

Abstract
A defective expression or activity of neurotrophic factors, such as brain- and glial-derived neurotrophic factors, contributes to neuronal damage in Huntington’s disease (HD). Here, we focused on transforming growth factor-β (TGF-β1), a pleiotropic cytokine with an established role in mechanisms of neuroprotection. Asymptomatic HD patients showed a reduction in TGF-β1 levels in the peripheral blood, which was related to trinucleotide mutation length and glucose hypometabolism in the caudate nucleus. Immunohistochemical analysis in post-mortem brain tissues showed that TGF-β1 was reduced in cortical neurons of asymptomatic and symptomatic HD patients. Both YAC128 and R6/2 HD mutant mice showed a reduced expression of TGF-β1 in the cerebral cortex, localized in neurons, but not in astrocytes. We examined the pharmacological regulation of TGF-β1 formation in asymptomatic R6/2 mice, where blood TGF-β1 levels were also reduced. In these R6/2 mice, both the mGlu2/3 metabotropic glutamate receptor agonist, LY379268, and riluzole failed to increase TGF-β1 formation in the cerebral cortex and corpus striatum, suggesting that a defect in the regulation of TGF-β1 production is associated with HD. Accordingly, reduced TGF-β1 mRNA and protein levels were found in cultured astrocytes transfected with mutated exon 1 of the human huntingtin gene, and in striatal knock-in cell lines expressing full-length huntingtin with an expanded glutamine repeat. Taken together, our data suggest that serum TGF-β1 levels are potential biomarkers of HD development during the asymptomatic phase of the disease, and raise the possibility that strategies aimed at rescuing TGF-β1 levels in the brain may influence the progression of HD.
Keywords: transforming growth factor-β, Huntington’s disease, brain cortex, peripheral markers, neurodegeneration, neurodysfunction
Introduction
Huntington’s disease (HD) is a hereditary progressive and invalidating disease caused by an expanded cytosine-adenine-guanosine (CAG) repeat in the gene encoding the ubiquitous protein, huntingtin [1, 2]. Mutated huntingtin causes neuronal and glial dysfunction in the striatum and cerebral cortex [3, 4] through a cascade of events that include excitotoxic mechanisms [5] and abnormal production and transport of neurotrophic factors [6]. Activation of astrocytes and microglia, which reflects a process of neuroinflammation, is already observed during the asymptomatic stage of HD, and correlates with the severity of HD progression [7–9]. Activated astrocytes and microglia critically regulate processes of neuronal death and survival by secreting glutamate, neurotrophic factors, and pro- and anti-inflammatory cytokines [10]. An imbalance between neurotoxic and neuroprotective factors may ultimately be responsible for neuronal dysfunction and cell death in HD. Here, we focused on transforming growth factor-β (TGF-β), which is increasingly recognized as an endogenous neuroprotective factor and has been implicated in the pathophysiology of chronic neurodegenerative disorders [11, 12]. Accordingly, an altered expression of type-II TGF-β receptor has been demonstrated in the brain of patients affected with Alzheimer’s disease (AD) [13–16], and a reduced TGF-β1 signalling increases amyloid deposition and neurodegeneration in transgenic AD mice [17]. In addition, TGF-β1 loss in transgenic mice enhances neuronal susceptibility to toxic insults [18]. TGF-β1 production is under the control of extracellular signals, which include oestrogens and excitatory amino acids acting at mGlu3 metabotropic glutamate receptors [12, 19]. Activation of glial mGlu3 receptors is neuroprotective via a paracrine mechanism mediated by TGF-β1[12, 20, 21]. Interestingly, the expression of mGlu3 receptors is substantially reduced in the striatum of transgenic R6/2 mice, which express exon 1 of the human HD gene with an expanded CAG repeat [22]. We suggested that early abnormalities in the production of TGF-β1 could contribute to the pathophysiology of neuronal death associated with HD. To examine this possibility, we measured TGF-β1 levels in the brain and serum of asymptomatic and symptomatic HD patients, as related to the stage and progression of the disease. In addition, we examined the expression and regulation of TGF-β1 in mutant HD mice or in cultured cells expressing a mutated huntingtin.
Subjects and methods
HD patients
All participants underwent genetic testing after informed consent and neurological examination, including motor, psychiatric, cognitive and functional assessments, by physicians with expertise in HD [23]. Age at onset of symptoms was calculated according to the initial neurological manifestations [23] and the predicted time to onset in years according to a CAG-based model, as described [24]. Motor symptoms, behavioural and cognitive changes were assessed clinically with the Unified Huntington’s Disease Rating Scale (UHDRS) [25] and Mini-Mental State Examination (MMSE) [26]. Patients’ consent was obtained according to the Declaration of Helsinki. All asymptomatic patients had no or minimal and nonspecific clinical motor or behavioural manifestations with cognitive scores almost unchanged over the previous 2 years [27]. These patients were not taking benzodiazepines or neuroleptics or other drugs for cardiological or psychiatric pathologies. The rate of independence decline and symptom progression was measured in loss of units per year using the Disability Scale (DS) [23, 28, 29], in patients with a disease history of at least 5 years. The DS combines patients’ independence and motor performance, thus taking into account the patients’ independence on neurological motor impairment [28]. The disease stage was calculated according to the total functional capacity score [29]. Most patients were taking benzodiazepines; some patients in stages IV-V of disease were receiving low doses of atypical neuroleptics (olanzapine, 2.5–10 mg, risperidone, 1–3 mg or tetrabenazine 12.5–25 mg), associated, in some patients, with benzodiazepines, lithium carbonate or valproate.
Positron-emission-tomography (PET) scanning in HD patients
Twenty-three asymptomatic and 55 symptomatic patients underwent static [fluorine-18]-fluoro- 36 32-deoxy-D-glucose (FDG) FDG-PET scan after injection of 300 MBq of FDG in the resting state, with eyes closed and ears plugged to reduce background noise [27, 30]. Images were acquired about 30 min. after tracer injection with an ECAT EXACT 47 scanner (CTI/Siemens, Knoxville, TN, USA). After a transmission scan lasting 5 min. with a Ge-68 rod source, an emission scan lasting 25 min. was done in two dimensional mode. Emission scan images were reconstructed using the back-projection method with a Shepp-Logan filter (cut-off frequency 0.35), resulting in 47 slices with a 128 × 128 matrix (pixel size 1.8 mm) and an interplane separation of 3.125 mm. The attenuation effects were corrected with measured transmission images. PET analysis was performed with regions of interest, defined in the normalized space of Talairach using the automated voxel identification routine of the Talairach Daemon software. Data were normalized by mean global counts (measured as [18F]-FDG uptake).
Measurements of TGF-β1 levels in human and mouse serum
To measure TGF-β1 levels in human serum, peripheral venous blood samples from HD patients and healthy controls were collected between 8 and 11 a.m. [31]. To minimize possible influence of patients’ circadian rhythms on plasma TGF-β1 concentrations, we also confirmed the cytokine levels in blood samples collected between 5 and 7 p.m. in a group of 10 patients and 10 control patients [31]. No age-related differences in TGF-β1 serum levels were noticed in control patients, as expected [32], thus excluding the potential bias due to the younger age of asymptomatic patients (Fig. S1). Blood samples were centrifuged at room temperature (3500 ×g for 10 min.) and plasma samples were stored at −80°C until processing. Asymptomatic (4 weeks) and symptomatic (12 weeks) transgenic R6/2 mice and asymptomatic YAC128 mice (2 months) were killed to collect blood and brain tissues. Each group included a total of 15 transgenic asymptomatic mice with respective number of wild-type control and age-matched mice. The mice were anaesthetized by intraperitoneal injection of 0.5 ml of Pentoject (sodium pentobarbitone, Animal Ltd., York, UK) and exsanguinated by intracardiac puncture, followed by cervical dislocation. Mouse blood was transferred into a standard 1.5 ml plastic eppendorf tube and allowed to stand for 2 hrs to clot. The blood was spun at 3700 ×g for 5 min. and the supernatants were stored at −80°C until processing. Serum TGF-β1 levels were measured using an ELISA kit (TGF-β1 Emax Immunoassay System kit Promega, Madison, WI, USA), according to the manufacturer’s instructions. Serum TGF-β1 concentrations were measured in four independent experiments. Medium TGF-β1 levels from knock-in cells were measured using the same kit and according to the manufacturer’s instructions.
Transgenic R6/2 and YAC128 mice
Breeding pairs of the R6/2 line of transgenic mice (containing 250 CAG repeats in exon-1 of human HD gene; strain name: B6CBATgN(HDexon1) 62Gpb/J) [33] and of the YAC line of transgenic mice (containing 128 CAG repeats in a YAC with human full length HD gene; strain name: FVB-Tg (YAC128)53Hay/J) [34], were purchased from the Jackson Laboratories (Bar Harbor, ME, USA). The lines were maintained by backcrossing to B6CBAF1 X C57BL/6 F1 and to FVB, respectively. The mice were maintained in the animal facilities of the Neurological Institute Neuromed (R6/2), of University of Alberta (YAC128) and of British Columbia (YAC128), and kept under environmentally controlled conditions (ambient temperature = 22°C, humidity = 40%) on a 12-hr light/dark cycle with food and water ad libitum. Experiments were performed following the Guidelines for Animal Care and Use of the National Institutes of Health. Hemizygous transgenic mice were used in all experiments, as was confirmed by PCR according to the protocol by Jackson Laboratories (http://jaxmice.jax.org/pub-cgi/protocols/protocols.sh?objtype1/4 protocol& protocol_ id ¼282). Mice were housed in groups of transgenic and non-transgenic littermates.
Measurements of TGF-β1 levels in mice
Wild-type and R6/2 mice at 4 weeks of age were treated with saline or LY379268 (10 mg/kg, i.p.) and killed 6 hrs later. Both cortical cortices and striata were dissected out and immediately homogenized in radioimmunoprecipitation assay (RIPA) buffer for measurements of TGF-β1 protein levels. The amount of TGF-β1 protein was assessed by Western blot analysis. Wild-type and R6/2 mice, 4 week old, were also treated with saline or riluzole (10 mg/kg, i.p.), a drug increasing patients’ brain [35] and serum [36] concentration of the brain-derived neurotrophic factor (BDNF), and killed 6 hrs later. Cerebral cortex and striatum were dissected out and used for TGF-β1 protein assessment. Brain areas were dissected out from transgenic mice and age-matched controls (4 weeks for R6/2 mice and 2 months for YAC128 mice). Tissues were homogenized at 4°C in a buffer composed of Tris-HCl pH 7.4, 10 mM; NaCl, 150 mM; ethylenediaminetetraacetic acid, 5 mM; phenylmethanesulphonylfluoride (PMSF), 10 mM; Triton X-100, 1%; leupeptin, 1 μg/ml; aprotinin, 1 μg/ml with a motor-driven Teflon-glass homogenizer (1700 rev./min.). Tissue extracts were used for protein determinations; 30 μg of proteins were resuspended in SDS-bromophenol blue reducing buffer with 40 mM dithiothreitol (DTT) and used for protein identification. Western blot analyses were carried out using 8% SDS, for mGlu2/3 receptors, and 12.5%, for TGF-β1, polyacrylamide gels run on a minigel apparatus (Biorad, Milano, Italy; Mini Protean II Cell); gels were electroblotted on ImmunBlot PVDF Membrane (Biorad) for 1 hr using a semi-dry electroblotting system (Biorad, Trans-blot system SD), and filters were blocked overnight in TTBS buffer (100 mM Tris-HCl; 0.9% NaCl, 0.1% Tween 20, pH 7.4) containing 5% non-fat dry milk. Blots were then incubated for 1 hr at room temperature with a primary polyclonal antibody (2 μg/ml) which recognizes mGlu2/3 receptors (Chemicon International, Inc., Tecumela, CA, USA), or overnight at 4°C with a monoclonal anti-human TGF-β1 antibody (1.5 μg/ml, Chemicon International, Inc.) and then incubated for 1 hr with the secondary antibody (1:5000 or 1:10,000, peroxidase-coupled antimouse, Amersham, Milano, Italy). Immunostaining was revealed by the enhanced chemiluminescence (ECL) Western blotting analysis system (Amersham). The blots were reprobed with anti-β-actin monoclonal antibody (1:250, Sigma, St. Louis, MO, USA).
Immunocytochemistry for TGF-β1 in human and mouse brain samples
We examined six cases of HD, one asymptomatic and eight control patients. One patient affected by multiple sclerosis was included as positive control expressing high levels of TGF-β1. Patients’ data were obtained from the databases of the department of Neuropathology of the Academical Medical Center (University of Amsterdam, UVA) in Amsterdam and the Department of Neurology of Leiden University Medical Center (LUMC). Informed consent was obtained for the use of brain tissue and for access to medical records for research purposes. Tissue was obtained and used in a manner compliant with the Declaration of Helsinki (Br Med J 1991; 302; 1194). Formalin-fixed, paraffin-embedded tissue was sectioned at 6 μm and mounted on organosilane-coated slides (Sigma). Representative sections of all specimens were processed for haematoxylin eosin, as well as for immunocytochemical reactions. The anti-TGF-β1 (LC1–30; purified rabbit antibody) was kindly provided by Dr. K. Flanders, (National Cancer Institute, Bethesda, MD, USA) [37, 38]. Immunocytochemistry was carried out on paraffin-embedded tissue as previously described [39]. The sections were incubated with the primary antibody (1:50) overnight at 4°C. Single-label immunocytochemistry was performed with avidin-biotin peroxidase method for the polyclonal goat antibodies and Powervision (Immunologic, Duiven, The Netherlands) for the monoclonal mouse and polyclonal rabbit antibodies. As chromogen, 3,3-diaminobenzidine was used. Sections incubated without the primary antibody were essentially blank.
Mouse brains were dissected out and immediately placed in a solution composed of ethyl alcohol (60%), acetic acid (10%) and chloroform (30%). Twenty hours later brains were placed in 70% ethanol until they were included in paraffin. Ten micrometre serial sections were cut and used for histological analysis. Sections were incubated overnight with rabbit polyclonal anti-TGF-β1(V) (1:5, Santa Cruz Biotechnology, Santa Cruz, CA, USA) antibodies, and then for 1 hr with secondary biotinylated anti-rabbit antibodies (1:200; Vector Laboratories, Burlingame, CA, USA). 3,3-Diaminobenzidine tetrachloride was used for detection (ABC Elite kit; Vector Laboratories). Control staining was performed without the primary antibodies. Double fluorescence immunohistochemistry was performed by incubating brain sections overnight with polyclonal anti-TGF-β1(V) (1:5) and monoclonal mouse anti-glial fibrillary acidic protein (GFAP) (1:100; Sigma-Aldrich; Milan, Italy) or mouse anti-NeuN (1:100; Chemicon International, Inc.) antibodies, and then for 1 hr with secondary Cy3 anti-rabbit (1:300; Chemicon International, Inc.) and fluorescein antimouse (1:100; Vector, Laboratories) antibodies.
In vitro experiments
Glial cell cultures were prepared from cortex of postnatal CD1 mice (1–3 days after birth), as previously described [40]. Dissociated cortical cells were grown in 60 mm dishes (Falcon Primaria, Lincoln Park, NJ, USA) using a plating medium of minimum essential medium (MEM)-Eagle’s salts supplemented with 10% of heat inactivated horse serum, 10% foetal bovine serum, 2 mM glutamine, 25 mM sodium bicarbonate and 21 mM glucose. Cultures were kept at 37°C in a humidified CO2 atmosphere. Confluent cells were trensfected (see below) and incubated with LY379268 (1 μM for 10 min.), and after the drug washout, cultures were kept for 6–8 hrs (for real time PCR analysis) or 20 hrs (for cytometry analysis) in the incubator.
Striatal knock-in cultures
Striatal knock-in cells, stably expressing full-length huntingtin STHdhQ7/Q7, (ST7/7Q, wild-type cells) or full-length mutated huntingtin STHdhQ111/Q111 (ST111/111Q, mhtt cells) established from HdhQ111 knock-in mice [41], were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% foetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine and 400 μg/ml geneticin (G-418), at 33°C in a humidified 5% CO2 atmosphere. Cultures cells were transfected (see below) and incubated with LY379268 (1 μM for 10 min.), and after the drug washout, cultures were kept for 6–8 hrs (for real time PCR analysis) or 20 hrs (for cytometry and ELISA analysis) in the incubator.
Cell transfections
All DNA constructs were transfected using Lipofectamine 2000 (Invitrogen, Stockholm, Sweden) as instructed by the manufacturer. Both ST7/7Q, ST111/111Q and primary astrocyte cells were transfected at 50% of confluence. DNA constructs were exon-1-Htt-25Q-EGFP which express 25 CAG triplets and exon-1-Htt-72Q-EGFP which express 72 CAG triplets. The expression of the green fluorescence protein was used as an index of successful transfection.
Real-time RT-PCR analysis
Total RNA isolated with TRIzol®LS (Invitrogen) was treated with DNase I (Qiagen, Milan, Italy) and retrotranscribed into cDNA by using SuperScript III Reverse Transcriptase (Invitrogen). Real-time RT-PCR was performed on a Step One PCR cycler (Applied Biosystems, Foster City, CA, USA). PCR was performed by using Power SYBR Green PCR Master Mix Kit (Applied Biosystems) according to the manufacturer’s instructions. Thermal cycler conditions were as follows: 5 min. at 50°C, 1 min. at 95°C, 40 cycles of denaturation (45 sec. at 95°C), and combined annealing/extension (1 min. at 59°C). The sequences of β-actin and TGF-β1 primers used are: β-actin Forward: 5′-ctggctcctagcaccatga-3′ and Reverse: 5′-tagagccaccaatccacaca-3′ TGF-β1 Forward: 5′-acaattcctggcgttacctt- 3′ and Reverse: 5′-ccacgtggagtttgttatct- 3′.
Gene expression measures were derived from samples run in triplicates. Relative expression was calculated by normalization to β-actin expression using the ΔΔCt method [42].
Measurements of TGF-β1 levels by fluorescent-activated cell sorting (FACS) analysis
Intracellular expression of TGF-β1 in primary astrocyte, ST7/7Q and ST111/111Q cell cultures, was performed by three colour-flow cytometry using a Becton-Dickinson FACS calibur flow cytometer (Franklin Lakes, NJ, USA). Cell suspension was fixed with 2% paraformaldehyde for 20 min. at room temperature, resuspended in permeabilization buffer (0.1%[w/v] saponin, 0.05%[w/v] NaN3 in phosphate-buffered solution [PBS]) and centrifuged at 200 g for 7 min. Cells were then incubated with PE-conjugated anti-TGF-β1 monoclonal antibody and phycoerythrin (PE)-conjugated IgG1 isotype (negative control) for 45 min. at room temperature (PharMingen, San Jose, CA USA). Cells were washed and resuspended with PBS before acquisition. The cytokine-positive cells were scored on the basis of isotype control.
Statistical analysis
Student’s t-test was used to compare between-group differences in the median age. Statistical differences in serum TGF-β1 levels among patients at different HD stage and versus controls were performed by Student’s t-test and anova. Linear dependence of serum TGF-β1 on CAG repeat expansion, time to onset in years, DS loss of units per year and on 18F-FDG uptake reduction in brain caudate, was determined by a simple regression model. Other statistical analyses were performed by Student’s t-test or one-way anova+ Fisher’s PLSD. Data were considered statistically significant at P < 0.05. Statistical analysis was performed with the StatView 5 software.
Results
Clinical, genetic, serum TGF-β1 levels and demographic data from all patients are summarized in the Table S1.
TGF-β1 serum levels in HD patients and healthy controls
Serum TGF-β1 levels in the total population of HD patients were lower than in non-HD controls (P= 0.0002) and this entirely reflected the drop in TGF-β1 levels found in the patients at the asymptomatic and first HD stages (Fig. 1A, B). No significant changes in serum TGF-β1 levels were found between controls and sympthomatic patients regardless of their clinical stage (Fig. 1B). In asymptomatic patients, we found a negative correlation between serum TGF-β1 levels and the number of CAG repeats in the huntingtin gene (Fig. 1C), whereas there was no correlation between TGF-β1 levels and the number of CAG repeats in symptomatic patients (Fig. 1D). We also found a significant correlation (P= 0.029) with the rate of progression in symptomatic patients (Fig. 1E). Asymptomatic HD patients, whose estimated age at onset of symptoms in years was predicted by a mathematical CAG-based model [24], showed a relative increase in serum cytokine levels while approaching manifest HD (P= 0.0096; Fig. 1F), although the mean serum concentrations of TGF-β1 were still reduced as compared to control patients (P= 0.0004).
Fig 1.
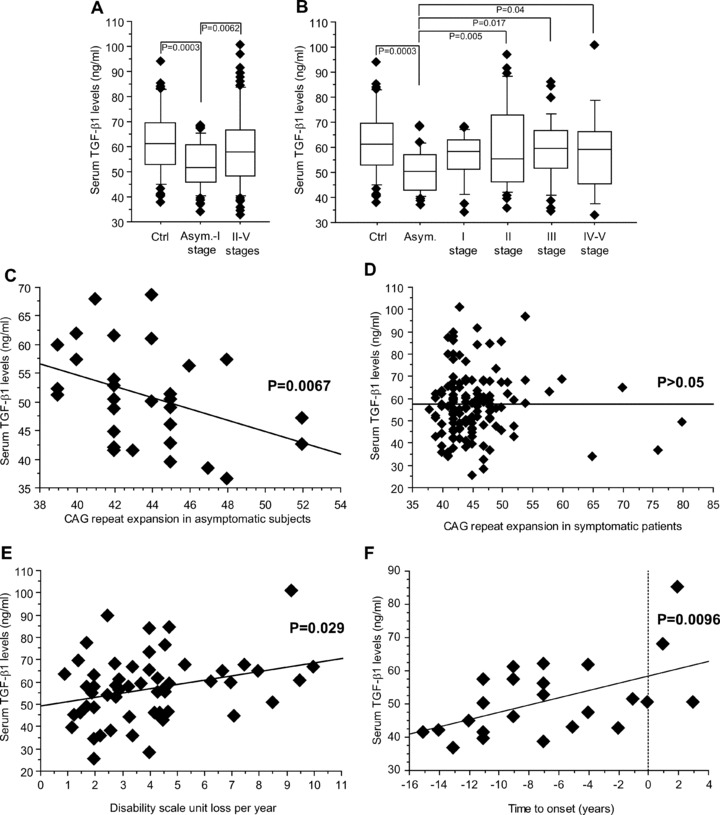
Serum TGF-β1 levels in HD patients. Reduced serum TGF-β1 levels in asymptomatic and stage-I HD patients (A). TGF-β1 levels in control (Ctrl) and all life stage patients are shown in (B). Statistical analysis was carried out by Student’s t-test in (A) and by one-way anova+ Fisher PLSD in (B). Linear dependence of serum TGF-β1 levels on expanded CAG repeat number in asymptomatic patients is shown in (C) (n= 30, R2= 0.16, P= 0.0067). The lack of correlation between serum TGF-β1 levels and the number of expanded CAG repeats in symptomatic patients is shown in (D). (E) Patients with disease length beyond 5 years and manifest HD (stages II-V) showed a slight but significant linear dependence of serum TGF-β1 levels on the rate of progression of the disease, calculated as loss of units of the DS per year (n= 54, R2= 0.10, P= 0.029). (F) Serum TGF-β1 in asymptomatic mutation carriers, whose predicted age at onset was estimated within further 15 years, showed an increase linearly correlating with the estimated time to onset (n= 24, R2= 0.27, P= 0.0096, simple regression analysis). The few patients with estimated time to onset higher than 0 on x-axis showed an age in years delayed to that expected on the basis of their CAG size.
We then examined the relation between changes in TGF-β1 serum levels and glucose metabolism in the caudate nucleus, as assessed by FDG-PET scan in both asymptomatic and symptomatic patients. In asymptomatic patients, serum TGF-β1 levels showed a positive correlation with glucose metabolism in the caudate nucleus (Fig. 2).
Fig 2.
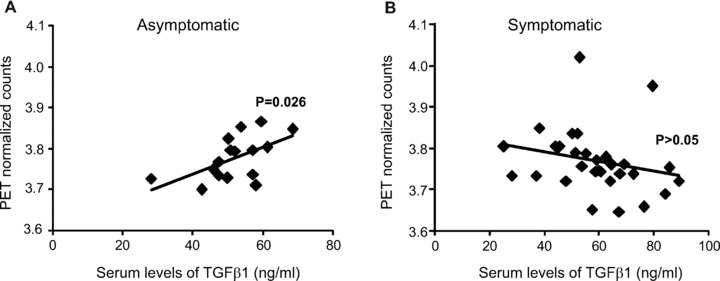
Correlation between serum TGF-β1 levels and glucose metabolism in patients at asymptomatic and symptomatic HD stages. The relation between serum TGF-β1 levels and glucose metabolism in the caudate nucleus of asymptomatic and symptomatic HD patients is shown in (A) and (B), respectively. Note that a positive correlation is found only in asymptomatic patients (n= 23, R2= 0.31, P= 0.026).
TGF-β1 expression in the HD brain
We examined the expression of TGF-β1 by immunohistochemistry in post-mortem brain samples of six symptomatic patients (grades III–IV), one asymptomatic patient who was clinically and genetically characterized as described [43], and eight controls (see Table 1). Expression was also examined in the cerebral cortex of one patient with multiple sclerosis, considered as a positive control because of the expected increase in microglia immunoreactivity. TGF-β1 immunoreactive neurons could be detected in both the cerebral cortex and caudate nucleus of all control patients, whereas no expression was found in cortical or striatal astrocytes, or in the white matter of control patients. The asymptomatic patient was devoid of TGF-β-immunoreactive neurons in the cerebral cortex, and did not show TGF-β1 immunoreactivity in the white matter (Fig. 3). The caudate nucleus of the asymptomatic patient was not available. In symptomatic patients no expression of TGF-β1 in cortical neurons and low-to-moderate TGF-β1 expression in cortical astrocytes, were detected. In contrast, TGF-β1 was highly expressed in the white matter and caudate nucleus of symptomatic patients, where reactive astrocytes and microglia showed a strong TGF-β1 immunoreactivity (Fig. 3; Table 1).
Table 1.
Pathological, genetic, clinical data and expression of TGF-β1 in cerebral cortex, white matter and caudate nucleus of eight control subjects, six patients with original pathological diagnosis of HD and one asymptomatic subject
| Patients | Age in years | Gender | Vonsattel et al.’s Grade | CAG repeats | Brain weight (g) | Clinical diagnosis of HD | TGF-β1immunoreactivity* | ||
|---|---|---|---|---|---|---|---|---|---|
| Cerebral cortex | White matter | Caudate nucleus | |||||||
| 1 | 59 | F | 0 | - | 1456 | - | 2 | 0 | 2 |
| 2 | 73 | M | 0 | - | 1389 | - | 2 | 0 | 1 |
| 3 | 66 | F | 0 | - | 1463 | - | 3 | 0 | 2 |
| 4 | 61 | M | 0 | - | 1510 | - | 3 | 0 | 1 |
| 5 | 68 | F | 0 | - | 1478 | - | 2 | 0 | 1 |
| 6 | 67 | M | 0 | - | 1470 | - | 2 | 0 | 1 |
| 7 | 59 | F | 0 | - | 1480 | - | 3 | 0 | 1 |
| 8 | 64 | M | 0 | - | 1420 | - | 2 | 0 | 2 |
| Mean ± S.E.M. | 64.6 ± 1.8 | 2.4 ± 0.2 | 1.4 ± 0.2 | ||||||
| 9 | 67 | M | III | 9/43 | 1185 | + | 1 | 2 | 3 |
| 10 | 61 | F | III | 22/44 | 1070 | + | 1 | 2 | 4 |
| 11 | 60 | F | III-IV | 20/46 | 825 | + | 2 | 3 | 3 |
| 12 | 61 | F | III | 25/44 | 800 | + | 1 | 3 | 3 |
| 13 | 59 | M | III | 19/42 | 1330 | + | 2 | 2 | 4 |
| 14 | 67 | M | III-IV | 20/45 | 1160 | + | 1 | 2 | 4 |
| Mean ± S.E.M. | 62.5 ± 1.6 | 1.3 ± 0.2 | 2.3 ± 0.2 | 3.5 ± 0.2 | |||||
| 15 | 39 | F | 0 | ND# | ND | At risk | 0 | 1 | |
Subjects 1–8= Healthy controls; subjects 9–14= Symptomatic HD patients, subjects 15= asymptomatic subjects (positive genetic test of HD and unreported CAG size #). HD: Huntington disease; (0)= negative; (1)= light; (2)= moderate; (3)= strong. *P <0.05 (Student’s t-test) HD versus non-HD patients.
Fig 3.
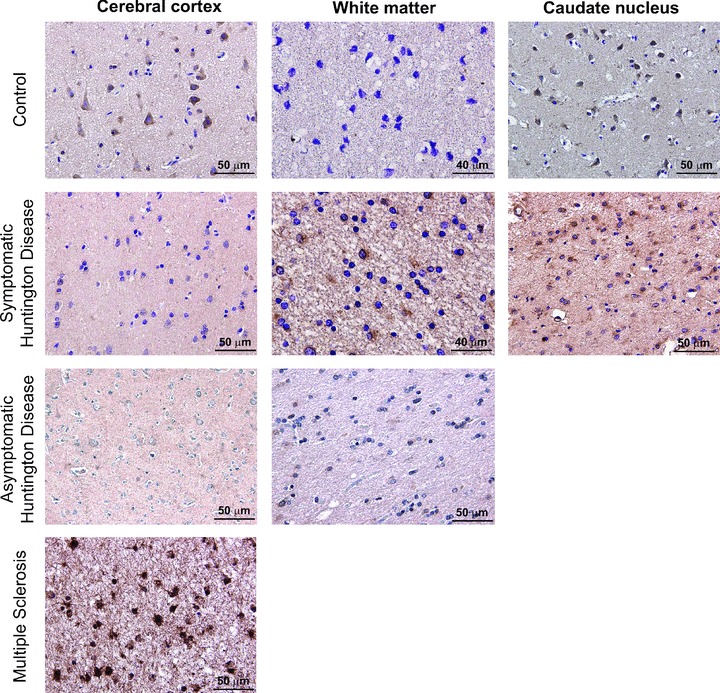
TGF-β1 Immunohistochemistry in post-mortem brain samples of HD and non-HD patients. Immunohistochemistry for TGF-β1 in representative post-mortem samples of the cerebral cortex, white matter and caudate nucleus of a control patient, a symptomatic patient, an asymptomatic patient and a patient with multiple sclerosis. Note the substantial loss of TGF-β1 in cortical neurons of the asymptomatic patient.
Reduced cortical TGF-β1 levels in asymptomatic HD mice
We examined the expression of TGF-β1 protein in the cerebral cortex and striatum of asymptomatic YAC128 and R6/2 mice (8 and 4 week old, respectively) and symptomatic R6/2 mice (12 week old). Immunoblot analysis showed a reduction in cortical TGF-β1 levels and no changes in striatal TGF-β1 levels in asymptomatic YAC128 and R6/2 mice (Figs 4A and 5A). Symptomatic R6/2 mice also showed a reduced expression of TGF-β1 levels in the cerebral cortex associated with a slight increase in TGF-β1 levels in the striatum (Fig. 5D). Immunohistochemical analysis confirmed the reduction in TGF-β1 levels in the cerebral cortex of asymptomatic R6/2 mice (Fig. 6A), and showed that TGF-β1 was exclusively localized in cortical neurons of both wild-type and R6/2 mice (Fig. 6B, C). TGF-β1 expression appeared to be also reduced in the CA1 region of the hippocampus and in the thalamus, but did not change in the striatum and cerebellum (Fig. 6A). TGF-β1 levels in peripheral blood were substantially reduced in asymptomatic R6/2 mice, but returned back to normal in symptomatic R6/2 mice (Fig. 5D). Serum TGF-β1 levels were unchanged in asymptomatic YAC128 mice (Fig. 4B). We also examined whether a defect in the pharmacological regulation of TGF-β1 formation occurred in asymptomatic R6/2 mice. First, we treated mice with the mGlu2/3 receptor agonist, LY379268 (10 mg/kg, i.p.), which is known to up-regulate the expression of TGF-β1 in brain tissues [12, 20]. A single injection of LY379268 increased TGF-β1 levels in the striatum and cerebral cortex of wild-type mice but did not change TGF-β1 levels in 4-week-old R6/2 mice (Fig. 5A). However, asymptomatic R6/2 mice also showed a reduced mGlu2/3 receptor expression in both cerebral cortex and striatum (Fig. 5B), a finding that might contribute to explain their lack of activity of LY379268. Interestingly, an increase in both cortical and striatal TGF-β1 levels was also observed in mice receiving a single i.p. injection with 10 mg/kg of riluzole (Fig. 5C), a drug that does not interact with mGlu receptors. Similar to LY379268, riluzole was inactive on cortical and striatal TGF-β1 levels in asymptomatic R6/2 mice (Fig. 5). Taken together these data suggest that HD mice have a defective regulation of TGF-β1 formation.
Fig 4.
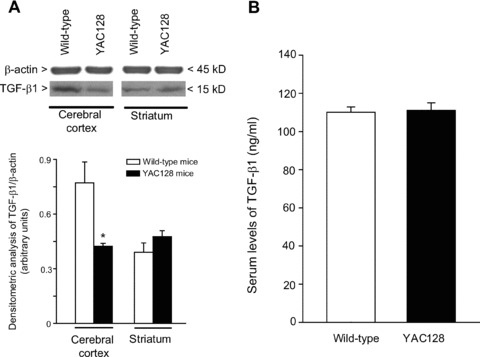
TGF-β1 in transgenic YAC128 mice brain and peripheral serum. Immunoblot analysis of TGF-β1 in the cerebral cortex and striatum of wild-type and asymptomatic YAC128 mice (8 weeks of age) is shown in (A). Densitometric values are means ± S.E.M. of five to seven determinations. *P < 0.05 (Student’s t-test) versus the corresponding values obtained in wild-type mice. Serum TGF-β1 levels in wild-type and asymptomatic YAC128 mice are shown in (B), where values are means ± S.E.M. of five to seven determinations.
Fig 5.
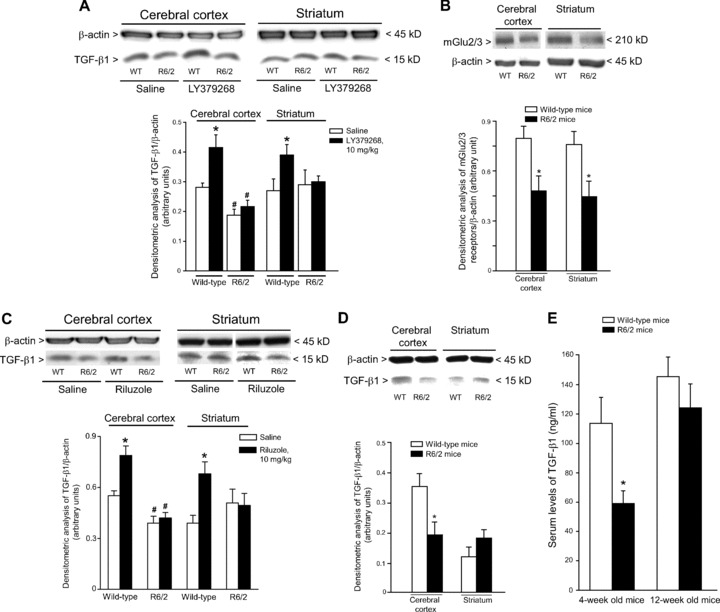
TGF-β1 levels in the cerebral cortex, striatum, and peripheral blood of asymptomatic or symptomatic R6/2 mice. Immunoblot analysis of TGF-β1 in the cerebral cortex and striatum of wild-type, and asymptomatic (4 weeks of age) R6/2 mice is shown in (A). Mice were also treated with a single i.p. injection of saline or LY379268 (10 mg/kg). Values are means ± S.E.M. of six determinations. P < 0.05 versus the respective values obtained in mice treated with saline (*) or versus the respective values obtained in wild-type mice (#) (one-way anova+ Fisher’s PLSD). A representative immunoblot is also shown. Immunoblot analysis of mGlu2/3 receptors in the cerebral cortex and striatum of asymptomatic R6/2 mice is shown in (B), where values are means ± S.E.M. of six determinations. *P < 0.05 versus the respective values obtained in wild-type mice (Student’s t-test). Immunoblot analysis of TGF-β1 in the cerebral cortex and striatum of wild-type and asymptomatic R6/2 mice treated with a single i.p. injection of saline or riluzole (10 mg/kg) is shown in (C). Values are means ± S.E.M. of five to six determinations. P < 0.05 versus the respective values obtained in mice treated with saline (*) or versus the respective values obtained in wild-type mice (#) (one-way anova+ Fisher’s PLSD). Immunoblot analysis of TGF-β1 in the cerebral cortex and striatum of wild-type and symptomatic (12 weeks of age) R6/2 mice is shown in (D). Values are means + S.E.M. of five to six determinations. *P < 0.05 (Student’s t-test) versus the respective values obtained in age matched wild-type mice. Serum TGF-β1 levels in wild-type, asymptomatic and symptomatic R6/2 mice are shown in (E), where values are means + S.E.M. of six determinations. P < 0.05 versus values obtained in age-matched wild-type mice (*) (Student’s t-test).
Fig 6.
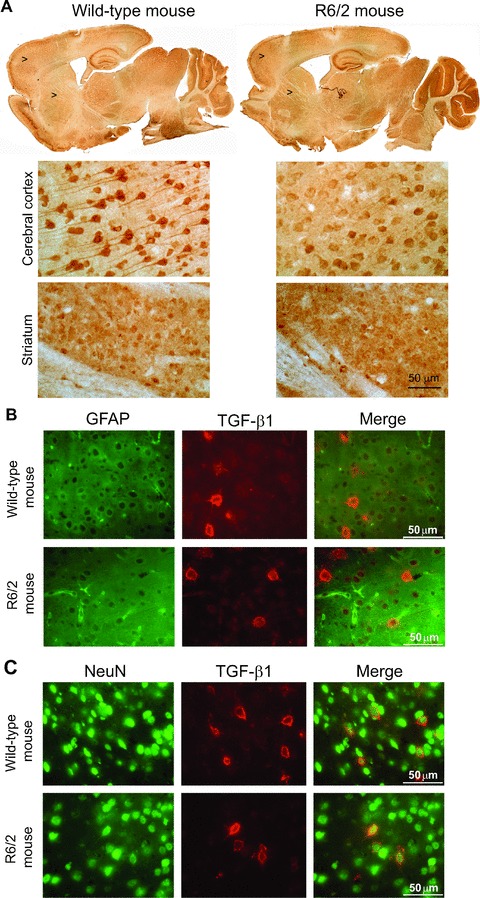
Immunohistochemical analysis of TGF-β1 in wild-type and asymptomatic R6/2 mice. Immunohistochemical analysis of TGF-β1 in a representative wild-type and asymptomatic R6/2 mouse is shown in (A). Arrowheads indicate the zone showed at higher magnification. Double fluorescent staining for TGF-β1 and GFAP, or TGF-β1 and NeuN in cortical neurons of a representative wild-type and asymptomatic R6/2 mouse is shown in (B) and (C), respectively. Note that TGF-β1 is expressed in neurons and not in astrocytes.
Reduced expression of TGF-β1 in cultured astrocytes expressing 72Q huntingtin exon 1 (exon-1 htt 72Q)
We used cultured cortical astrocytes to examine whether the presence of huntingtin with an expanded Q repeat could cause a defect in the production and/or regulation of TGF-β1. Cultures were transfected with exon 1 of the human huntingtin gene encoding for an expanded 72Q repeat or an unexpanded 25Q repeat (the latter used as a control). Real-time PCR analysis showed that basal TGF-β1 mRNA levels were lower in cultures expressing huntingtin with the expanded Q repeats. Addition of the mGlu2/3 receptor agonist, LY379268, enhanced TGF-β1 mRNA levels in control cultures, as expected [12, 20], but, interestingly, was ineffective in cultures expressing mutated huntingtin (Fig. 7A). Similar results were obtained by cytofluorimetric analysis, which showed a lower percentage of TGF-β1-expressing cells in cultures transfected with huntingtin exon-1 containing the expanded Q repeat both under basal conditions and in response to LY379268 (Fig. 7B,C).
Fig 7.
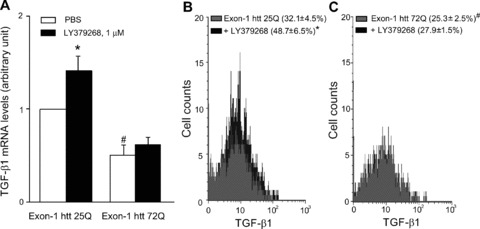
Analysis of TGF-β1 in astrocytes expressing exon-1 htt 72Q. Real Time RT-PCR of TGF-β1 mRNA analysis (A) and FACS analysis of intracellular TGF-β1 protein (B, C) in astrocytes transfected with exon-1 of human huntingtin (htt) containing an expanded 72Q repeat or an unexpanded 25Q repeat used as a control, under basal conditions and after treatment with LY379268, 1 μM for 10 min. The mean fluorescence intensity (MFI) of TGF-β1 in astrocytes transfected with exon-1 htt 25Q or exon-1 htt 72Q and treated with PBS or LY379268 was unchanged (MFI: 22.4 ± 5.8, 22.1 ± 5.7, 24.5 ± 6.7, 20.6 ± 6.3, respectively). Values are means ± S.E.M. of five to six determinations. P < 0.05 versus the respective values obtained in transfected astrocytes treated with PBS (*) or versus the respective values obtained in astrocytes transfected with exon-1 htt 25Q (#) (one-way anova+ Fisher’s PLSD).
Reduced expression of TGF-β1 in striatal-derived knock-in cells expressing full-length huntingtin with an expanded Q repeat
We used immortalized knock-in cells derived from striatal neurons expressing full-length (ST7/7Q, wild-type cells) or full-length mutated huntingtin (ST111/111Q, mhtt cells). Mhtt cells showed lower basal levels of TGF-β1 mRNA than wild-type cells (Fig. 8A). Interestingly, wild-type cells expressed detectable levels of mGlu2/3 receptors (not shown) and responded to LY379268 with a substantial rise in TGF-β1 mRNA levels, an effect that was largely attenuated in mhtt cells (Fig. 8B). This defective production and regulation of TGF-β1 was confirmed at protein level by combining cytofluorimetric analysis of TGF-β1-expressing cells (Fig. 8C, D) and in response to LY379268 (Fig. 8B, C) and measurements of TGF-β1 released into the cultured medium under basal conditions and in response to a pulse with LY379268 (Fig. 8E). Both wild-type and mhtt cells expressed mGlu2/3 receptors, as shown by immunoblot analysis exon 1 of huntingtin, and we measured TGF-β1 mRNA levels by real-time PCR. In wild-type knockin cells, transfection with the expanded exon 1 (72Q) substantially reduced TGF-β1 mRNA levels (as compared with the unexpanded exon-1). In contrast, expression of the expanded exon-1 did not further reduce TGF-β1 mRNA levels in mhtt knockin cells (Fig. 8F), suggesting a saturating effect produced by the mutated full-length huntingtin present in these cells. All results from human tissues, cell and animal models are summarized in Table 2.
Fig 8.
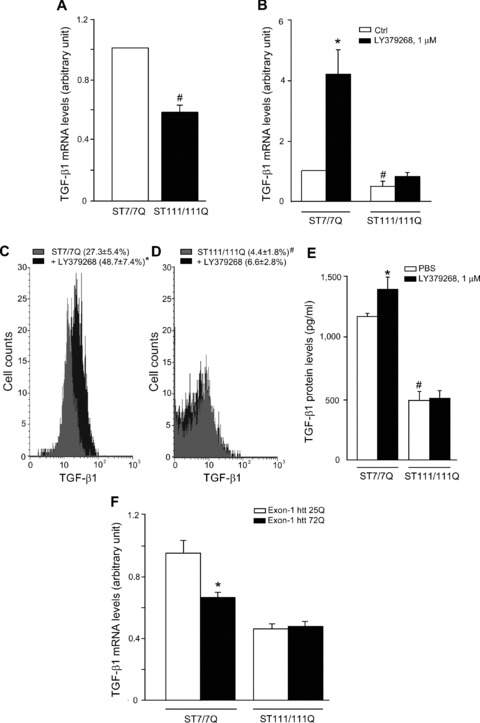
Analysis of TGF-β1 in striatal-derived knock-in cells expressing full-length huntingtin with an expanded Q repeat. Real Time RT-PCR of TGF-β1 mRNA analysis (A, B) in ST7/7Q, wild-type cells or full-length mutated huntingtin ST111/111Q, mhtt cells, under basal conditions and after treatment with LY379268, 1 μM for 10 min. FACS analysis of intracellular TGF-β1 protein (C) and ELISA analysis of extracellular TGF-β1 protein wild-type and mhtt cells, under basal conditions and after treatment with LY379268, 1 μM for 10 min (D). The mean fluorescence intensity (MFI) of TGF-β1 in ST7/7Q cells (MFI: 18.1 ± 4.3, 18.0 ± 5.6, respectively) or in ST111/111Q cells (MFI: 27.3 ± 7.3, 18.0 ± 8.0, respectively) treated with PBS or LY379268 was unchanged (E). Values are means ± S.E.M. of five to six determinations. P < 0.05 versus the respective values obtained in wild-type cells treated with LY379268 (*) or versus the respective values obtained in ST7/7Q cells (#) (one-way anova+ Fisher’s PLSD). Real Time RT-PCR of TGF-β1 mRNA analysis (F) in ST7/7Q and ST111/111Q knock-in cells transfected with the unexpanded exon-1 htt 25Q or the expanded exon-1 htt 72Q. Values are means ± S.E.M. of five to six determinations. P < 0.05 versus the respective values obtained in ST7/7Q cells transfected with exon-1 htt 25Q (one-way anova+ Fisher’s PLSD).
Table 2.
Human data and animal or in vitro models used for the study of TGF-β1 in HD
| HD patients or animal models | Serum | PET scan analysis | CAG repeat length | DS loss per year | Time to onset | Western | Immunohystochemistry | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Ctx | Str | Ctx | Str | |||||||
| Human beings | Asymptomatic | ↓ | + | – | + | + | ↓ | |||
| Symptomatic | n.c. | n.c. | n.c. | n.c. | ↓ | ↑ | ||||
| R6/2 Mice | Asymptomatic | ↓ | ↓ | ↔ | ↓ | ↔ | ||||
| Symptomatic | ↔ | ↓ | ↔ | |||||||
| YAC Mice | Asymptomatic | ↔ | ↓ | ↔ | ||||||
| In vitro models | TGF-β1 mRNA levels | TGF-β1 protein Levels | TGF-β1 mRNA levels after transfection (htt exon-1) | TGF-β1 protein levels after transfection (htt exon-1) | ||||||
| 25Q | 72Q | 25Q | 72Q | |||||||
| Knock-in cell lines | ST 7/7Q | ↔ | ↔ | ↔ | ↓ | |||||
| ST 111/111Q | ↓ | ↓ | ↔ | ↔ | ||||||
| Cell culture | Astrocytes | ↔ | ↓ | ↔ | ↓ | |||||
(+) positive correlation with serum TGF-β1 levels; (–) inverse correlation with serum TGF-β1 levels; (n.c.) lack of correlation with serum TGF-β1 levels; (↓) decrease; (↑) increase; (⇆) unchanged. Ctx: cerebral cortex; Str: striatum.
Discussion
A defective production and/or activity of neurotrophic factors has been implicated in the pathophysiology of HD. Mutated huntingtin looses the ability to increase BDNF gene transcription [44, 45] and to enhance intracellular trafficking of BDNF [46, 47]. In addition, strategies that increase brain levels of BDNF or glial-derived neurotrophic factor (GDNF) are neuroprotective in HD models [36, 48–58]. Recent evidence suggest that the study of neurotrophic factors in HD can be extended to the peripheral blood in an attempt to obtain accessible indicators of disease progression and drug efficacy. Blood levels of BDNF mRNA are progressively reduced in rodent HD models, and can be restored by the neuro protective drug, CEP-1347 [59]. In HD patients, serum BDNF levels are lower than in age-matched controls, and the reduction correlates with the length of CAG repeats in the huntingtin gene, and duration and severity of HD [31]. Thus, the reduction of BDNF levels in peripheral blood may reflect the reduced cortico-striatal production of BDNF associated with HD [60–62]. Our data provide the first evidence that changes in TGF-β1 are associated with HD and raise the intriguing possibility that a defective regulation of this factor contributes to the pathophysiology of neurodegeneration associated with HD. This hypothesis is supported by the emerging role of TGF-β1 in mechanisms of neuroprotection (see below), and by the evidence that TGF-β1 is required for the neuroprotective activity of GDNF [63] and of BDNF [64, 65]. In HD patients, we found serum TGF-β1 levels reduced in asymptomatic and first HD stages, and particularly in asymptomatic patients with lower brain glucose metabolism. As brain glucose metabolism progressively decreases during the asymptomatic phase of HD [27, 30, 66] we expect that blood TGF-β1 levels are maximally reduced in the proximity of the clinical onset of HD. We could examine brain TGF-β1 expression only in one asymptomatic HD patient. Interestingly, TGF-β1 was absent in cortical neurons of this patient, whereas it was always detected in cortical neurons of age-matched controls. Data obtained in post-mortem samples of symptomatic HD patients confirmed the reduction of TGF-β1 levels in cortical neurons. In contrast, the examination of the white matter and the caudate nucleus of symptomatic patients were confounded by the high expression of TGF-β1 in reactive astrocytes and microglia, which overwhelmed any possible change in neuronal TGF-β1 expression. This was not unexpected because TGF-β1 has a prominent role in the regulation of the immune response, and is up-regulated in processes of neuroinflammation (see the strong immunoreactivity in the cerebral cortex of the patient with multiple sclerosis in Fig. 3). The substantial increase in TGF-β1 associated with reactive gliosis might, in part, contribute to explain why blood TGF-β1 increase from the low levels at asymptomatic stage to normality in the symptomatic HD phase, finally correlating with HD severity and progression rate in severely affected patients. Recent evidence indicates that microglia is activated in the striatum of asymptomatic HD gene carrier individuals, and activation correlates with their predicted 5-year probability of developing HD [67]. It is possible that the amount of TGF-β1 produced by activated microglia is not sufficient to compensate for the loss of TGF-β1 in neurons during the asymptomatic phase of the disease, and, therefore, low serum TGF-β1 levels are detected in spite of microglia activation in asymptomatic HD patients. It should be highlighted that TGF-β1 can be produced outside the CNS during inflammation and platelet aggregation [32], and whether these processes are altered in the asymptomatic phase of HD and peripheral pathology is contributing to HD severity, is currently under study [68–70]. However, mutant huntingtin might also directly affect peripheral TGF-β1 production by cells of the immune system. It should be highlighted that plasma levels of the pro-inflammatory cytokine, interleukin-6 (IL-6), are higher before clinical onset of HD [68]. Reduction of TGF-β1 may be causally related to the increase in IL-6 because targeted disruption of the TGF-β1 gene in mice results into an enhanced expression of pro-inflammatory cytokines in peripheral organs [71]. It is possible that a reduced expression of the anti-inflammatory cytokine, TGF-β1, in the context of an increased expression of pro-inflammatory cytokines, such as IL-6 [68], contributes to immune dysfunction in asymptomatic HD patients.
The reduction in cortical TGF-β1 levels associated with the asymptomatic phase of HD was confirmed using R6/2 and YAC128 mice. YAC128 mice express the full length human huntingtin gene containing 128 CAG repeats, whereas R6/2 mice express exon 1 of the human huntingtin gene with 250 CAG repeats [33, 34]. We only examined asymptomatic HD mice to avoid the bias of the extensive atrophy of the striatum and cortex observed in symptomatic YAC128 mice [34, 72], and the atrophy of striatal neurons observed in symptomatic R6/2 mice [73].
Interestingly, both strains of asymptomatic HD mice showed reduced TGF-β1 levels in cortical neurons. This reduction might reflect an intrinsic defect of TGF-β1 formation, or, alternatively, a defect in mechanisms that regulate TGF-β1 formation in response to environmental factors. Data obtained in transfected astrocytes or in mhtt knock-in cells suggest that expression of mutated huntingtin causes a reduced expression of the TGF-β1 gene. However, both HD mice and cells expressing pathological huntingtin showed also an impaired response to extracellular stimuli that enhance TGF-β1 production. Previous studies have shown that activation of mGlu2/3 metabotropic glutamate receptors [see 74 for a review] enhances TGF-β1 formation in cultured astrocytes and brain tissue [20]. Activation of these receptors with compound LY379268 failed to stimulate TGF-β1 production in R6/2 mice and in astrocytes or knockin cells expressing huntingtin with an expanded CAG repeat. As these cells showed normal levels of mGlu2/3 receptors (although receptors were reduced in R6/2 mice), we can conclude that the intracellular machinery regulating the production of TGF-β1 in response to receptor activation is defective in the presence of mutated huntingtin. The hypothesis that the defect is downstream of mGlu2/3 receptors was supported by the use of riluzole, a drug that is known to enhance the formation of a number of trophic factors, including NGF, BDNF and GDNF [35, 36, 75]. Interestingly, riluzole was able to enhance the formation of TGF-β1 in wild-type mice, but was inactive in R6/2 mice.
Knowing that TGF-β1 is anterogradely transported by neurons [76], we speculate that, in the early phase of HD, the striatum is deprived of the amount of TGF-β1 that is physiologically secreted by cortico-striatal neurons. This might increase the vulnerability of striatal neurons because TGF-β1 regulates a number of intracellular events that are relevant to processes of neurodegeneration/ neuroprotection. TGF-β1 protects neurons against different insults, including excitotoxicity, hypoxia, ischemia, deprivation of trophic factors and Aβ toxicity [77, 78]. TGF-β1 has also a constitutive role in the suppression of inflammation, and appears to control the degree of microglial activation in the CNS [79]. TGF-β1 acts synergistically with other neurotrophins and is required for a full neuroprotective activity of BDNF, and GDNF [63–65], and enhances the expression of BDNF and TrkB in neuronal cultures [64]. At cellular level, TGF-β1 prevents apoptotic neuronal death by inhibiting caspase-3 [80], enhancing the expression of the anti-apoptotic proteins, Bcl-2 and Bcl-xl [81], and inhibiting the pro-apoptotic protein, Bad [82]. The anti-apoptotic activity of TGF-β1 in cultured neurons involves the activation of the mitogen-activated protein kinase pathway and the phosphatidylinositol-3-kinase pathway [83]. Established target genes of TGF-β are the ‘check points’ p27 and p21, which produce cell cycle arrest by inhibiting the activity of cyclin-dependent kinases [84, 85]. A lack of TGF-β1 might facilitate the activation of an abortive mitotic cycle in neurons, an event that has been implicated in the pathophysiology of neuronal death associated with chronic neurodegenerative disorders, including HD [86–90]. In addition, the lack of TGF-β1 might enhance the expression of type-2 cyclooxygenase, an enzyme that contributes to mechanisms of neuronal degeneration [91–93].
At this stage, we cannot prove whether decreased TGF-β1 dosage in human serum may represent an asymptomatic marker or its relative increase in advanced stages may suggest a novel marker of HD progression and severity. Additional cross-sectional and longitudinal studies on large populations of HD patients clinically stratified according to HD stage are required to demonstrate such role of TGF-β1 as potential biomarker.
In conclusion, our data show that (i) a reduction of TGF-β1 levels in peripheral blood occurs during the asymptomatic phase of HD and correlates with the reduction in brain glucose metabolism; (ii) HD is associated with an early loss of TGF-β1 in cortical neurons and (iii) this loss might reflect a defect in the intrinsic TGF-β1-producing machinery that makes the factor unresponsive to different stimulating agents. If the early loss in TGF-β1 contributes to the pathophysiology of neuronal death associated with HD, then strategies that rescue TGF-β1 levels should limit the progression of HD-related pathology. This hypothesis can be tested in HD mice using either by implantable cells engineered to overexpress TGF-β1 or viral vectors encoding TGF-β1.
Acknowledgments
We thank Dr. Marcy MacDonald, Molecular Neurogenetics Unit, Center for Human Genetic Research, Massachusetts General Hospital, Boston, MA, USA, for kindly providing striatal knock-in cell cultures; Drs. Mahmoud A. Pouladi and Michael R. Hayden, Centre for Molecular Medicine and Therapeutics, University of British Columbia, Vancouver, BC, Canada, for providing data on serum TGF-β1 levels from YAC128 mice and controls; Dr. Andrea Ciammola, Department of Neurology and Laboratory of Neuroscience, Dino Ferrari Center, University of Milan – IRCCS Istituto Auxologico Italiano, Milan, Italy for providing serum samples from healthy control persons; the European Huntington’s Disease (EURO-HD) Network, all patients and their families (Associazione Italiana Corea di Huntington-Neuromed), the financial support of 5x1000 from AICH-Neuromed (FS), the financial support of 5x1000 from IRCCS-Neuromed (SD), the Italian Society of Hospital Neurologists (SNO, ‘lascito Gobessi’) and the Italian Health Ministry (FS, COFIN 2006; finalizzato ex art.56 2007), for their financial support.
Disclosure
The authors report no conflicts of interest.
Supporting Information
Correlation between age and serumTGF-β1 levels in control subjects, asymptomatic andsymptomatic subjects. (A) Age of controls subjects andTGF-β1 levels (n = 47; R2= 0.02; P = 0.3315); (B). Age of asymptomaticsubjects and TGF-β1 levels (n = 30;R2 = 0.006; P = 0.6779); (C) Age ofsymptomatic patients and TGF-β1 levels (n =95; R2 = 0.01; P = 0.7703). Statisticalanalysis was carried out by ANOVA + Fisher PLSD. Data wereconsidered statistically significant at P < 0.05. Statistical analysis was performed with the StatView 5 software.
Demographics of Huntington’s diseasesubjects and healthy controls
References
- 1.Sathasivam K, Hobbs C, Turmaine M, et al. Formation of polyglutamine inclusions in non-CNS tissue. Hum Mol Genet. 1999;8:813–22. doi: 10.1093/hmg/8.5.813. [DOI] [PubMed] [Google Scholar]
- 2.The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–83. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 3.Vonsattel JP, Myers RH, Stevens TJ, et al. Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985;44:559–77. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 4.de la Monte SM, Vonsattel JP Richardson EPJr. Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington’s disease. J Neuropathol Exp Neurol. 1988;47:516–25. doi: 10.1097/00005072-198809000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Beal MF, Kowall NW, Ellison DW, et al. Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature. 1986;321:168–71. doi: 10.1038/321168a0. [DOI] [PubMed] [Google Scholar]
- 6.Ross CA. Huntington’s disease: new paths to pathogenesis. Cell. 2004;118:4–7. doi: 10.1016/j.cell.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 7.Sapp E, Kegel KB, Aronin N, et al. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J Neuropathol Exp Neurol. 2001;60:161–72. doi: 10.1093/jnen/60.2.161. [DOI] [PubMed] [Google Scholar]
- 8.Pavese N, Gerhard A, Tai YF, et al. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology. 2006;66:1638–43. doi: 10.1212/01.wnl.0000222734.56412.17. [DOI] [PubMed] [Google Scholar]
- 9.Tai YF, Pavese N, Gerhard A, et al. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain. 2007;130:1759–66. doi: 10.1093/brain/awm044. [DOI] [PubMed] [Google Scholar]
- 10.Shin JY, Fang ZH, Yu ZX, et al. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol. 2005;171:1001–12. doi: 10.1083/jcb.200508072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krieglstein K, Henheik P, Farkas L, et al. Glial cell line-derived neurotrophic factor requires transforming growth factor-beta for exerting its full neurotrophic potential on peripheral and CNS neurons. J Neurosci. 1998;18:9822–34. doi: 10.1523/JNEUROSCI.18-23-09822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bruno V, Battaglia G, Casabona G, et al. Neuroprotection by glial metabotropic glutamate receptors is mediated by transforming growth factor-beta. J Neurosci. 1998;18:9594–600. doi: 10.1523/JNEUROSCI.18-23-09594.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee HG, Ueda M, Zhu X, et al. Ectopic expression of phospho-Smad2 in Alzheimer’s disease: uncoupling of the transforming growth factor-beta pathway. J Neurosci Res. 2006;84:1856–61. doi: 10.1002/jnr.21072. [DOI] [PubMed] [Google Scholar]
- 14.Tesseur I, Zou K, Esposito L, et al. Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer’s pathology. J Clin Invest. 2006;116:3060–9. doi: 10.1172/JCI27341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ueberham U, Ueberham E, Gruschka H, et al. Altered subcellular location of phosphorylated Smads in Alzheimer’s disease. Eur J Neurosci. 2006;24:2327–34. doi: 10.1111/j.1460-9568.2006.05109.x. [DOI] [PubMed] [Google Scholar]
- 16.Chalmers KA, Love S. Neurofibrillary tangles may interfere with Smad 2/3 signaling in neurons. J Neuropathol Exp Neurol. 2007;66:158–67. doi: 10.1097/nen.0b013e3180303b93. [DOI] [PubMed] [Google Scholar]
- 17.Tesseur I, Wyss-Coray T. A role for TGF-beta signaling in neurodegeneration: evidence from genetically engineered models. Curr Alzheimer Res. 2006;3:505–13. doi: 10.2174/156720506779025297. [DOI] [PubMed] [Google Scholar]
- 18.Brionne TC, Tesseur I, Masliah E, et al. Loss of TGF-beta 1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron. 2003;40:1133–45. doi: 10.1016/s0896-6273(03)00766-9. [DOI] [PubMed] [Google Scholar]
- 19.Sortino MA, Chisari M, Merlo S, et al. Glia mediates the neuroprotective action of estradiol on beta-amyloid-induced neuronal death. Endocrinology. 2004;145:5080–86. doi: 10.1210/en.2004-0973. [DOI] [PubMed] [Google Scholar]
- 20.D’Onofrio M, Cuomo L, Battaglia G, et al. Neuroprotection mediated by glial group-II metabotropic glutamate receptors requires the activation of the MAP kinase and the phosphatidylinositol-3-kinase pathways. J Neurochem. 2001;78:435–45. doi: 10.1046/j.1471-4159.2001.00435.x. [DOI] [PubMed] [Google Scholar]
- 21.Corti C, Battaglia G, Molinaro G, et al. The use of knockout mice unravels distinct roles for mGlu2 and mGlu3 metabotropic glutamate receptors in mechanisms of neurodegeneration/neuroprotection. J Neurosci. 2007;27:8297–308. doi: 10.1523/JNEUROSCI.1889-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cha JH, Kosinski CM, Kerner JA, et al. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington disease gene. Proc Natl Acad Sci USA. 1998;95:6480–5. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Squitieri F, Gellera C, Cannella M, et al. Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course. Brain. 2003;126:946–55. doi: 10.1093/brain/awg077. [DOI] [PubMed] [Google Scholar]
- 24.Langbehn DR, Brinkman RR, Falush D, et al. A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clin Genet. 2004;65:267–77. doi: 10.1111/j.1399-0004.2004.00241.x. [DOI] [PubMed] [Google Scholar]
- 25.Huntington Study Group. The Unified Huntington’s Disease Rating Scale: reliability and consistency. Huntington Study Group. Mov Disord. 1996;11:136–42. doi: 10.1002/mds.870110204. [DOI] [PubMed] [Google Scholar]
- 26.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;2:189–98. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 27.Ciarmiello A, Cannella M, Lastoria S, et al. Brain white-matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington’s disease. J Nucl Med. 2006;47:215–22. [PubMed] [Google Scholar]
- 28.Myers RH, Sax DS, Koroshetz WJ, et al. Factors associated with slow progression in Huntington’s disease. Arch Neurol. 1991;48:800–4. doi: 10.1001/archneur.1991.00530200036015. [DOI] [PubMed] [Google Scholar]
- 29.Marder K, Zhao H, Myers RH, et al. Rate of functional decline in Huntington’s disease. Huntington Study Group. Neurology. 2000;54:452–8. doi: 10.1212/wnl.54.2.452. [DOI] [PubMed] [Google Scholar]
- 30.Squitieri F, Ciarmiello A, Di Donato S, et al. The search for cerebral biomarkers of Huntington’s disease: a review of genetic models of age at onset prediction. [Review] Eur J Neurol. 2006;13:408–15. doi: 10.1111/j.1468-1331.2006.01264.x. [DOI] [PubMed] [Google Scholar]
- 31.Ciammola A, Sassone J, Cannella M, et al. Low brain-derived neurotrophic factor (BDNF) levels in serum of Huntington’s disease patients. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:574–7. doi: 10.1002/ajmg.b.30501. [DOI] [PubMed] [Google Scholar]
- 32.Grainger DJ, Mosedale DE, Metcalfe JC. TGF-beta in blood: a complex problem. Cytokine Growth Factor Rev. 2000;11:133–45. doi: 10.1016/s1359-6101(99)00037-4. [DOI] [PubMed] [Google Scholar]
- 33.Mangiarini L, Sathasivam K, Seller M, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 34.Slow EJ, van Raamsdonk J, Rogers D, et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–67. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- 35.Katoh-Semba R, Asano T, Ueda H, et al. Riluzole enhances expression of brain-derived neurotrophic factor with consequent proliferation of granule precursor cells in the rat hippocampus. FASEB J. 2002;16:1328–30. doi: 10.1096/fj.02-0143fje. [DOI] [PubMed] [Google Scholar]
- 36.Squitieri F, Ciammola A, Colonnese C, et al. Neuroprotective effects of riluzole in Huntington’s disease. Eur J Nucl Med Mol Imaging. 2008;35:221–2. doi: 10.1007/s00259-007-0615-y. [DOI] [PubMed] [Google Scholar]
- 37.Flanders KC, Thompson NL, Cissel DS, et al. Transforming growth factor-beta 1: histochemical localization with antibodies to different epitopes. J Cell Biol. 1989;108:653–60. doi: 10.1083/jcb.108.2.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Groot CJ, Montagne L, Barten AD, et al. Expression of transforming growth factor (TGF)-beta1, -beta2, and -beta3 isoforms and TGF-beta type I and type II receptors in multiple sclerosis lesions and human adult astrocyte cultures. J Neuropathol Exp Neurol. 1999;58:174–87. doi: 10.1097/00005072-199902000-00007. [DOI] [PubMed] [Google Scholar]
- 39.Aronica E, Leenstra S, Jansen GH, et al. Expression of brain-derived neurotrophic factor and tyrosine kinase B receptor proteins in glioneuronal tumors from patients with intractable epilepsy: colocalization with N-methyl-D-aspartic acid receptor. Acta Neuropatho. 2001;101:383–92. doi: 10.1007/s004010000296. [DOI] [PubMed] [Google Scholar]
- 40.Rose K, Goldberg MP, Choi DW. Cytotoxicity in murine neocortical cell cultures. In: Tyson CA, Frazier J, editors. Methods in toxicology. In vitro biological system. San Diego: San Diego Academics; 1992. pp. 46–60. [Google Scholar]
- 41.Trettel F, Rigamonti D, Hilditch-Maguire P, et al. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- 42.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 43.Maat-Schieman M, Roos R, Losekoot M, et al. Neuronal intranuclear and neuropil inclusions for pathological assessment of Huntington’s disease. Brain Pathol. 2007;17:31–7. doi: 10.1111/j.1750-3639.2006.00040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuccato C, Ciammola A, Rigamonti D, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. 2001;293:493–8. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- 45.Zuccato C, Tartari M, Crotti A, et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
- 46.Gauthier LR, Charrin BC, Borrell-Pagès M, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–38. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 47.del Toro D, Canals JM, Ginés S, et al. Mutant huntingtin impairs the post-Golgi trafficking of brain-derived neurotrophic factor but not its Val66Met polymorphism. J Neurosci. 2006;26:12748–57. doi: 10.1523/JNEUROSCI.3873-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bemelmans AP, Horellou P, Pradier L, et al. Brain-derived neurotrophic factor-mediated protection of striatal neurons in an excitotoxic rat model of Huntington’s disease, as demonstrated by adenoviral gene transfer. Hum Gene Ther. 1999;10:2987–97. doi: 10.1089/10430349950016393. [DOI] [PubMed] [Google Scholar]
- 49.Pérez-Navarro E, Arenas E, Marco S, et al. Intrastriatal grafting of a GDNF-producing cell line protects striatonigral neurons from quinolinic acid excitotoxicity in vivo. Eur J Neurosci. 1999;11:241–9. doi: 10.1046/j.1460-9568.1999.00433.x. [DOI] [PubMed] [Google Scholar]
- 50.Duan W, Guo Z, Jiang H, et al. Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proc Natl Acad Sci USA. 2003;100:2911–6. doi: 10.1073/pnas.0536856100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kells AP, Fong DM, Dragunow M, et al. AAV-mediated gene delivery of BDNF or GDNF is neuroprotective in a model of Huntington disease. Mol Ther. 2004;9:682–8. doi: 10.1016/j.ymthe.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 52.Borrell-Pagès M, Canals JM, Cordelières FP, et al. Cystamine and cysteamine increase brain levels of BDNF in Huntington disease via HSJ1b and transglutaminase. J Clin Invest. 2006;116:1410–24. doi: 10.1172/JCI27607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McBride JL, During MJ, Wuu J, et al. Structural and functional neuroprotection in a rat model of Huntington’s disease by viral gene transfer of GDNF. Exp Neurol. 2003;181:213–23. doi: 10.1016/s0014-4886(03)00044-x. [DOI] [PubMed] [Google Scholar]
- 54.McBride JL, Ramaswamy S, Gasmi M, et al. Viral delivery of glial cell line-derived neurotrophic factor improves behavior and protects striatal neurons in a mouse model of Huntington’s disease. Proc Natl Acad Sci USA. 2006;103:9345–50. doi: 10.1073/pnas.0508875103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lynch G, Kramar EA, Rex CS, et al. Brain-derived neurotrophic factor restores synaptic plasticity in a knock-in mouse model of Huntington’s disease. J Neurosci. 2007;27:4424–34. doi: 10.1523/JNEUROSCI.5113-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pineda JR, Rubio N, Akerud P, et al. Neuroprotection by GDNF-secreting stem cells in a Huntington’s disease model: optical neuroimage tracking of brain-grafted cells. Gene Ther. 2007;14:118–28. doi: 10.1038/sj.gt.3302847. [DOI] [PubMed] [Google Scholar]
- 57.DeMarch Z, Giampà C, Patassini S, et al. Beneficial effects of rolipram in the R6/2 mouse model of Huntington’s disease. Neurobiol Disease. 2008;30:375–87. doi: 10.1016/j.nbd.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 58.Gharami K, Xie Y, An JJ, et al. Brain-derived neurotrophic factor over-expression in the forebrain ameliorates Huntington’s disease phenotypes in mice. J Neurochem. 2008;105:369–79. doi: 10.1111/j.1471-4159.2007.05137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Conforti P, Ramos C, Apostol BL, et al. Blood level of brain-derived neurotrophic factor mRNA is progressively reduced in rodent models of Huntington’s disease: restoration by the neuroprotective compound CEP-1347. Mol Cell Neurosci. 2008;39:1–7. doi: 10.1016/j.mcn.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 60.Ferrer I, Goutan E, Marín C, et al. Brain-derived neurotrophic factor in Huntington disease. Brain Res. 2000;866:257–61. doi: 10.1016/s0006-8993(00)02237-x. [DOI] [PubMed] [Google Scholar]
- 61.Zuccato C, Liber D, Ramos C, et al. Progressive loss of BDNF in a mouse model of Huntington’s disease and rescue by BDNF delivery. Pharmacol Res. 2005;52:133–9. doi: 10.1016/j.phrs.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 62.Zuccato C, Marullo M, Conforti P, et al. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington’s disease. Brain Pathol. 2008;18:225–38. doi: 10.1111/j.1750-3639.2007.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schober A, Peterziel H, von Bartheld CS, et al. GDNF applied to the MPTP-lesioned nigrostriatal system requires TGF-beta for its neuroprotective action. Neurobiol Dis. 2007;25:378–91. doi: 10.1016/j.nbd.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 64.Sometani A, Kataoka H, Nitta A, et al. Transforming growth factor-beta1 enhances expression of brain-derived neurotrophic factor and its receptor, TrkB, in neurons cultured from rat cerebral cortex. J Neurosci Res. 2001;66:369–76. doi: 10.1002/jnr.1229. [DOI] [PubMed] [Google Scholar]
- 65.Lu J, Wu Y, Sousa N, et al. SMAD pathway mediation of BDNF and TGF beta 2 regulation of proliferation and differentiation of hippocampal granule neurons. Development. 2005;132:3231–42. doi: 10.1242/dev.01893. [DOI] [PubMed] [Google Scholar]
- 66.Harris GJ, Codori AM, Lewis RF, et al. Reduced basal ganglia blood flow and volume in pre-symptomatic, gene-tested persons at-risk for Huntington’s disease. Brain. 1999;122:1667–78. doi: 10.1093/brain/122.9.1667. [DOI] [PubMed] [Google Scholar]
- 67.Tai YF, Pavese N, Gerhard A, et al. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain. 2007;130:1759–66. doi: 10.1093/brain/awm044. [DOI] [PubMed] [Google Scholar]
- 68.Björkqvist M, Wild EJ, Thiele J, et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med. 2008;205:1869–77. doi: 10.1084/jem.20080178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van der Burg JM, Björkqvist M, Brundin P. Beyond the brain: widespread pathology in Huntington’s disease. Lancet Neurol. 2009;8:765–74. doi: 10.1016/S1474-4422(09)70178-4. [DOI] [PubMed] [Google Scholar]
- 70.Squitieri F, Falleni A, Cannella M, et al. Abnormal morphology of peripheral cell tissues from patients with Huntington disease. J Neural Transm. 2010;117:77–83. doi: 10.1007/s00702-009-0328-4. [DOI] [PubMed] [Google Scholar]
- 71.Shull MM, Ormsby I, Kier AB, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–9. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Van Raamsdonk JM, Murphy Z, Slow EJ, et al. Selective degeneration and nuclear localization of mutant huntingtin in the YAC128 mouse model of Huntington disease. Hum Mol Genet. 2005;14:3823–35. doi: 10.1093/hmg/ddi407. [DOI] [PubMed] [Google Scholar]
- 73.Ferrante RJ, Andreassen OA, Jenkins BG, et al. Neuroprotective effects of creatine in a transgenic mouse model of Huntington’s disease. J Neurosci. 2000;20:4389–97. doi: 10.1523/JNEUROSCI.20-12-04389.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schoepp DD, Jane DE, Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–76. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- 75.Mizuta I, Ohta M, Ohta K, et al. Riluzole stimulates nerve growth factor, brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor synthesis in cultured mouse astrocytes. Neurosci Lett. 2001;310:117–20. doi: 10.1016/s0304-3940(01)02098-5. [DOI] [PubMed] [Google Scholar]
- 76.Jiang Y, McLennan IS, Koishi K, et al. Transforming growth factor-beta 2 is anterogradely and retrogradely transported in motoneurons and up-regulated after nerve injury. Neuroscience. 2000;97:735–42. doi: 10.1016/s0306-4522(00)00084-1. [DOI] [PubMed] [Google Scholar]
- 77.Wyss-Coray T. Tgf-Beta pathway as a potential target in neurodegeneraation and Alzheimer’s. Curr Alzh Res. 2006;3:191–5. doi: 10.2174/156720506777632916. [DOI] [PubMed] [Google Scholar]
- 78.Vivien D, Ali C. Transforming growth factor-β signalling in brain disorder. Cytokine Growth Factor Rev. 2006;17:121–8. doi: 10.1016/j.cytogfr.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 79.Brionne TC, Tesseur I, Masliah E, et al. Loss of TGF-β1 leads to increate neuronal cell death and microgliosis in mouse brain. Neuron. 2003;40:1133–45. doi: 10.1016/s0896-6273(03)00766-9. [DOI] [PubMed] [Google Scholar]
- 80.Zhu Y, Ahlemeyer B, Bauerbach E, et al. TGF-beta1 inhibits caspase 3 activation and neuronal apoptosis in rat hippocampal cultures. Neurochem Int. 2001;38:227–35. doi: 10.1016/s0197-0186(00)00084-x. [DOI] [PubMed] [Google Scholar]
- 81.Prehn JH, Bindokas VP, Jordan J, et al. Protective effect of transforming growth factor-beta1 on beta-amyloid neurotoxicity in rat hippocampal neurons. Mol. Pharmacol. 1996;49:319–28. [PubMed] [Google Scholar]
- 82.Zhu Y, Yang GY, Ahlemeyer B, et al. Transforming growth factor-beta1 increases Bad phosphorylation and protects neurons against damage. J Neurosci. 2002;22:3898–909. doi: 10.1523/JNEUROSCI.22-10-03898.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhu Y, Culmsee C, Klumpp S, et al. Neuroprotection by transforming growth factor-beta1 involves activation of nuclear factor-kappaB through phosphatidylinositol-3-OH kinase/Akt and mitogen-activated protein kinase-extracellular-signal regulated kinase 1,2 signaling pathways. Neuroscience. 2004;123:897–906. doi: 10.1016/j.neuroscience.2003.10.037. [DOI] [PubMed] [Google Scholar]
- 84.Datto MB, Li Y, Panus JF, et al. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc Natl Acad Sci USA. 1995;92:5545–9. doi: 10.1073/pnas.92.12.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ravitz MJ, Yan S, Dolce C, et al. Differential regulation of p27 and cyclin D1 by TGF-beta and EGF in C3H 10T1/2 mouse fibroblasts. J Cell Physiol. 1996;168:510–20. doi: 10.1002/(SICI)1097-4652(199609)168:3<510::AID-JCP3>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 86.Freeman RF, Estus S, Johnson EM., Jr Analysis of cell-related gene expression in postmitotic neurons: selective induction of cyclin D1 during programmed cell death. Neuron. 1994;12:343–55. doi: 10.1016/0896-6273(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 87.Herrup K, Busser JC. The induction of multiple cell cycle events precedes target-related neuronal death. Development. 1995;121:2385–95. doi: 10.1242/dev.121.8.2385. [DOI] [PubMed] [Google Scholar]
- 88.Copani A, Uberti D, Sortino MA, et al. Activation of cell-cycle-associated proteins in neuronal death: a mandatory or dispensable path. Trends Neurosci. 2001;24:25–31. doi: 10.1016/s0166-2236(00)01663-5. [DOI] [PubMed] [Google Scholar]
- 89.Curtis MA, Penney EB, Pearson AG, et al. Increased cell proliferation and neurogenesis in the adult human Huntington’s disease brain. Proc Natl Acad Sci USA. 2003;100:9023–7. doi: 10.1073/pnas.1532244100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Akashiba H, Ikegaya Y, Nishiyama N, et al. Differential involvement of cell cycle reactivation between striatal and cortical neurons in cell death induced by 3-nitropropionic acid. J Biol Chem. 2008;283:6594–606. doi: 10.1074/jbc.M707730200. [DOI] [PubMed] [Google Scholar]
- 91.Yamagata K, Andreasson KI, Kaufmann WE, et al. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–86. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- 92.Adams J, Collaço-Moraes Y, de Belleroche J. Cyclooxygenase-2 induction in cerebral cortex: an intracellular response to synaptic excitation. J Neurochem. 1996;66:6–13. doi: 10.1046/j.1471-4159.1996.66010006.x. [DOI] [PubMed] [Google Scholar]
- 93.Tocco G, Freire-Moar J, Schreiber SS, et al. Maturational regulation and regional induction of cyclooxygenase-2 in rat brain: implications for Alzheimer’s disease. Exp Neurol. 1997;144:339–49. doi: 10.1006/exnr.1997.6429. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Correlation between age and serumTGF-β1 levels in control subjects, asymptomatic andsymptomatic subjects. (A) Age of controls subjects andTGF-β1 levels (n = 47; R2= 0.02; P = 0.3315); (B). Age of asymptomaticsubjects and TGF-β1 levels (n = 30;R2 = 0.006; P = 0.6779); (C) Age ofsymptomatic patients and TGF-β1 levels (n =95; R2 = 0.01; P = 0.7703). Statisticalanalysis was carried out by ANOVA + Fisher PLSD. Data wereconsidered statistically significant at P < 0.05. Statistical analysis was performed with the StatView 5 software.
Demographics of Huntington’s diseasesubjects and healthy controls