebrates | Found in vertebrates |
| Is germ-line encoded | Develops upon activation and selection |
| The innate immune system constitutes the ‘first line’ of defence in the vertebrates | The adaptive immune system is the ‘second line’ of defence in vertebrates |
| It reflects an immediate response to danger signals by monocytes, granulocytes, NK-cells, but also by ‘non-professional’ immune cells, such as epithelial cells, EC and SMC | The maximal response of the adaptive system occurs, after it has been primed, thus, it needs some time to respond |
| There is no antigen-specific memory, but an unspecific “innate-immunovascular-memory” has been proposed (compare [37]) | The adaptive immune system is characterized by the presence of a memory against antigens, which is conferred by lymphocytes, such as T cells and B cells |
| It recognizes ‘types’ of antigen (PAMP; DAMP) by PRRs such as CLRs, RLRs, pentraxins, TLRs, NLRs | It recognizes unique epitopes on the specific antigens by antibodies or receptors |
| The innate ‘specificity’ is limited to PAMP/DAMP recognition | |
| The innate immune system is responsible for the defence during infection and ‘self’-immunity (danger-related). The activated inflammatory pathways finally may lead to the activation of the adaptive immune system | The adaptive immune system is highly specific |
| The cytokine system, the chemokine system and the complement system, as well as phagocytic cells are constituents of the innate immune system | |
| The innate immune system is evolutionary older than the adaptive immune system |
CLR: C-type lectin receptor; DAMP: damage-associated molecular pattern; EC: endothelial cell; NK: natural killer; NLR: NOD-like receptor; PAMP: pathogen-associated molecular pattern; PRR: pattern-recognition receptor; RLR: RIG-like receptor; SMC: smooth muscle cell; TLR: toll-like receptor.
Table 2.
The various groups of pattern-recognition receptors (PRRs)
| Group | Class | Name | Function |
|---|---|---|---|
| Pentraxins | Short pentraxins | CRP SAP | Present in the plasma, produced in the liver, e.g. after IL-6 stimulation |
| Long pentraxins | PTX3 | Produced at the site of inflammation, produced upon TLR activation, binds fungi and influenza virus | |
| Activation of complement, clearance of debris, pathogen recognition, apoptotic cell recognition | |||
| NOD | NOD 1-5 CIITA | Cytosolic proteins, recognize bacterial ligands such as peptidoglycans, MDP | |
| NLR | NLRP | NLRP 1-14 NALP | Cytosolic proteins, recognize bacterial ligands such as peptidoglycans, MDP |
| IPAF | IPAF NAIP | Cytosolic proteins, recognize bacterial ligands such as peptidoglycans, MDP | |
| RLR | RLR | RIG-IMDA5 LGP2 PKR | Cytosolic proteins, recognize viral forms of RNA |
| DAI AIM2 | Cytosolic proteins, recognize viral forms of DNA | ||
| CLR | CLR | Dectin’sMINCLE | Cell surface proteins, recognize fungal ligands, carbohydrates on viruses, fungi, bacteria |
| TLR | TLR | 1, 2, 4, 5, 6, 10, 11 | Cell membrane associated |
| 3, 7, 8, 9 | Endosomal membrane associated | ||
| Various bacterial and viral components are recognized, including acylated lipopetides, lipoproteins, lipoteichoic acids, peptidoglycan, proteins, lipopolysaccharides, various forms of RNA and DNA. | |||
AIM2: absent in melanoma 2; CIITA: class II major histocompatibility complex transactivator; CLR: C-type lectin receptor; CRP: C-reactive protein; DAI: DNA-dependent activator of IFN-regulatory factors; IL-6: interleukin-6; IPAF: ICE-protease-activating factor; LGP2: laboratory of genetics and physiology 2; MDA5: melanoma differentiation-associated gene 5; MDP: muramyl dipeptide; MINCLE: macrophage-inducible C-type lectin; NAIP: neuronal apoptosis inhibitory protein; NALP: NACHT-, LRR- and PYD-containing protein; NLR: NOD-like receptor; NLRP: nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain containing; NOD: nucleotide-binding oligomerization domain; PKR: protein kinase regulated by RNA; PTX3: pentraxin 3; RIG-I: retinoic acid-inducible gene I; RLR: RIG-like receptor; SAP: serum amyloid A; TLR: toll-like receptor
PAMP and DAMP
Many infectious ‘agents’ activate the cells via the PRR by pathogen-associated molecular pattern (PAMP) containing molecules. PAMPs are highly conserved structures of microorganisms, or other ‘danger signals’, indicating ‘a need for’ defence. PRR can also detect endogenous signals, derived from damaged cells, containing damage-associated molecular patterns (DAMPs). Among the PRR activation pathways are the pentaxin or pentraxin molecules, including the acute phase molecules and serum proteins C-reactive protein (CRP) or serum amyloid P (SAP). The PAMPs and DAMPs are involved in pathogen recognition and removal of apoptotic cells. On the other hand, there are PRRs present in the cytosol of cells. They include the nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) [21], which contribute to the activation of inflammatory pathways by the inflammasome. Other cytosolic PRRs are the retinoic acid-inducible gene I (RIG)-like receptors (RLRs), which can detect viral DNA and RNA. Finally, numerous bacterial or viral components are recognized by the C-type lectin receptors (CLRs) and by membrane-associated cell-surface molecules, the TLRs. CLRs and TLRs preferentially recognize fungal or bacterial/viral ligands, respectively. However, some endosomal TLR molecules may also be activated by endogenous nucleic acids, delivered by accessory molecules, such as the advanced glycation end-product receptor, certain Fcγ receptors (Fc; antibody fragment, crystallizable) or antimicrobial peptides. The pentraxins, NLR and TLR are discussed below.
Pentraxins
The pentraxins (or pentaxins) are soluble proteins present in the plasma. CRP and SAP belong to the short pentraxins, whereas PTX3 is the prototype of a long pentraxin [22]. The short pentraxins are mainly produced in the liver, whereas PTX3 is produced by many cells including monocytes, fibroblast or vascular cells, and its activation is induced by inflammatory compounds, such as IL-1, TNF, LPS or oxidized LDL (oxLDL). PTX3 is a multifunctional protein; it contributes to activation of ECM deposition and inflammation by binding to various ligands, including complement factors, ECM, growth factors, membrane components or pathogens. Subsequently, PTX3 activates the complement cascade, as well as the removal of pathogens or cellular debris. This molecule has been detected in the atherosclerotic plaque in monocytes, EC and in SMC. Very recently this molecule has been described as a marker of inflammation in patients with heart failure and myocardial infarction [22, 23].
NOD-like proteins/receptors (NLRs)
The NOD-like proteins/receptors belong to a group of cytoplasmatic proteins containing a nucleotide-binding oligomerization domain (NOD) [21, 24]. They bind pathogen-derived molecules and either activate mitogen-activated protein kinases or NF-κB (NOD1; NOD2), or caspase-1 (NALP1 [NACHT, LRR and PYD containing protein]; NALP3; IPAF [ICE-protease-activating factor]) by initiating the assembly of the inflammasome. The latter is a multiprotein complex important for activation of IL-1, IL-18 and IL-33 [25, 26]. The activation of the inflammatory network by NLRs contributes to cardiovascular diseases. For example, it has been described that NOD1 agonists, but not NOD2 agonists, activate SMCs. These data indicated the presence of NOD1 pathways in SMCs, possibly resulting in nitric oxide production and vascular dysfunction.
toll-like receptors (TLRs)
The toll-like receptors are related to the Drosophila receptor toll, and on the other hand, to the IL-1 receptors [27]. The TLRs and the IL-1 receptors share with toll a signal pathway related to NF-κB and I-κB, termed dorsal and cactus in the fly. All three pathways (toll, TLR, IL-1) contribute to defence mechanisms in the respective organism. TLR-4 is the receptor for Gram-negative endotoxin (LPS) [28]. There are 10 human and 13 murine TLRs described, besides many parallel or different TLRs in other species [29]. The TLR molecules contain extracellular leucine-rich repeats (LRR), a transmembrane domain and an intracellular TIR-(toll/IL-1-receptor)-domain. Various adapter molecules including MyD88, NOD, MyD88-adapter-like (MAL), TIR-domain-containing adaptor-protein-inducing IFN-β (TRIF) or TRAM (TRIF-related adaptor molecule) mediate the intracellular signalling of the respective TLR molecules. The various TLRs, depending on their different PAMP specificity, will bind different activators of microbial/viral origin (exogenous), as well as endogenous activators (danger signals; DAMP) of the host [30].
Evidence for a role of TLRs in cardiovascular diseases has been provided by showing that TLR-4 is present in the failing myocardium and in the atherosclerotic plaque [31]. It has been proposed that, besides endotoxin, other molecules, such as endogenous host molecules like heat shock proteins, the fibronectin EDA domain (FNEDA), hyaluronic acids, lipoteichoic acids and even modified forms of LDL, as well as saturated fatty acids, are recognized by TLR-4. In cell culture experiments LPS-induced expression of TLR-4 on SMCs has also been described [32]. In addition to TLR-4 mRNA, arterial SMCs express mRNA for TLR-3, -6 and to some degree for TLR-1, -5, but not for TLR-7, TLR-8, or TLR-9 and TLR-2. Not only expression of TLR molecules has been investigated. Also, transfection experiments with high-cholesterol-fed rabbits showed that co-transfection with TLR-2 and TLR-4 synergistically enhanced atherosclerosis, as well as NF-κB, MCP-1, vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule (ICAM-1) expression [33]. The same authors also showed in vitro that co-transfection with TLR-2 and TLR-4 in cultured SMCs increased the expression of NF-κB.
Knockout of pattern recognition molecules reduces atherosclerosis
The above information indicates that plaque tissue and vascular cells express various types of TLR and that the cells can be activated via these molecules. The contribution of TLRs has been shown in atherosclerosis-prone C57BL/6 mice lacking TLR-4 (ApoE−/− TLR-4−/−) or the TLR-4 downstream signalling molecule MyD88 (ApoE−/− MyD88−/−). These mice developed smaller plaque area, although hypercholesterolemia was not reduced. The mice also had lower numbers of monocytes, lipids and cyclooxygenase (COX)-2 immunoreactivity in their plaques [34]. The data indicated a reduction of inflammatory components, despite a lack of reduction of circulating cholesterol or lipoprotein. In line with this, MyD88-deficient (ApoE−/− MyD88−/−), but not CD14-deficient (ApoE−/− CD14−/−) mice expressed reduced lesion area and monocyte recruitment [35]. The lower recruitment of monocytes into the aorta was probably caused by a reduced production of chemokines in the aortic vessel wall, indicated also by reduced chemokine serum levels and reduced chemokine in vitro production. The authors, however, did not identify which component of the diet, causing the hyperlipidaemia, may use the MyD88 signalling pathway. More recently, a contribution of TLR-2 in atherosclerosis has been shown in LDLr−/− TLR2−/− mice [36]. These mice had lower cholesterol and lesion area. Furthermore, bone marrow transplantation experiments suggested, that vascular TLR-2 expression was important for atherogenesis in this model. The authors also suggested that recurrent microbial infection may influence disease severity by TLR-2/TLR-1 activation.
Taken together, the data show that the innate TLR molecules may contribute to atherogenesis through activation of cells in the vessel wall by infectious agents (exogenous activation), but also may activate cells through endogenous, host-derived ligands. The activation of cells by PAMP- or DAMP-containing ligands finally results in the production of inflammatory mediators such as cytokines. These potent cell activators contribute to vascular pathology in many ways. Once produced, innate cytokines such as IL-1 and TNF perpetuate the inflammatory response by autocrine and synergistic mechanisms (compare Fig. 1– yellow/green crosshatched arrow).
Innate cytokines
Activation of local cellular responses in various ways may initiate atherogenesis [37]. Among others, activation of cells via innate pathways in the initial steps of atherosclerosis may result in cytokine production. The latter are potent mediators of inflammation. They have the capacity to regulate many of the functions/dysfunctions important for atherogenesis [20]. Among these functions are expression of adhesion molecules, chemokine production, activation of penetration and migration, stimulation of cell growth or synthesis of new products, including matrix degrading enzymes and cytokines. All these functions of cardiovascular and invading cells collectively contribute and lead – in the worst case – to coronary plaque development and life threatening plaque rupture.
Experiments using double-deficient mice for a variety of cytokines or cytokine receptors (e.g. ApoE/cytokine or LDLR/cytokine) have shown the significance of these mediators for cardiovascular diseases. A great number of knockout experiments showed a partial reduction of atherosclerosis, pointing to the necessity of interaction of several cytokine/inflammatory pathways for a complete reduction of atherosclerosis.
The innate cytokines IL-1 and TNF-α
Two central mediators in the cytokine network, relevant in innate pathways and inflammation, are IL-1 and TNF-α. IL-1 was originally described as a monocyte product. Its production is activated by various stimuli including bacterial components [38]. IL-1 is multifunctional, involved for example in fever, synthesis of acute phase proteins, proliferation of various cells, as well as in activation of T or B cells. Many functions of IL-1 are mediated by secondary cytokines, such as IL-6 or chemokines. The two isoforms of IL-1 have been cloned two decades ago [39, 40]. The IL-1 isoforms IL-1α and IL-1β are produced as 31 kD precursor molecules. IL-1β is enzymatically processed by caspase-1 (IL-1β converting enzyme, ICE; activated in the inflammasome) into its 17 kD mature form. IL-1α can be expressed on the cell surface of monocytes [41, 42]. It is also found in a functionally active form on cardiovascular cells, such as ECs, SMCs and heart cells [43, 44]. In contrast to the IL-1α precursor, the IL-1β precursor is not (or million-fold less) active [45]. In SMC IL-1β is present, but not processed [46]; however, leucocytes can activate the precursor present in SMC in a cell number dependent fashion [47]. In monocytes most of the IL-1α activity remains cell-associated, whereas most of the IL-1β is released.
In the vessel wall monocytes (as well as ECs or SMCs) may express IL-1 [48]. IL-1 activates many functions important for atherogenesis in ECs and SMCs. The expression of adhesion molecules on ECs or SMCs, necessary for invasion of leucocytes into the vessel wall, is potently induced by IL-1. Also, production of chemokines, such as IL-8, MCP-1 or fractalkine, necessary for invasion of leucocytes and retaining these cells in the wall is regulated by IL-1. Within the vessel wall IL-1 may activate proliferation of the local smooth muscle cells, an effect which is probably mediated or amplified by induction of platelet-derived growth factor (PDGF). IL-1 also induces nitric oxide production in SMC, which may in turn contribute to regulation of proliferation. Activation of the production of many other molecules, including IL-6 [49], vascular endothelial growth factor (VEGF) [50], transforming growth factor (TGF) [51] or endothelin [52], may further enhance the inflammatory load in the vessel wall. In addition, IL-1 and TNF also affect functions separate from inflammation, such as contraction of vascular cells [53].
Activation of ECM production, its degradation and re-assemblement is of great importance for atherogenesis. In human beings the development of a diffuse intimal thickening (DIT) has been suggested to be present before atherosclerosis develops [54]. The enhanced amount of ECM is thought to contribute to initiation of atherosclerosis through enhanced lipoprotein retention (response to retention hypothesis) [55]. Cytokine-mediated regulation of enzyme [56] and/or matrix production [57] may be the initial step of this procedure, started by the innate mechanisms discussed above. In other words, upon a – still undefined – initial activation by innate pathways, ECM production is up-regulated in response to IL-1, TGF, PDGF or TNF stimulation in the vessel wall [58]. ECM components can increase the retention of LDL or lipoproteins [59]. In addition, components of the ECM can also bind a variety of cytokines [60]. ECM or factors released from it, such as cytokines or modified LDL, can alter various cell functions. Deposition of matrix components, which in turn may stabilize or enhance LDL, SMC or monocyte retention, contributes to the ‘immunovascular memory’ effect. The “immunovascular memory” proposal includes that invading cells, ECM and the substances attached to it, may represent a reservoir for activation of cells invading the vessel wall in the future. Thereby, monocytes or SMCs invading the intima at these later time-points are more potently restrained and activated by the existing components (growth factors, modified LDL, macrophages, SMCs, ECM), thus establishing an unspecific ‘memory’ effect.
TNF-α shares many characteristics with IL-1. It is a pleiotropic cytokine, it is also produced by cardiovascular cells, and it also can signal via NF-κB. Like IL-1 it is also produced preferentially by monocytic cells. TNF is produced as a 26 kD membrane-associated molecule and transformed by a protease (TNF-converting enzyme, TACE, also known as ADAM17) into its 17 kD soluble form. It is binding to its receptor as a homotrimer. Depending on the presence of the TNF-receptor subtypes I (p55) or II (p75) it induces NF-κB and/or the death pathway, or only the NF-κB pathway, respectively. It is a member of a family of molecules, including CD40L, FasL or CD70, which are preferentially membrane bound. Their function in many cases needs cell-cell contact, and they are capable of signalling in two directions (forward, by the ligand itself: death, survival, differentiation, inflammation; reverse, by the respective receptor: proliferation, cytokine secretion, oxidative burst, class switch) [61].
TNF−/− ApoE−/− mice are less atherosclerotic
Because of their potent multifunctional activity IL-1, as well as TNF, may have an important role in vessel wall function and dysfunction. This suggestion is substantiated by investigations using mice deficient for these cytokines, their receptors or the IL-1 receptor antagonist. ApoE−/− TNF-α−/− mice have less atherosclerosis than control ApoE-deficient mice [62]. In these mice the plaque area was approximately 30% smaller, and ICAM-1, VCAM-1 or MCP-1 RNA was reduced in the aorta. In addition, scavenger receptor expression on macrophages, as well as macrophage oxLDL uptake in the double-knockout cells were reduced, although the cholesterol level was not altered. Likewise, in a TNF-receptor p75−/− ApoE−/− model the lesion area was also reduced [63]. Interestingly, blocking another TNF-receptor family member related to the adaptive immune system, important for T cell activation, the OX40, also resulted in reduction of lesion area [62].
IL-1−/− ApoE−/− mice are less atherosclerotic
In ApoE IL-1β double-deficient mice also 30% reduction of the lesion area was observed, accompanied by a reduced VCAM-1 and MCP-1 level, despite no changes in body weight or lipid levels [64]. More recently, bone marrow transplantation experiments from IL-1α−/− or IL-1β−/− mice into irradiated wild-type mice also showed reduced lesion area and plasma serum amyloid A (SAA) [65]. IL-1 activities are mediated by the IL-1-receptor type 1. Lack of the IL-1-receptor type 1 gene in ApoE+/− mice resulted in an enormous reduction of lesion size, both in uninfected mice (78%), as well as in Porphyromonasgin givalis-infected mice (97%) [66]. Further evidence for a role of IL-1 in vessel wall inflammation came from experiments with the IL-1-receptor antagonist. IL-1 receptor antagonist (IL-1ra)-deficient mice presented with enhanced neointimal thickening after injury [67], a result that was verified by the finding that IL-1-receptor type 1-deficient mice, as well as IL-1β-deficient mice expressed reduced neointima/media in ligation-induced neointima formation [68]. In line with the above, atherosclerosis-prone mice (ApoE-deficient) containing less IL-1ra (IL-1ra+/−) expressed a significantly increased lesion size, as compared to IL-1ra+/+ mice, in keeping with LDLrec−/− IL-1ra−/− mice [69]. On the other hand, application of IL-1ra or TNF-binding protein to diet-treated ApoE−/− mice resulted in reduced lesion area. TNF-binding protein had a lower effect compared with IL-1ra and was inactive in male mice [70]. Finally, investigating a homozygous IL-1ra knockout, instead of a heterozygous, Merhi-Soussi and colleagues found massive inflammation and early mortality in the ApoE double-deficient mice [71]. They suggested that the ratio of IL-1 and IL-1ra may be of great importance for the development of atherosclerosis.
Thus, TNF-α and IL-1 may contribute potently to atherosclerosis. In analogy, as discussed in the field of immunosenescence, multiple subsequent infectious or inflammatory incidents (‘multiple hits’) may cause an enhanced ‘inflammatory/pathogen burden’, resulting in enhanced risk of cardiovascular diseases [72].
Monocyte chemoattractant protein-1 and fractalkine
A variety of chemokines are produced in the vessel wall which may contribute to atherogenesis. Among them are fractalkine and MCP-1. MCP-1 is expressed in atherosclerosis. A role for MCP-1 has been shown in ApoE−/− CCR2−/− mice, lacking the MCP-1 receptor CCR2, because these mice developed smaller lesions [73]. Likewise, a knockout of MCP-1 in LDL-receptor-deficient mice also showed reduced atherosclerosis [74]. In accordance with this, mice overexpressing MCP-1 had larger lesions and higher numbers of invading monocytes, compared with control mice. Furthermore, mice overexpressing ApoB, characterized by low enhancement of cholesterol, but increased diet-induced atherosclerosis, if crossed with MCP-1-deficient mice, were protected from lesion development.
The chemokine fractalkine is unique, because it can function as a cell-associated adhesion molecule and, in its soluble form, as a chemokine. It is expressed on ECs, SMCs and in atherosclerotic lesions. Interestingly, soluble IL-6-receptor inhibited the expression of fractalkine in ECs [75]. The cleavage and release of fractalkine from the cell surface is processed by enzymes such as TACE or ADAM-10. Fractalkine receptor−/− ApoE−/− mice had reduced lesion formation, and less monocytes in the lesions. SMCs, but not monocytes, of these mice potently expressed fractalkine [76]. On the other hand, fractalkine-deficient mice also had reduced atherosclerosis, but in this model atherosclerosis was more pronounced at the brachiocephalic artery, as compared with the aortic root.
IL-6 activates innate pathways and is produced by cardiovascular cells
Among the secondary cytokines produced in response to IL-1 or TNF-α IL-6 is of major interest, because this cytokine is an important activator of the acute phase response [77]. IL-6 activates the production of acute phase response proteins in hepatocytes. Some of these molecules (CRP, SAP) belong to the innate PRR called pentaxins or pentraxins, present in the plasma (compare above) [22]. Among its multiple functions, CRP can bind to various pathogens, and it is activating the complement system, another facet of the innate defence system. IL-6 potently activates production of these acute phase molecules [78]. IL-6 is produced by a variety of cells, including monocytes [79], epithelial cells [80] and cardiovascular cells [49, 81]. In cardiovascular cells IL-6 is induced by a variety of stimuli, with IL-1 as one of the most potent [49]. In our hands, IL-6 is produced much more potently in SMCs than in monocytes or other cells, and IL-1 is the most potent stimulus tested so far. MCP-1 or oncostatin M have also been shown to induce IL-6 production in SMCs [82], and vice versa, IL-6 induced MCP-1 via STAT3-mediated mechanisms [83]. CRP also induced MCP-1 and IL-6 production in SMCs [84]. An autocrine loop for stimulation of SMCs by endogenous IL-6, involving trans-signalling mechanisms, has also been suggested [85].
Functions of IL-6 important for atherogenesis
Besides the autocrine effect of IL-6 on SMC activation discussed above [85], stimulation of a number of SMC functions by IL-6 has been described. Importantly, IL-6 induced the proliferation of rat SMCs, but not in human aortic and venous SMCs (unpublished observation). It reduces the contractility of SMCs [86], it may alter prostaglandin-E2 production [87], stimulate SMC migration [88] or induce matrix metalloproteinase (MMP)-9 secretion or MMP-1 production [89]. Some clinical data also point to a role of IL-6 in cardiovascular diseases. Thus, IL-6 or CRP are of potential value as markers for cardiovascular diseases, such as coronary artery disease and atherosclerosis [90], and the expression of IL-6 has been detected in the atherosclerotic lesion [91].
Role of trans-signalling in cardiovascular inflammation
IL-6 trans-signalling is responsible for pro-inflammatory activities of IL-6. On its target cells IL-6 first binds to the membrane-associated IL-6 receptor (IL-6R). The complex of IL-6 and IL-6R associates with the signal-transducing membrane protein gp130, thereby inducing its dimerization and initiation of signalling (Fig. 1) [92]. The glycoprotein gp130 is expressed by all cells in the body, whereas IL-6R is mainly expressed by hepatocytes, monocytes/macrophages and some leucocytes. A naturally occurring soluble form of the IL-6R (sIL-6R), which has been found in various body fluids, is preferentially generated by proteolysis of the membrane protein [93] and in human beings, but not in mice, by translation from an alternatively spliced mRNA [94]. Interestingly, the sIL-6R together with IL-6 stimulates cells, which only express gp130, a process, which has been named trans-signalling (Fig. 2) [95, 96]. Moreover, it has been shown that the sIL-6R strongly sensitizes target cells, which do express the membrane bound IL-6R [97]. Importantly, the autocrine loop for the stimulation of SMCs by IL-6, has been suggested to involve trans-signalling mechanisms [85].
Fig 2.
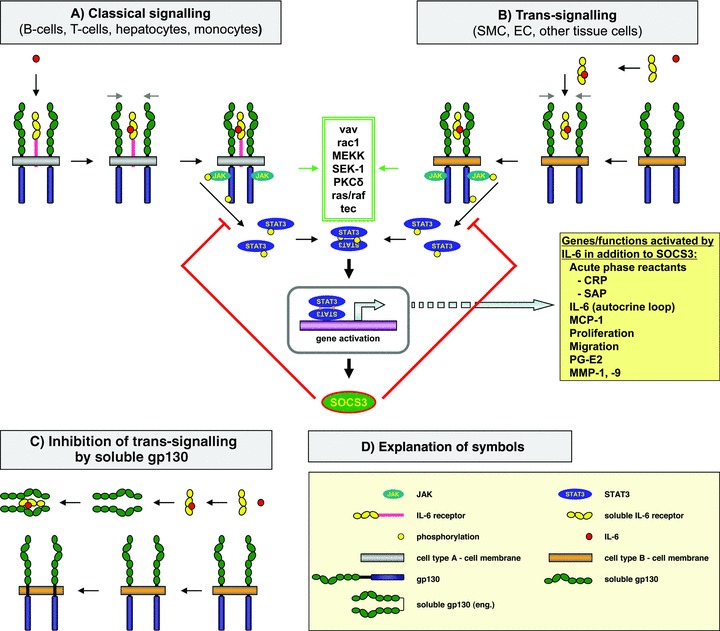
IL-6 ‘classical’ signalling, IL-6 ‘trans-signalling’ and inhibition of ‘trans-signalling’. (A) In the classical signalling IL-6 binds to the membrane-bound IL-6 receptor. (B) In trans-signalling IL-6 binds to the soluble IL-6 receptor, previously released from some cells. This complex is interacting with membrane-associated gp130. In both, classical and trans-signalling, the signal-transducer gp130 is recruited and STAT3 phosphorylation is activated. This can be achieved by PKC-γ. Other signal pathways may also be activated (box with green double frame), resulting in STAT3 phosphorylation. Subsequently, STAT3 activates gene expression in the nucleus. Among the genes activated by IL-6 is SOCS3, a down-regulator of IL-6 signalling. Some additional genes and functions relevant for atherosclerosis are mentioned in the yellow box. (C) Inhibition of trans-signalling by soluble gp130. Soluble gp130 is generated by alternative splicing. This molecule can bind the complex of soluble IL-6 receptor and IL-6, but not IL-6 alone. Thus, soluble gp130 is a selective inhibitor of IL-6 trans-signalling. (D) The definitions of the different symbols are provided in the yellow box. The definitions of the different symbols include another type of soluble gp130 [soluble gp130 (eng.)], which refers to a dimeric sgp130 engineered by molecular biological methods, which is a more effective inhibitor of trans-signalling than natural soluble gp130.
The gp130 protein is also present in a soluble form in body fluids. In contrast to the IL-6 receptor it is preferentially produced following alternative splicing [98, 99]. The function of the soluble form of gp130 (sgp130) was analysed using a soluble gp130 fusion protein, in which the extracellular portion of gp130 was fused to the constant portion of a human IgG1 antibody protein, resulting in a dimeric sgp130 molecule. It turned out that sgp130 inhibited IL-6 trans-signalling, mediated by IL-6 and the sIL-6R, without interfering with IL-6 responses via the membrane bound IL-6R (classical signalling). Therefore, it was postulated that sgp130 can act as a natural inhibitor of trans-signalling by IL-6/sIL-6R complexes [100].
IL-6 not only has pro-inflammatory properties but also has been shown to be involved in regenerative and metabolic functions [101, 102]. Although blockade of IL-6 is generally regarded to be beneficial in the case of chronic inflammatory states, in a model of inflammation-induced colon cancer it has recently been shown that IL-6 deficient mice were more inflamed [103], suggesting that IL-6 also has anti-inflammatory activities[104].
Using the recombinant sgp130Fc protein and mice transgenic for sgp130Fc, it has been shown that IL-6 trans-signalling, but not IL-6 classic signalling via the membrane bound IL-6R, is needed for the maintenance of chronic inflammatory states [105, 106], such as inflammatory bowel disease [107, 108], peritonitis [109], rheumatoid arthritis [110] and colon cancer [111, 112]. It was concluded that the pro-inflammatory activities of IL-6 are mainly executed via IL-6 trans-signalling [113], pointing to a therapeutic potential of the sgp130Fc protein for the treatment of inflammatory diseases [114]. It remains to be seen, whether inhibition of IL-6 trans-signalling with the sgp130Fc protein shows beneficial effects in the clinic.
The role of IL-6 receptor components was also analysed in hypertension and vascular hypertrophy in mice. Angiotensin II caused hypertension and cardiac/aortic hypertrophy in wild-type, but not in IL-6−/− mice. Recombinant sgp130Fc blocked angiotensin II hypertension, but not hypertrophy, in wild-type mice. Angiotensin II infusion activated STAT3 in the heart of wild-type mice and was unaffected by sgp130Fc. These data show that IL-6 trans-signalling was required for angiotensin II-dependent hypertension, but hypertrophy and cardiac STAT3 activation were mediated via membrane bound IL-6R. These findings indicate that IL-6 responses in a single disease context can be governed by both modes of IL-6 signalling. Blockade of IL-6 signalling therefore should have therapeutic potential for the treatment of hypertension and cardiac hypertrophy [115].
Animal experiments suggest a role for the IL-6 system in atherosclerosis
Evidence for an involvement of IL-6 in atherosclerosis is provided by experiments injecting IL-6 to several types of male mice fed normal or high fat diets [116]. Atherosclerosis-resistant non-obese diabetic mice developed larger lesions upon IL-6 treatment. On the other hand, ApoE-deficient mice treated with an IL-6-reducing agent (Am80) had smaller lesions than untreated mice [117]. In contrast, atherosclerosis-prone C57Bl/6 and ApoE-deficient mice showed a reduction of lesion size by elevated levels of IL-6. In line with this, Schieffer et al. showed reduced MMP-9 expression, reduced monocyte recruitment and increased lesion size in mice lacking ApoE and IL-6 [118], suggesting an atheroprotective role of IL-6. Furthermore, female ovariectomized ApoE−/− IL-6−/− mice, fed 1 year on normal diet, also developed larger lesions than IL-6-expressing wild-type mice [119]. Experiments using gp130-deficient mice provided additional evidence for a role of IL-6 in atherosclerosis. gp130 is a key part of the IL-6-receptor system and its removal leads to premature death. However, a hepatocyte-specific gp130 deletion indicated that an acute phase response protein may contribute to an enhanced atherosclerosis lesion size [120]. This study suggested a role for serum amyloid A in CCL2 (chemokine (C_C-motif) ligand 2; MCP-1) induction and subsequent attraction of monocytes. Furthermore, a role for CRP was suggested by experiments with atherosclerosis-prone mice crossed with CRP-transgenic mice (>34% lesion size) [121]. Experiments with mast cell-deficient (Ldl−/−; Kit W-sh/W-sh) mice provided further evidence for a role of IL-6 [18]. These mice developed less atherosclerosis. Adoptive transfer of mast cells from wild-type or TNF-deficient mice restored atherogenesis, whereas mast cells from IL-6 or IFN-deficient mice did not, due to the lack of enzyme (cathepsin; MMP) induction by IL-6 in the latter. The above data indicate an important role of the IL-6 system in atherosclerosis; however, the contribution of SMC- or EC-derived IL-6 still needs to be investigated in more detail.
Cytokine-mediated interaction of vessel wall cells and leucocytes
Vessel wall cells, in addition to the classical cytokine producers, the leucocytes, can also produce cytokines, such as IL-1 or IL-6 [49, 122]. It has been shown in the literature that cytokines stimulate cardiovascular cells to modulate an array of functions, including proliferation, contraction, migration or synthesis of new mediators or enzymes. In the atherosclerotic process, upon an undefined or multi-facetted activation, emigrating cells, such as monocytes, T cells or mast cells, interact with the endothelium. Subsequently, these cells contact the cells present in sub-endothelial layers, which may include SMCs, and in later phases leucocytes. These cells may potently interact in the vessel wall by means of cytokines, thereby perpetuating the atherosclerotic process (compare Fig. 1).
Endothelial cell (EC)-monocyte interaction
Interaction of these cells, independent which cell (i.e. patrolling monocytes or activated ECs) initiates this interaction, may result in subsequent enhancement of cellular responses. Thus, enhanced production of the monocyte attractant MCP-1 in both the ECs and the migrating monocytes as well as enhanced IL-8 production has been shown [123]. Monocytes and ECs also produce PDGF upon interaction, probably by an IL-1- and TNF-dependent pathway [124]. Interaction of ECs and monocytes also results in production of the potent cell activator granulocyte macrophage-colony stimulating factor. It can also lead to reduced nitric oxide production, and lower dilatation [125]. Monocyte-EC cocultures also produced enhanced tissue factor, which may be of importance for thromboembolic problems. Also of potential importance for later steps in atherosclerosis, MMP-1 is up-regulated in monocyte-EC cocultures [126].
Smooth muscle cell (SMC)-EC interaction
Upon migration of SMCs into the sub-endothelial space interaction of ECs and SMCs may result in further activation. In a model using microporous membranes the adhesion of monocytes was increased upon coculture of ECs with SMCs [127]. It has also been described that the proliferation, collagen production or chemokine production, was elevated upon EC-SMC interaction. TGF-β production is regulated depending on the type of coculture (bilayer or mix), and may have impact on adhesion molecule expression on SMC, or nitric oxide production.
SMC-monocyte and SMC-T cell interaction
Following invasion of the neointima monocytes may interact with the local SMCs in various ways. Enhanced VEGF was observed in monocyte-SMC interaction, which was partially mediated by soluble cytokines, such as IL-6 [128]. PDGF- and hepatocyte growth factor-production were also enhanced in coculture experiments [129]. These molecules possibly contribute to cell growth in the developing plaque. However, interaction may also activate counter-balancing mechanisms [130]. On the other hand, the production of enzymes was enhanced in coculture models [89]. Furthermore, an enhanced nitric oxide production upon interaction was detected [131]. Not only monocyte-SMC interactions have been studied. In the later phases of atherosclerosis T cells are potently involved. Thus, it has been shown that T cells and SMCs of several tissues interact in various ways [132]. Very recently, we have shown a dramatic up-regulation of IL-6 and MCP-1 production in a SMC-mononuclear cell coculture, which was mediated by soluble factors; however, the leucocytes involved were monocytes and not T cells [133]. This synergistic cytokine production was potently reduced by antiphlogistics and statins [134].
As indicated above, experimental evidence exists for a variety of cytokine-mediated interactions of immigrant cells and vascular vessel wall cells. These interactions and the resulting alterations in homeostasis (compare Fig. 1– triple-headed green arrow and box) have to be balanced very precisely, in order to maintain a ‘normal’ vessel. In case of enhanced ‘inflammatory burden’ caused by an increased number of subsequent activations, too high frequency of activation or too potent activation, the control mechanisms may no longer ensure the vascular homeostasis. Malfunction may result from this and finally result in plaque development or protrusion. Better understanding of the multiple interactions in the vessel wall may provide information about potential therapeutic points of vantage.
Summary and conclusion
There is a broad range of evidence that inflammation is essential for atherogenesis. Still, the initial activators and the sequence of the following steps are incompletely evaluated. Critical ‘risk factors’ for atherosclerosis have been defined and each of these risk factors may cause atherosclerosis in consequence of one or more molecular reasons (i.e. activators) inherent in it (compare Fig. 1– blue boxes and arrow). Some of the various activators, representative for the many risk factors, may activate inflammation by means of innate receptors or alter the functionality of these receptors.
The involvement of innate pathways can be deduced from data showing that in mice MyD88 deficiency (IL-1/TLR signalling pathway) potently reduced atherosclerosis. These data indicate that the same receptors which help us to survive after infection (by recognizing PAMPs), can interfere with regulation of inflammation involved in atherosclerosis. Thus, it is not surprising that some researchers are convinced that microorganisms may be involved in atherogenesis.
Microorganisms (through PAMP), however, are not the only initiators of atherosclerosis (compare Fig. 1– red arrows), and they are not essential for atherosclerosis. Other compounds, host-derived or modified by the host (i.e. DAMPs), may use the same innate pathways [10] in a non-infectious, sterile mode [135]. Examples for such compounds are crystals (urate crystals in gout; cholesterol crystals in atherosclerosis), particles (such as silica or asbestos in lung diseases), DNA or others. Cell death in the vessel wall, following ischemia or toxins, also has the potential to initiate sterile inflammation.
The above pathways, may they activate the endothelium directly (from the lumen) or by vessel wall-derived activation, finally lead to enhanced infiltration into the vessel wall of the different cell types. Subsequently, interaction(s) of the invading and local cells in the vessel wall can further enhance the inflammatory response (compare Fig. 1– triple-headed green arrow and box) and result in enhanced cytokine and chemokine production, expression of more adhesion molecules, stimulation of enzyme production or arachidonic compounds, etc. (compare Fig. 1– green/yellow cross-hatched arrow; as well as yellow arrow). Cholinergic regulation through the vagal inflammatory reflex [136] may influence this situation by keeping inflammation under control; however, after down-regulation of vagal activity the cytokine inhibition may fade and inflammation may increase. All these processes, initiated to a large degree by innate pathways, perpetuate atherogenesis by cytokine-mediated regulation of cell functions, and, finally, reach a point, where repair processes no longer can keep the inflammation and tissue damage under control, resulting in plaque appearance and, eventually, rupture of the plaque.
In conclusion, inflammatory pathways, including innate mechanisms and inflammatory cytokines, potently contribute to atherogenesis. The present data suggest that not one separate and singular incident, but rather many subsequent and probably different interferences in and on the vessel wall may result in altered vessel wall function. Enhanced accumulation of monocytes/macrophages and SMCs, as well as increased levels of ECM components, may serve as an “immunovascular memory” leading to an ever-growing response to reiterative invasion. The subsequently increased interaction of local vessel wall cells with invading leucocytes may further enhance and reinforce inflammation in the vessel wall by potently regulating local cytokine production. Thus, regulation of cytokine-mediated inflammation in the early and later phases of atherogenesis in the vessel wall by anti-inflammatory drugs will be a target of further research.
Future perspectives
Although it has been substantiated by many investigations that inflammation is involved in atherogenesis, we are still far from understanding the whole complexity of the pathways being involved in atherogenesis. In particular, the early steps of atherosclerosis are less well delineated. The above summary intended to present some of the pathways strenuous in early atherosclerosis. Among the points to be addressed more intensively in the future are the following questions:
Which initiator(s) starts atherogenesis?
Is there more than one starting point?
What are the specific roles of the various cytokines in the vessel wall?
Are treatments addressing different phases of atherogenesis of advantage?
What about prevention?
What are the expectations?
Which initiator(s) starts atherogenesis?
It has been unequivocally proven by in vitro experiments, transgenic experiments and clinical data that innate molecules and cytokines are involved in atherogenesis. However, the knowledge about the initiation of these processes and the sequence of steps is still limited (compare Fig. 1). Are endogenous activators relevant (i.e. cytokines, plasma components, autoantigens), or are exogenous components (microorganisms, dietary components) the cause of the initiation of the expression of the adhesion molecules on the endothelium and the intravascular chemotactic gradient within the vessel wall? Do multiple mechanisms synergize (compare next paragraph)? Can the interaction of invading monocytes and local vessel wall cells increase the inflammatory potential? The exciting microscopical and imaging methods available today may enable the investigation of the initiatory steps of atherogenesis in more detail, as recently shown for cholesterol crystals [11].
Is there more than one starting point?
In diet-induced animal atherosclerosis it is without dispute that dietary components initiate atherogenesis. However, do activating components of other risk factors cooperate or synergize with the dietary pathway, or is the dietary pathway the central player? Many mouse models, however, show that atherosclerosis is reduced in knockout mice, despite any effect on the lipid levels, indicating a complex interaction of the inflammatory and dietary pathways in atherogenesis. Thus, in the future multiple components representing separate risk factors should be employed experimentally at the same time.
What are the specific roles of the various cytokines in the vessel wall?
A role of cytokines and innate receptors in atherogenesis has been proven in animal experiments. The capacity of the vessel wall cells to produce cytokines and to react to cytokines or innate activators has also been shown in many in vitro experiments. Thus, we know the cells can do, but, do they do it really, and in what sequence, and using which cytokine? In addition, not all cytokines produced by vessel wall cells have clearly identified roles in the vessel wall during atherogenesis. For example, IL-6 is very potently produced by SMC in vitro[49], but its role in the vessel wall is still discussed controversially. Does IL-6 even move out of the wall, due to its highly elevated production, thereby contributing to enhanced plasma IL-6 levels, and, subsequently, to enhanced levels of CRP? Molecular biological and imaging methods may contribute to answer such questions.
Are treatments addressing different phases of atherogenesis of advantage?
For the understanding of atherosclerosis, and, finally, development of therapeutic approaches, comprehension of the initiation period, as well as the stable plaque and the vulnerable plaque period is necessary. The different phases of atherosclerosis may be responsive to different treatments, because different cell types and molecular mechanisms appear to be involved in the different phases. In particular, the initiation process is less well understood. Imaging methods, following the fade of various molecules or cells, or analysing functional aspects, such as enzymatic activity, are excellent analysis tools to be used more frequently in the future.
Despite all efforts, not all possible inflammatory targets identified in the past have been useful for therapeutic approaches, probably because of the redundancy of the cytokine system. However, some of these inflammatory substances still may serve as biomarkers. For example, it has been shown that CRP measurement increases the sensitivity of existing scores, as shown for the Framingham score. Thus, digging for additional innate/inflammatory molecules still appears to be auspicious.
What about prevention?
Besides each and any effort in therapy and prevention, in the first run improved information of the general public about positive effects of ‘good’ lifestyle (weight, diet, physical activity, no smoking), needs to be meliorated. Furthermore, identification of (a specific) ‘ultimate’ genetic atherosclerosis marker(s), or (an unspecific) inflammatory recognition algorithm(s), may help to identify people eligible for prophylactic therapy.
What are the expectations?
As indicated in this summary myriads of publications have indicated the capacity of inflammatory molecules to regulate atherogenesis and that vessel wall cells are sources and responders to such molecules by themselves. From our point of view, the primary activators of these processes have not been identified in their full spectrum. In addition, the composition and sequence of the inflammatory pathways outside and inside the vessel wall during atherogenesis, remains to be determined more clearly. We expect that from such comprehension of ‘fine-tuning’ of atherogenesis many new therapeutic targets will be derived in the future.
Acknowledgments
Parts of the studies were supported by grants of the Deutsche Forschungsgemeinschaft to H.L. (Lo385/4–1 and Lo385/5–1), and by a grant of the BMBF to H.L. (Myocardial Hypertrophy – Project 06). S.R.-J. was supported by the Deutsche Forschungsgemeinschaft (SFB877, project A1) and by the Cluster of Excellence ‘Inflammation at Interfaces’.
Conflict of interest
S.R.-J. is the inventor on the patent describing sgp130Fc and is a shareholder of the CONARIS Research Institute Kiel, Germany.
References
- 1.Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999;340:115–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 2.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–19. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, Cliff WJ, Schoefl GI, et al. Plasma protein insudation as an index of early coronary atherogenesis. Am J Pathol. 1993;143:496–506. [PMC free article] [PubMed] [Google Scholar]
- 4.Auffray C, Fogg D, Garfa M, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–70. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 5.Audoy-Remus J, Richard JF, Soulet D, et al. Rod-Shaped monocytes patrol the brain vasculature and give rise to perivascular macrophages under the influence of proinflammatory cytokines and angiopoietin-2. J Neurosci. 2008;28:10187–99. doi: 10.1523/JNEUROSCI.3510-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woollard KJ, Geissmann F. Monocytes in atherosclerosis: subsets and functions. Nat Rev Cardiol. 2010;7:77–86. doi: 10.1038/nrcardio.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tacke F, Alvarez D, Kaplan TJ, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–94. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiechl S, Egger G, Mayr M, et al. Chronic infections and the risk of carotid atherosclerosis: prospective results from a large population study. Circulation. 2001;103:1064–70. doi: 10.1161/01.cir.103.8.1064. [DOI] [PubMed] [Google Scholar]
- 9.Wiedermann CJ, Kiechl S, Dunzendorfer S, et al. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: prospective results from the Bruneck Study. J Am Coll Cardiol. 1999;34:1975–81. doi: 10.1016/s0735-1097(99)00448-9. [DOI] [PubMed] [Google Scholar]
- 10.Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010 doi: 10.1155/2010/672395. 10.1155/2010/672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–61. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sherer Y, Shoenfeld Y. Mechanisms of disease: atherosclerosis in autoimmune diseases. Nat Clin Pract Rheumatol. 2006;2:99–106. doi: 10.1038/ncprheum0092. [DOI] [PubMed] [Google Scholar]
- 13.Swirski FK, Libby P, Aikawa E, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ingersoll MA, Spanbroek R, Lottaz C, et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood. 2010;115:e10–9. doi: 10.1182/blood-2009-07-235028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu H, Gower RM, Wang H, et al. Functional role of CD11c+ monocytes in atherogenesis associated with hypercholesterolemia. Circulation. 2009;119:2708–17. doi: 10.1161/CIRCULATIONAHA.108.823740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mosig S, Rennert K, Krause S, et al. Different functions of monocyte subsets in familial hypercholesterolemia: potential function of CD14+ CD16+ monocytes in detoxification of oxidized LDL. FASEB J. 2009;23:866–74. doi: 10.1096/fj.08-118240. [DOI] [PubMed] [Google Scholar]
- 17.Llodra J, Angeli V, Liu J, et al. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci USA. 2004;101:11779–84. doi: 10.1073/pnas.0403259101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun J, Sukhova GK, Wolters PJ, et al. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat Med. 2007;13:719–24. doi: 10.1038/nm1601. [DOI] [PubMed] [Google Scholar]
- 19.Loppnow H. Cytokines: classification, receptors, mechanisms of action. Internist. 2001;42:13–27. doi: 10.1007/s001080050720. [DOI] [PubMed] [Google Scholar]
- 20.Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev. 2006;86:515–81. doi: 10.1152/physrev.00024.2005. [DOI] [PubMed] [Google Scholar]
- 21.Fritz JH, Ferrero RL, Philpott DJ, et al. NOD-like proteins in immunity, inflammation and disease. Nat Immunol. 2006;7:1250–7. doi: 10.1038/ni1412. [DOI] [PubMed] [Google Scholar]
- 22.Mantovani A, Garlanda C, Doni A, et al. Pentraxins in innate immunity: from C-reactive protein to the long pentraxin PTX3. J Clin Immunol. 2008;28:1–13. doi: 10.1007/s10875-007-9126-7. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki S, Takeishi Y, Niizeki T, et al. Pentraxin 3, a new marker for vascular inflammation, predicts adverse clinical outcomes in patients with heart failure. Am Heart J. 2008;155:75–81. doi: 10.1016/j.ahj.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 24.Franchi L, Park JH, Shaw MH, et al. Intracellular NOD-like receptors in innate immunity, infection and disease. Cell Microbiol. 2008;10:1–8. doi: 10.1111/j.1462-5822.2007.01059.x. [DOI] [PubMed] [Google Scholar]
- 25.Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production. Nat Rev Immunol. 2010;10:210–5. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 26.Alnemri ES. Sensing cytoplasmic danger signals by the inflammasome. J Clin Immunol. 2010;30:512–9. doi: 10.1007/s10875-010-9419-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gay NJ, Keith FJ. Drosophila toll and IL-1 receptor. Nature. 1991;351:355–6. doi: 10.1038/351355b0. [DOI] [PubMed] [Google Scholar]
- 28.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila toll protein signals activation of adaptive immunity. Nature. 1997;388:394–7. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 29.Roach JC, Glusman G, Rowen L, et al. The evolution of vertebrate toll-like receptors. Proc Natl Acad Sci USA. 2005;102:9577–82. doi: 10.1073/pnas.0502272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22:240–73. doi: 10.1128/CMR.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu XH, Shah PK, Faure E, et al. toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. 2001;104:3103–8. doi: 10.1161/hc5001.100631. [DOI] [PubMed] [Google Scholar]
- 32.Lin FY, Chen YH, Tasi JS, et al. Endotoxin induces toll-like receptor 4 expression in vascular smooth muscle cells via NADPH oxidase activation and mitogen-activated protein kinase signaling pathways. Arterioscler Thromb Vasc Biol. 2006;26:2630–7. doi: 10.1161/01.ATV.0000247259.01257.b3. [DOI] [PubMed] [Google Scholar]
- 33.Shinohara M, Hirata K, Yamashita T, et al. Local overexpression of toll-like receptors at the vessel wall induces atherosclerotic lesion formation: synergism of TLR-2 and TLR-4. Arterioscler Thromb Vasc Biol. 2007;27:2384–91. doi: 10.1161/ATVBAHA.106.139253. [DOI] [PubMed] [Google Scholar]
- 34.Michelsen KS, Wong MH, Shah PK, et al. Lack of toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci USA. 2004;101:10679–84. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Björkbacka H, Kunjathoor VV, Moore KJ, et al. Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat Med. 2004;10:416–21. doi: 10.1038/nm1008. [DOI] [PubMed] [Google Scholar]
- 36.Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by toll-like receptor 2. J Clin Invest. 2005;115:3149–56. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loppnow H, Werdan K, Buerke M. Vascular cells contribute to atherosclerosis by cytokine- and innate-immunity-related inflammatory mechanisms. Innate Immunity. 2008;14:63–87. doi: 10.1177/1753425908091246. [DOI] [PubMed] [Google Scholar]
- 38.Loppnow H, Brade H, Dürrbaum I, et al. IL-1 induction capacity of defined lipopolysaccharide partial structures. J Immunol. 1989;142:3229–38. [PubMed] [Google Scholar]
- 39.Auron PE, Webb AC, Rosenwasser LJ, et al. Nucleotide sequence of human monocyte interleukin-1 precursor cDNA. Proc Natl Acad Sci USA. 1984;81:7907–11. [PubMed] [Google Scholar]
- 40.Lomedico PT, Gubler U, Hellmann CP, et al. Cloning and expression of murine interleukin-1 cDNA in Escherichia coli. Nature. 1984;312:458–62. doi: 10.1038/312458a0. [DOI] [PubMed] [Google Scholar]
- 41.Singer II, Scott S, Hall GL, et al. Interleukin-1ß is localized in the cytoplasmatic ground substance but is largely absent from the golgi apparatus and plasma membranes of stimulated human monocytes. J Exp Med. 1988;167:389–407. doi: 10.1084/jem.167.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kurt-Jones EA, Beller DI, Mizel SB, et al. Identification of a membrane-associated interleukin-1 in macrophages. Proc Natl Acad Sci USA. 1985;82:1204–8. doi: 10.1073/pnas.82.4.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Loppnow H, Libby P. Functional significance of human vascular smooth muscle cell-derived interleukin-1 in paracrine and autocrine regulation pathways. Exp Cell Res. 1992;198:283–90. doi: 10.1016/0014-4827(92)90381-h. [DOI] [PubMed] [Google Scholar]
- 44.Westphal E, Chen L, Pilowski C, et al. Endotoxin-activated cultured neonatal rat cardiomyocytes express functional surface-associated interleukin-1α. J Endotoxin Res. 2007;13:25–34. doi: 10.1177/0968051907078609. [DOI] [PubMed] [Google Scholar]
- 45.Black RA, Kronheim SR, Cantrell M, et al. Generation of biologically active interleukin-1β by proteolytic cleavage of the inactive precursor. J Biol Chem. 1988;263:9437–42. [PubMed] [Google Scholar]
- 46.Schönbeck U, Herzberg M, Petersen A, et al. Human vascular smooth muscle cells express interleukin-1β-converting enzyme (ICE), but inhibit processing of the interleukin-1β precursor by ICE. J Exp Med. 1997;185:1287–94. doi: 10.1084/jem.185.7.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Westphal E, Herzberg M, Neumann I, et al. Neutrophils process IL-1ß and IL-18 precursors in a caspase-1-like fashion – processing is inhibited by human vascular smooth muscle cells. Eur Cytokine Netw. 2006;17:19–28. [PubMed] [Google Scholar]
- 48.Moyer CF, Sajuthi D, Tulli H, et al. Synthesis of IL-1α and IL-1ß by arterial cells in atherosclerosis. Am J Pathol. 1991;138:951–60. [PMC free article] [PubMed] [Google Scholar]
- 49.Loppnow H, Libby P. Proliferating or interleukin-1-activated human vascular smooth muscle cells secrete copious interleukin-6. J Clin Invest. 1990;85:731–8. doi: 10.1172/JCI114498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li J, Perrella MA, Tsai JC, et al. Induction of vascular endothelial growth factor gene expression by interleukin-1 beta in rat aortic smooth muscle cells. J Biol Chem. 1995;270:308–12. doi: 10.1074/jbc.270.1.308. [DOI] [PubMed] [Google Scholar]
- 51.Yue TL, Wang XK, Olson B, et al. Interleukin-1 beta (IL-1 beta) induces transforming growth factor-beta, (TGF-beta 1) production by rat aortic smooth muscle cells. Biochem Biophys Res Commun. 1994;204:1186–92. doi: 10.1006/bbrc.1994.2588. [DOI] [PubMed] [Google Scholar]
- 52.Yoshizumi M, Kurihara H, Morita T, et al. Interleukin-1 increases the production of endothelin by cultured endothelial cells. Biochem Biophys Res Commun. 1990;166:324–9. doi: 10.1016/0006-291x(90)91948-r. [DOI] [PubMed] [Google Scholar]
- 53.Beasley D, Cohen RA, Levinsky NG. Interleukin-1 inhibits contraction of vascular smooth muscle. J Clin Invest. 1989;83:331–5. doi: 10.1172/JCI113879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakashima Y, Wight TN, Sueishi K. Early atherosclerosis in humans: role of diffuse intimal thickening and extracellular matrix proteoglycans. Cardiovasc Res. 2008;79:14–23. doi: 10.1093/cvr/cvn099. [DOI] [PubMed] [Google Scholar]
- 55.Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15:551–61. doi: 10.1161/01.atv.15.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sukhova GK, Shi GP, Simon DI, et al. Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J Clin Invest. 1998;102:576–83. doi: 10.1172/JCI181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schlumberger W, Thie M, Rauterberg J, et al. Collagen synthesis in cultured aortic smooth muscle cells: modulation by collagen lattice culture, TGF-ß1, and EGF. Arterioscler Thromb. 1991;11:1660–6. doi: 10.1161/01.atv.11.6.1660. [DOI] [PubMed] [Google Scholar]
- 58.Katsuda S, Kaji T. Atherosclerosis and extracellular matrix. J Atheroscler Thromb. 2003;10:267–74. doi: 10.5551/jat.10.267. [DOI] [PubMed] [Google Scholar]
- 59.Gustafsson M, Boren J. Mechanism of lipoprotein retention by the extracellular matrix. Curr Opin Lipidol. 2004;15:505–14. doi: 10.1097/00041433-200410000-00003. [DOI] [PubMed] [Google Scholar]
- 60.Schönherr E, Hausser HJ. Extracellular matrix and cytokines: a functional unit. Dev Immunol. 2000;7:89–101. doi: 10.1155/2000/31748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun M, Fink PJ. A new class of reverse signaling costimulators belongs to the TNF family. J Immunol. 2007;179:4307–12. doi: 10.4049/jimmunol.179.7.4307. [DOI] [PubMed] [Google Scholar]
- 62.Ohta H, Wada H, Niwa T, et al. Disruption of tumor necrosis factor-alpha gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis. 2005;180:11–7. doi: 10.1016/j.atherosclerosis.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 63.Chandrasekharan UM, Mavrakis L, Bonfield TL, et al. Decreased atherosclerosis in mice deficient in tumor necrosis factor-alpha receptor-II (p75) Arterioscler Thromb Vasc Biol. 2007;27:e16–7. doi: 10.1161/01.ATV.0000255551.33365.22. [DOI] [PubMed] [Google Scholar]
- 64.Kirii H, Niwa T, Yamada Y, et al. Lack of interleukin-1β decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:656–60. doi: 10.1161/01.ATV.0000064374.15232.C3. [DOI] [PubMed] [Google Scholar]
- 65.Kamari Y, Werman-Venkert R, Shaish A, et al. Differential role and tissue specificity of interleukin-1alpha gene expression in atherogenesis and lipid metabolism. Atherosclerosis. 2007;195:31–8. doi: 10.1016/j.atherosclerosis.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 66.Chi H, Messas E, Levine RA, et al. Interleukin-1 receptor signaling mediates atherosclerosis associated with bacterial exposure and/or a high-fat diet in a murine apolipoprotein E heterozygote model – pharmacotherapeutic implications. Circulation. 2004;110:1678–85. doi: 10.1161/01.CIR.0000142085.39015.31. [DOI] [PubMed] [Google Scholar]
- 67.Isoda K, Shiigai M, Ishigami N, et al. Deficiency of interleukin-1 receptor antagonist promotes neointimal formation after injury. Circulation. 2003;108:516–8. doi: 10.1161/01.CIR.0000085567.18648.21. [DOI] [PubMed] [Google Scholar]
- 68.Chamberlain J, Evans D, King A, et al. Interleukin-1beta and signaling of interleukin-1 in vascular wall and circulating cells modulates the extent of neointima formation in mice. Am J Pathol. 2006;168:1396–403. doi: 10.2353/ajpath.2006.051054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Devlin CM, Kuriakose G, Hirsch E, et al. Genetic alterations of IL-1 receptor antagonist in mice affect plasma cholesterol level and foam cell lesion size. Proc Natl Acad Sci USA. 2002;99:6280–5. doi: 10.1073/pnas.092324399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Elhage R, Maret A, Pieraggi M-T, et al. Differential effects of IL-1 receptor antagonist and TNF binding protein on fatty-streak formation in apolipoprotein E-deficient mice. Circulation. 1998;97:242–4. doi: 10.1161/01.cir.97.3.242. [DOI] [PubMed] [Google Scholar]
- 71.Merhi-Soussi F, Kwak BR, Magne D, et al. Interleukin-1 plays a major role in vascular inflammation and atherosclerosis in male apolipoprotein E-knockout mice. Cardiovasc Res. 2005;66:583–93. doi: 10.1016/j.cardiores.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 72.Rupprecht HJ, Blankenberg S, Bickel C, et al. Impact of viral and bacterial infectious burden on long-term prognosis in patients with coronary artery disease. Circulation. 2001;104:25–31. doi: 10.1161/hc2601.091703. [DOI] [PubMed] [Google Scholar]
- 73.Boring L, Gosling J, Cleary M, et al. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–7. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 74.Gu L, Okada Y, Clinton SK, et al. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell. 1998;2:275–81. doi: 10.1016/s1097-2765(00)80139-2. [DOI] [PubMed] [Google Scholar]
- 75.Matsumiya T, Imaizumi T, Fujimoto K, et al. Soluble interleukin-6 receptor alpha inhibits the cytokine-Induced fractalkine/ CX3CL1 expression in human vascular endothelial cells in culture. Exp Cell Res. 2001;269:35–41. doi: 10.1006/excr.2001.5300. [DOI] [PubMed] [Google Scholar]
- 76.Lesnik P, Haskell CA, Charo IF. Decreased atherosclerosis in CX3CR1-/- mice reveals a role for fractalkine in atherogenesis. J Clin Invest. 2003;111:333–40. doi: 10.1172/JCI15555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kopf M, Baumann H, Freer G, et al. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature. 1994;368:339–42. doi: 10.1038/368339a0. [DOI] [PubMed] [Google Scholar]
- 78.Geiger T, Andus T, Klapproth J, et al. Induction of rat acute phase proteins by IL-6 in vivo. Eur J Immunol. 1988;18:717–21. doi: 10.1002/eji.1830180510. [DOI] [PubMed] [Google Scholar]
- 79.Gauldie J, Richards C, Harnish D, et al. Interferon-β2/BSF-2 shares identity with monocyte derived hepatocyte-stimulating factor and regulates the major acute phase protein response in liver cells. Proc Natl Acad Sci USA. 1987;84:7251–5. doi: 10.1073/pnas.84.20.7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.von Patay B, Loppnow H, Feindt J, et al. Catecholamines and lipopolysaccharide synergistically induce the release of interleukin-6 from thymic epithelial cells. J Neuroimmunol. 1997;86:182–9. doi: 10.1016/s0165-5728(98)00051-4. [DOI] [PubMed] [Google Scholar]
- 81.Loppnow H, Libby P. Adult human vascular endothelial cells express the IL-6 gene differentially in response to LPS or IL-1. Cell Immunol. 1989;122:493–503. doi: 10.1016/0008-8749(89)90095-6. [DOI] [PubMed] [Google Scholar]
- 82.Viedt C, Vogel J, Athanasiou T, et al. Monocyte chemoattractant protein-1 induces proliferation and interleukin-6 production in human smooth muscle cells by differential activation of nuclear factor-kappaB and activator protein-1. Arterioscler Thromb Vasc Biol. 2002;22:914–20. doi: 10.1161/01.atv.0000019009.73586.7f. [DOI] [PubMed] [Google Scholar]
- 83.Watanabe S, Mu W, Kahn A, et al. Role of JAK/STAT pathway in IL-6-induced activation of vascular smooth muscle cells. Am J Nephrol. 2004;24:387–92. doi: 10.1159/000079706. [DOI] [PubMed] [Google Scholar]
- 84.Hattori Y, Matsumura M, Kasai K. Vascular smooth muscle cell activation by C-reactive protein. Cardiovasc Res. 2003;58:186–95. doi: 10.1016/s0008-6363(02)00855-6. [DOI] [PubMed] [Google Scholar]
- 85.Klouche M, Bhakdi S, Hemmes M, et al. Novel path to activation of vascular smooth muscle cells: up-regulation of gp130 creates an autocrine activation loop by IL-6 and its soluble receptor. J Immunol. 1999;163:4583–9. [PubMed] [Google Scholar]
- 86.Ohkawa F, Ikeda U, Kawasaki K, et al. Inhibitory effect of interleukin-6 on vascular smooth muscle contraction. Am J Physiol. 1994;266:H898–902. doi: 10.1152/ajpheart.1994.266.3.H898. [DOI] [PubMed] [Google Scholar]
- 87.Lahiri T, Laporte JD, Moore PE, et al. Interleukin-6 family cytokines: signaling and effects in human airway smooth muscle cells. Am J Physiol. 2001;280:L1225–32. doi: 10.1152/ajplung.2001.280.6.L1225. [DOI] [PubMed] [Google Scholar]
- 88.Wang Z, Newman WH. Smooth muscle cell migration stimulated by interleukin-6 is associated with cytoskeletal reorganization. J Surg Res. 2003;111:261–6. doi: 10.1016/s0022-4804(03)00087-8. [DOI] [PubMed] [Google Scholar]
- 89.Zhu Y, Hojo Y, Ikeda U, et al. Interaction between monocytes and vascular smooth muscle cells enhances matrix metalloproteinase-1 production. J Cardiovasc Pharmacol. 2000;36:152–61. doi: 10.1097/00005344-200008000-00003. [DOI] [PubMed] [Google Scholar]
- 90.Kinlay S, Egido J. Inflammatory biomarkers in stable atherosclerosis. Am J Cardiol. 2006;98:2P–8P. doi: 10.1016/j.amjcard.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 91.Schieffer B, Schieffer E, Hilfiker-Kleiner D, et al. Expression of angiotensin II and interleukin-6 in human coronary atherosclerotic plaques: potential implications for inflammation and plaque instability. Circulation. 2000;101:1372–8. doi: 10.1161/01.cir.101.12.1372. [DOI] [PubMed] [Google Scholar]
- 92.Taga T, Kishimoto T. gp130 and the interleukin-6 family of cytokines. Annu Rev Immunol. 1997;15:797–819. doi: 10.1146/annurev.immunol.15.1.797. [DOI] [PubMed] [Google Scholar]
- 93.Müllberg J, Schooltink H, Stoyan T, et al. The soluble interleukin-6 receptor is generated by shedding. Eur J Immunol. 1993;23:473–80. doi: 10.1002/eji.1830230226. [DOI] [PubMed] [Google Scholar]
- 94.Lust JA, Donovan KA, Kline MP, et al. Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine. 1992;4:96–100. doi: 10.1016/1043-4666(92)90043-q. [DOI] [PubMed] [Google Scholar]
- 95.Rose-John S, Heinrich PC. Soluble receptors for cytokines and growth factors: generation and biological function. Biochem J. 1994;300:281–90. doi: 10.1042/bj3000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rose-John S, Scheller J, Elson G, et al. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: role in inflammation and cancer. J Leukoc Biol. 2006;80:227–36. doi: 10.1189/jlb.1105674. [DOI] [PubMed] [Google Scholar]
- 97.Peters M, Jacobs S, Ehlers M, et al. The function of the soluble interleukin-6 (IL-6) receptor in vivo: sensitization of human soluble IL-6 receptor transgenic mice towards IL-6 and prolongation of the plasma half-life of IL-6. J Exp Med. 1996;183:1399–406. doi: 10.1084/jem.183.4.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Althoff K, Mullberg J, Aasland D, et al. Recognition sequences and structural elements contribute to shedding susceptibility of membrane proteins. Biochem J. 2001;353:663–72. doi: 10.1042/0264-6021:3530663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Diamant M, Rieneck K, Mechti N, et al. Cloning and expression of an alternatively spliced mRNA encoding a soluble form of the human interleukin-6 signal transducer gp130. FEBS Lett. 1997;412:379–84. doi: 10.1016/s0014-5793(97)00750-3. [DOI] [PubMed] [Google Scholar]
- 100.Jostock T, Müllberg J, Özbek S, et al. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur J Biochem. 2001;268:160–7. doi: 10.1046/j.1432-1327.2001.01867.x. [DOI] [PubMed] [Google Scholar]
- 101.Cressman DE, Greenbaum LE, DeAngelis RA, et al. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 1996;274:1379–83. doi: 10.1126/science.274.5291.1379. [DOI] [PubMed] [Google Scholar]
- 102.Wallenius V, Wallenius K, Ahren B, et al. Interleukin-6-deficient mice develop mature-onset obesity. Nat Med. 2002;8:75–9. doi: 10.1038/nm0102-75. [DOI] [PubMed] [Google Scholar]
- 103.Grivennikov S, Karin E, Terzic J, et al. IL-6 and STAT3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–13. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rose-John S, Mitsuyama K, Matsumoto S, et al. Interleukin-6 trans-signaling and colonic cancer associated with inflammatory bowel disease. Curr Pharm Des. 2009;15:2095–103. doi: 10.2174/138161209788489140. [DOI] [PubMed] [Google Scholar]
- 105.Chalaris A, Rabe B, Paliga K, et al. Apoptosis is a natural stimulus of IL-6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils. Blood. 2007;110:1748–55. doi: 10.1182/blood-2007-01-067918. [DOI] [PubMed] [Google Scholar]
- 106.Rabe B, Chalaris A, May U, et al. Transgenic blockade of interleukin-6 transsignaling abrogates inflammation. Blood. 2008;111:1021–8. doi: 10.1182/blood-2007-07-102137. [DOI] [PubMed] [Google Scholar]
- 107.Atreya R, Mudter J, Finotto S, et al. Blockade of interleukin-6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat Med. 2000;6:583–8. doi: 10.1038/75068. [DOI] [PubMed] [Google Scholar]
- 108.Mitsuyama K, Matsumoto S, Rose-John S, et al. STAT3 activation via interleukin-6 trans-signalling contributes to ileitis in SAMP1/Yit mice. Gut. 2006;55:1263–9. doi: 10.1136/gut.2005.079343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hurst SM, Wilkinson TS, McLoughlin RM, et al. IL-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity. 2001;14:705–14. doi: 10.1016/s1074-7613(01)00151-0. [DOI] [PubMed] [Google Scholar]
- 110.Nowell MA, Williams AS, Carty SA, et al. Therapeutic targeting of IL-6 trans-signaling counteracts STAT3 control of experimental inflammatory arthritis. J Immunol. 2009;182:613–22. doi: 10.4049/jimmunol.182.1.613. [DOI] [PubMed] [Google Scholar]
- 111.Becker C, Fantini MC, Schramm C, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21:491–501. doi: 10.1016/j.immuni.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 112.Matsumoto S, Hara T, Mitsuyama K, et al. Essential roles of IL-6 trans-signaling in colonic epithelial cells, induced by the IL-6/soluble-IL-6 receptor derived from lamina propria macrophages, on the development of colitis-associated premalignant cancer in a murine model. J Immunol. 2010;184:1543–51. doi: 10.4049/jimmunol.0801217. [DOI] [PubMed] [Google Scholar]
- 113.Rose-John S, Waetzig GH, Scheller J, et al. The IL-6/sIL-6R complex as a novel target for therapeutic approaches. Expert Opin Ther Targets. 2007;11:613–24. doi: 10.1517/14728222.11.5.613. [DOI] [PubMed] [Google Scholar]
- 114.Melton L, Coombs A. Actemra poised to launch IL-6 inhibitors. Nat Biotechnol. 2008;26:957–9. doi: 10.1038/nbt0908-957. [DOI] [PubMed] [Google Scholar]
- 115.Coles B, Fielding CA, Rose-John S, et al. Classic interleukin-6 receptor signaling and interleukin-6 trans-signaling differentially control angiotensin II-dependent hypertension, cardiac signal transducer and activator of transcription-3 activation, and vascular hypertrophy in vivo. Am J Pathol. 2007;171:315–25. doi: 10.2353/ajpath.2007.061078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Huber SA, Sakkinen P, Conze D, et al. Interleukin-6 exacerbates early atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 1999;19:2364–7. doi: 10.1161/01.atv.19.10.2364. [DOI] [PubMed] [Google Scholar]
- 117.Takeda N, Manabe I, Shindo T, et al. Synthetic retinoid Am80 reduces scavenger receptor expression and atherosclerosis in mice by inhibiting IL-6. Arterioscler Thromb Vasc Biol. 2006;26:1177–83. doi: 10.1161/01.ATV.0000214296.94849.1c. [DOI] [PubMed] [Google Scholar]
- 118.Schieffer B, Selle T, Hilfiker A, et al. Impact of interleukin-6 on plaque development and morphology in experimental atherosclerosis. Circulation. 2004;110:3493–500. doi: 10.1161/01.CIR.0000148135.08582.97. [DOI] [PubMed] [Google Scholar]
- 119.Elhage R, Clamens S, Besnard S, et al. Involvement of interleukin-6 in atherosclerosis but not in the prevention of fatty streak formation by 17beta-estradiol in apolipoprotein E-deficient mice. Atherosclerosis. 2001;156:315–20. doi: 10.1016/s0021-9150(00)00682-1. [DOI] [PubMed] [Google Scholar]
- 120.Luchtefeld M, Schunkert H, Stoll M, et al. Signal transducer of inflammation gp130 modulates atherosclerosis in mice and man. J Exp Med. 2007;204:1935–44. doi: 10.1084/jem.20070120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Paul A, Ko KW, Li L, et al. C-reactive protein accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;109:647–55. doi: 10.1161/01.CIR.0000114526.50618.24. [DOI] [PubMed] [Google Scholar]
- 122.Libby P, Ordovas JM, Birinyi LK, et al. Inducible interleukin-1 gene expression in human vascular smooth muscle cells. J Clin Invest. 1986;78:1432–8. doi: 10.1172/JCI112732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lukacs NW, Strieter RM, Elner V, et al. Production of chemokines, interleukin-8 and monocyte chemoattractant protein-1, during monocyte: endothelial cell interactions. Blood. 1995;86:2767–73. [PubMed] [Google Scholar]
- 124.Funayama H, Ikeda U, Takahashi M, et al. Human monocyte-endothelial cell interaction induces platelet-derived growth factor expression. Cardiovasc Res. 1998;37:216–24. doi: 10.1016/s0008-6363(97)00224-1. [DOI] [PubMed] [Google Scholar]
- 125.Marczin N, Antonov A, Papapetropoulos A, et al. Monocyte-induced downregulation of nitric oxide synthase in cultured aortic endothelial cells. Arterioscler Thromb Vasc Biol. 1996;16:1095–103. doi: 10.1161/01.atv.16.9.1095. [DOI] [PubMed] [Google Scholar]
- 126.Hojo Y, Ikeda U, Takahashi M, et al. Matrix metalloproteinase-1 expression by interaction between monocytes and vascular endothelial cells. J Mol Cell Cardiol. 2000;32:1459–68. doi: 10.1006/jmcc.2000.1179. [DOI] [PubMed] [Google Scholar]
- 127.Kinard F, Jaworski K, Sergent-Engelen T, et al. Smooth muscle cells influence monocyte response to LDL as well as their adhesion and transmigration in a coculture model of the arterial wall. J Vasc Res. 2001;38:479–91. doi: 10.1159/000051081. [DOI] [PubMed] [Google Scholar]
- 128.Hojo Y, Ikeda U, Maeda Y, et al. Interaction between human monocytes and vascular smooth muscle cells induces vascular endothelial growth factor expression. Atherosclerosis. 2000;150:63–70. doi: 10.1016/s0021-9150(99)00370-6. [DOI] [PubMed] [Google Scholar]
- 129.Okada M, Hojo Y, Ikeda U, et al. Interaction between monocytes and vascular smooth muscle cells induces expression of hepatocyte growth factor. J Hypertens. 2000;18:1825–31. doi: 10.1097/00004872-200018120-00017. [DOI] [PubMed] [Google Scholar]
- 130.Proudfoot D, Fitzsimmons C, Torzewski J, et al. Inhibition of human arterial smooth muscle cell growth by human monocyte/macrophages: a co-culture study. Atherosclerosis. 1999;145:157–65. doi: 10.1016/s0021-9150(99)00028-3. [DOI] [PubMed] [Google Scholar]
- 131.Ikeda U, Maeda Y, Funayama H, et al. Monocyte-vascular smooth muscle cell interaction enhances nitric oxide production. Cardiovasc Res. 1998;37:820–5. doi: 10.1016/s0008-6363(97)00265-4. [DOI] [PubMed] [Google Scholar]
- 132.Kosuge H, Suzuki J, Haraguchi G, et al. Critical role of inducible costimulator signaling in the development of arteriosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:2660–5. doi: 10.1161/01.ATV.0000245805.52081.ca. [DOI] [PubMed] [Google Scholar]
- 133.Chen L, Frister A, Wang S, et al. Interaction of vascular smooth muscle cells and monocytes by soluble factors synergistically enhances interleukin-6 and MCP-1 production. Am J Physiol. 2009;296:H987–96. doi: 10.1152/ajpheart.01158.2008. [DOI] [PubMed] [Google Scholar]
- 134.Loppnow H, Zhang L, Buerke M, et al. Statins potently reduce the cytokine-mediated IL-6 release in SMC / MNC cocultures. J Cell Mol Med. 2010 doi: 10.1111/j.1582-4934.2010.01036.x. 10.1111/j.1582-4934.2010.01036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rock KL, Latz E, Ontiveros F, et al. The sterile inflammatory response. Annu Rev Immunol. 2010;28:321–42. doi: 10.1146/annurev-immunol-030409-101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tracey KJ. Reflex control of immunity. Nat Rev Immunol. 2009;9:418–28. doi: 10.1038/nri2566. [DOI] [PMC free article] [PubMed] [Google Scholar]