

Abstract
To test the hypothesis that combinatorial interference of toll-like receptor 2 (TLR2) and TLR4 is superior to isolated interference of TLR2 or TLR4 in stabilizing atherosclerotic plaques, lentiviruses carrying small interfering RNA of TLR2 or TLR4 were constructed and proved efficacious for knocking down mRNA and protein expression of TLR2 or TLR4 significantly in vitro. One hundred and fifty apolipoprotein E−/− mice fed a high-fat diet were divided into the control, mock, TLR2i, TLR4i and TLR2 + 4i subgroups and a constrictive collar was placed around carotid artery of these mice to induce plaque formation. TLR2i and TLR4i viral suspension was transfected into carotid plaques, respectively, in TLR2i and TLR4i subgroups, or in combination in TLR2 + 4i subgroup. Four weeks after lentivirus transfection, mRNA and protein expression of TLR2 or TLR4 was attenuated markedly in carotid plaques, leading to reduced local inflammatory cytokine expression and plaque content of lipid and macrophages, increased plaque content of collagen and lowered plaque vulnerability index. Factorial ANOVA analysis revealed that there was a synergistic effect between TLR4i and TLR2i in stabilizing plaques. In conclusion, combinatorial interference of TLR2 and TLR4 reduces local inflammation and stabilizes plaques more effectively than interference of TLR2 or TLR4 alone.
Keywords: atherosclerosis, inflammation, gene therapy, receptors, vulnerable plaques
Introduction
Toll-like receptors (TLRs) are a group of receptors that play a key role in innate immune signalling and initiating inflammatory response. Activation of one or more TLRs causes recruitment and activation of adaptor proteins such as myeloid differentiation factor 88 (MyD88) and members of the interleukin (IL)-1R-associated kinase family, followed by activation of tumour necrosis factor (TNF) receptor-associated factor 6 and nuclear factor-κB, with ultimate release of a large number of inflammatory cytokines, including IL-1β, IL-6, TNF-α, monocyte chemoattractant protein-1 (MCP-1) and matrix metalloproteinases (MMPs) [1–3]. Recent evidence indicates that several TLRs are involved in atherosclerosis and the most relevant subtypes are TLR2 and TLR4 [4, 5], which are expressed in most vascular wall cells, such as endothelial cells [6], smooth muscle cells (SMCs) [7], macrophages [8] and adventitial fibroblasts [9]. Activation of these cells as a response to self- and non-self ligands appears to be mediated by TLR2 and TLR4 [10, 11], and apolipoprotein E (ApoE)−/− mice with TLR2 or TLR4 gene knockout exhibited attenuated atherosclerosis [12, 13]. Our recent study demonstrated that combinatorial interference of TLR1 and TLR2 resulted in an enhanced improvement of plaque stability in ApoE−/− mice but such an effect was only additive rather than synergistic, probably because TLR1 and TLR2 share the same signalling pathway upon activation [14]. On the other hand, the downstream signalling pathway of TLR2 activation is MyD88 dependent, whereas that of TLR4 activation is both MyD88 dependent and MyD88 independent [1, 2]. Therefore, it is likely that TLR2 and TLR4 may contribute independently to the formation and progression of atherosclerotic plaques and thus we suggested that TLR2 and TLR4 may have a synergistic effect on the vulnerability of atherosclerotic plaques and combinatorial interference of TLR2 and TLR4 may stabilize plaques more efficiently than interference of TLR2 or TLR4 alone. To test this hypothesis, we constructed lentiviral vectors, which can efficiently deliver small interfering RNAs (siRNAs) due to their stable transduction of both dividing and non-dividing cells [15], and aimed at knocking down TLR2 and/or TLR4 to elucidate their individual and synergistic roles in stabilizing atherosclerotic plaques in ApoE−/− mice.
Materials and methods
Detailed materials and methods are described in the supplemental data.
Cell culture
RAW264.7 and 293FT cell lines were cultured using previously reported method.
Preparation of lentiviral vectors
The preparation of lentiviral vectors expressing TLR2 and TLR4 involved use of the BLOCK-It Lentiviral Pol II miR RNAi Expression System with green fluorescence protein (GFP) (catalogue no. K4948–00; Invitrogen, Carlsbad, CA, USA). A scrambled siRNA sequence (named mock-siRNA) with no known homology to mammal genes served as a control. All vectors were combined with a GFP expression cassette.
Animal protocol
One hundred and fifty male ApoE−/− mice received a high-fat diet and underwent constrictive collar placement around the right common carotid artery after anaesthesia with an intraperitoneal injection of pentobarbital sodium, using the method of von der Thüsen and his colleagues [16] .The mice were then randomly divided into treatment group (n= 90) and non-treatment group (n= 60), and were maintained on a high-fat diet for 12 weeks.
At the end of week 8, after the proximal right common carotid artery and the distal right internal and external carotid arteries were temporarily ligated, lentiviral suspension was instilled into the right common carotid artery and was left in situ for 15 min. [17]. Mice in the treatment group were further randomly divided into the TLR2i, TLR4i and TLR2 + 4i subgroups (n= 30, each), which received an infusion of 10 μl of TLR2i viral suspension, an infusion of 10 μl of TLR4i viral suspension and combined infusion of 5 μl of TLR2i and 5 μl of TLR4i viral suspension, respectively. Mice in the non-treatment group were randomly divided into the mock and control subgroups (n= 30, each). The mock subgroup received an infusion of 10 μl of mock viral suspension and the control subgroup had their collars removed without virus infusion. The animal experimental protocol conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996) and was approved by the Animal Care Committee of Shandong University.
Measurement of serum lipid levels
Serum levels of total cholesterol and triglycerides were measured by an enzymatic assay (Zhejiang Dongou Biotechnology, Wenzhou, China).
Histological analysis
In all mice, the right common carotid artery together with its bifurcation was carefully excised and stored. Each vessel throughout the entire length of the common carotid artery underwent histological analysis. The carotid artery was cross-sectioned into pieces 6 μm thick at 50 μm intervals. Sections were stained with haematoxylin and eosin for histological analysis. The plaque was subdivided into a fibrous cap and a necrotic core on the basis of haematoxylin and eosin staining for extracellular matrix, picrosirius red staining for collagen and Oil-red O staining for lipids, respectively. Corresponding sections on separate slides were immunostained with monoclonal antibodies. Sections reacted with non-immune IgG, secondary antibody only and no primary and secondary antibodies were used as negative controls.
An automated image analysis system (Image-Pro Plus 5.0, Media Cybernetics, Silver Spring, MD, USA) was used for quantitative measurements. The vulnerability index was calculated by the following formula: positive staining area of (macrophages + lipid)/positive staining area of (α-SMCs + collagen) [18].
Quantitative real-time RT-PCR analysis
The mRNA expression levels of β-actin, TLR2, TLR4, IL-1β, IL-6, TNF-α, MCP-1, MMP-2 and MMP-9 were quantitatively analysed using real time RT-PCR.
Western blot analysis
The protein expression levels of β-actin, TLR2 and TLR4 in plaque tissue, β-actin, TLR2, TLR4, IL-1β, IL-6, TNF-α and MCP-1 in RAW264.7 cells were assayed by Western blot analysis.
Statistical analysis
Data were expressed as mean ± S.D. Multiple comparisons were analysed by least significance difference post hoc tests. Factorial ANOVA was used to analyse the individual and interactive effects of two independent variables, TLR2i and TLR4i. With the statistical design of 2 × 2 factorial, the main effects of TLR2i, main effects of TLR4i and the interaction between TLR2i and TLR4i were compared. In such an analysis, if a significant interaction is present, the effects between TLR2i and TLR4i should be interpreted as synergistic rather than additive. All analyses involved use of SPSS 11.5 (SPSS, Inc., Chicago, IL, USA). P < 0.05 was considered statistically significant.
Results
RNAi efficiency in vitro
To evaluate the efficacy of gene silencing in vitro, TLR2 and TLR4 protein expression levels in RAW264.7 cells were measured and shown to be substantially reduced by TLR2i and TLR4i, respectively (Fig. 1A). The protein expression levels of IL-1β, IL-6, TNF-α and MCP-1 in the TLR2i and TLR4i cell groups were lower than the corresponding measurements in the control or mock cell groups. Moreover, the TLR2 + 4i cell group showed a significantly lower protein expression level of IL-1β, IL-6, TNF-α, MCP-1 than the other four cell groups (Fig. 1B).
Fig 1.
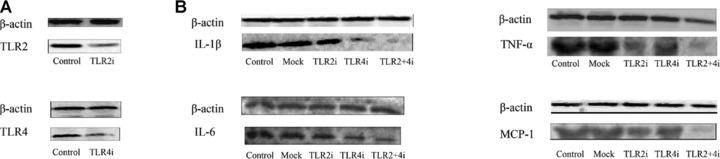
Efficiency of RNAi in vitro. (A) The protein expression levels of TLR2 in the control and TLR2i groups (upper panel) and the protein expression levels of TLR4 in the control and TLR4i groups (lower panel). (B) The protein expression levels of IL-1β, IL-6, TNF-α and MCP-1 in the control, mock, TLR2i, TLR4i and TLR2 + 4i groups.
Lentivirus transfection efficiency in vivo
A local virus delivery to the carotid artery of ApoE−/− mice has proven an efficient approach to transferring gene into atherosclerotic plaque [17]. Since GFP expression provides a convenient monitor for checking the transfection efficiency of lentivirus, GFP fluorescence in the carotid plaque was examined serially, which revealed a successful transfection of siRNA 1 week after transfection (at the end of week 9, Fig. 2), strong fluorescence in the carotid plaque 2 weeks after transfection (at the end of week 10) and visible fluorescence 4 weeks after transfection (at the end of week 12). In our preliminary study, we measured the ratio of the GFP+ staining area to the plaque area in the mock, TLR2i, TLR4i and TLR2 + 4i groups 2 weeks after transfection and used this ratio to quantitate the transduction efficiency. The results showed that this ratio was 0.77 ± 0.13, 0.79 ± 0.08, 0.75 ± 0.10 and 0.78 ± 0.15 in the mock, TLR2i, TLR4i and TLR2 + 4i groups, respectively (P > 0.05), indicating that the difference in the transduction efficiency among the four groups was not significant. These results demonstrated an efficient in vivo transfection of lentivirus by siRNA in atherosclerotic plaque.
Fig 2.
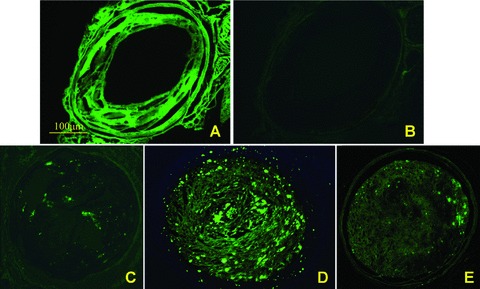
Efficiency of lentivirus transfection in atherosclerotic plaques. (A) and (B) were images from the same cryosection viewed under fluorescence microscopy in different conditions to identify GFP expression. (A) At baseline, background autofluorescence of the plaque was visible; (B) after use of SkyBlue 6B, the background autofluorescence was eliminated. (C), (D) and (E) were fluorescence images of plaques obtained 1 week, 2 weeks and 4 weeks after transfection, respectively. GFP fluorescence in the carotid plaques was visible 1 week after transfection (at week 9), and stronger fluorescence was observed 2 weeks after transfection (at week 10). Four weeks after transfection (at week 12) when the study was terminated, GFP was still visible, albeit weak. Scale bar = 100 μm.
Serum lipid levels
There was no significant difference in body weight between the treatment group (30.7 ± 3.2 g) and the non-treatment group (30.0 ± 3.1 g), indicating that virus transfection was safe in these animals. The serum levels of total cholesterol and triglycerides in the treatment group (31.1 ± 2.1 mmol/l and 2.99 ± 0.98 mmol/l, respectively) did not differ significantly from those in the non-treatment group (29.8 ± 2.8 mmol/l and 2.95 ± 0.89 mmol/l, respectively), indicating that the therapeutic effects of gene interference was independent of serum lipid levels.
TLR2 and TLR4 expression in carotid plaques
To examine the efficacy of lentivirus-mediated gene silencing in vivo, the levels of mRNA and protein expression of TLR2 and TLR4 in carotid plaques were measured (Fig. 3). In comparison with the control subgroup, the TLR2 mRNA expression level in the TLR2i subgroup and TLR2 + 4i subgroup was reduced by 50% and 59%, respectively (both P < 0.05), and the TLR2 protein expression levels in the TLR2i subgroup and TLR2 + 4i subgroup were lowered by 44% and 51%, respectively (both P < 0.05). In contrast, there was no significant difference in the TLR2 mRNA or protein expression levels among the control, mock and TLR4i subgroups.
Fig 3.
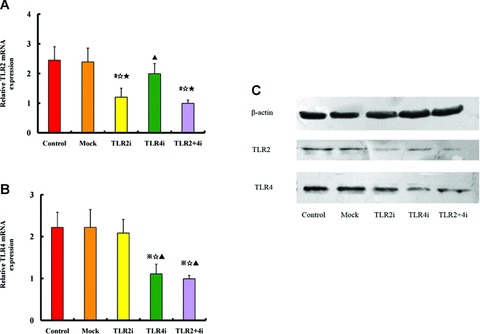
TLR2 and TLR4 gene expression in atherosclerosis plaques after gene knockdown with siRNA-lentivirus. (A) RT-PCR quantification of TLR2 mRNA expression in the control, mock and three gene interference subgroups of mice; (B) RT-PCR quantification of TLR4 mRNA expression in the control, mock and three gene interference subgroups of mice (n= 12 per group). #P < 0.05 versus control, ⋆P < 0.05 versus mock, ▴P < 0.05 versus TLR2i, ⋆P < 0.05 versus TLR4i. (C) Western blots analysis of TLR2 and TLR4 protein expression in the control, mock and three gene interference subgroups of mice.
Compared with the control subgroup, the TLR4 mRNA expression level was decreased by 48% and 53% in the TLR4i and TLR2 + 4i subgroups, respectively (both P < 0.05), and the TLR4 protein level was lowered by 43% and 47% in the TLR4i and TLR2 + 4i groups, respectively (both P < 0.05). In contrast, the control subgroup showed no difference in TLR4 mRNA and protein expression levels from the mock or TLR2i subgroups.
Plaque morphology
No significant difference in plaque area and the ratio of plaque area/internal elastic lamina area were found between the treatment and non-treatment subgroups (Fig. 4A and B). The plaques of both groups consisted of a distinctive fibrous cap overlying a necrotic core, and the cap/core area ratio in the control, mock, TLR2i, TLR4i and TLR2 + 4i subgroups was 0.16 ± 0.021, 0.162 ± 0.028, 0.272 ± 0.053, 0.274 ± 0.032 and 0.289 ± 0.024, respectively, which was significantly higher in the TLR2i, TLR4i and TLR2 + 4i subgroups than the control or mock subgroups (all P < 0.05, Fig. 4C). The fibrous cap thickness in plaques of control, mock, TLR2i, TLR4i and TLR2 + 4i subgroups was 10.01 ± 0.97 μm, 10.13 ± 1.42 μm, 17.17 ± 3.2 μm, 15.52 ± 1.18 μm and 17.33 ± 0.83 μm, respectively, and was significantly increased in the TLR2i, TLR4i and TLR2 + 4i subgroups compared with the control or mock subgroups (all P < 0.05). In comparison with the control group, the relative increase in the fibrous cap thickness in plaques of the TLR2i, TLR4i and TLR2 + 4i subgroups was 70.4%, 54.7% and 73.6%, respectively. Furthermore, TLR2i and TLR4i had a synergistic effect on increasing fibrous cap thickness (P < 0.05, Fig. 4D).
Fig 4.
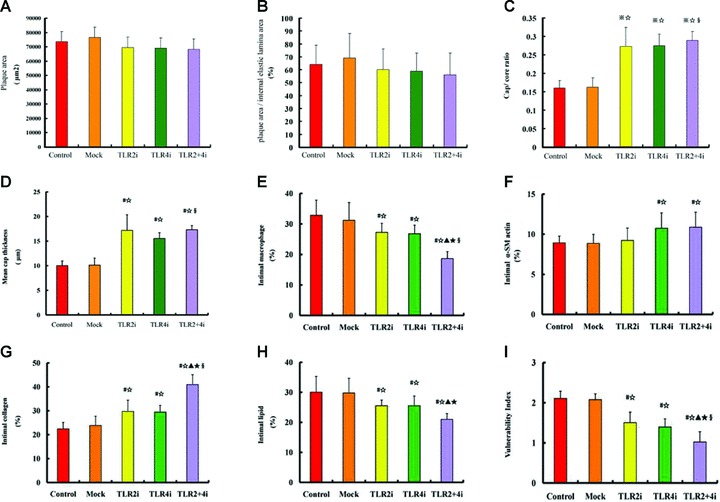
Comparison of plaque morphological parameters among five subgroups of mice. (A) Plaque area in the control, mock and three gene interference subgroups; (B) The ratio of plaque area/internal elastic lamina area in the control, mock and three gene interference subgroups; (C) Cap/core area ratio in the control, mock and three gene interference subgroups; (D) Fibrous cap thickness in the control, mock and three gene interference subgroups; (E)–(H) Relative contents of macrophages, α-actin+ SMCs, collagen and lipids, respectively, in plaques of the control, mock, TLR2i, TLR4i and TLR2 + 4i subgroups; (I) Plaque vulnerability index in the control, mock and TLR2i, TLR4i and TLR2 + 4i subgroups of mice (n= 12 per group). #P < 0.05 versus control, ⋆P < 0.05 versus mock, ▴P < 0.05 versus TLR2i, ⋆P < 0.05 versus TLR4i, §P < 0.05, interaction between TLR2i and TLR4i.
Plaque composition
The relative content of macrophages, SMCs, collagen and lipids in plaques was measured by histological and immunohistochemical staining (Figs 4E–I and 5). The relative content of macrophages in plaques of control, mock, TLR2i, TLR4i and TLR2 + 4i subgroups was 32.8%, 31.1%, 27.2%, 26.8% and 18.6%, respectively, and were significantly lower in the three gene-knockdown subgroups than the control or mock subgroups (all P < 0.05). In comparison with the control group, the relative reduction of the macrophage content in plaques of the TLR2i, TLR4i and TLR2 + 4i subgroups was 15.4%, 16.4% and 41.5%, respectively (Fig. 4E).
Fig 5.
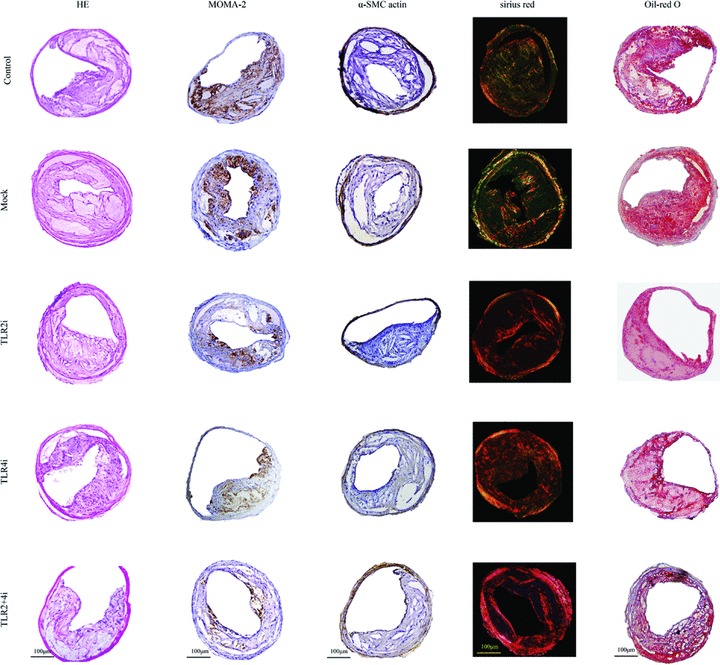
Histopathological staining showing plaque composition in the control, mock and three gene interference subgroups of mice. Cross-sections of carotid arteries in different groups were stained for haematoxylin and eosin, macrophages (MOMA-2) and vascular SMCs (α-actin), collagen (sirius red) and lipids (Oil-red O). Scale bar = 100 μm.
The relative content of α-actin+ staining SMCs in plaques of control, mock, TLR2i, TLR4i and TLR2 + 4i subgroups was 8.9%, 8.9%, 9.2%, 10.7% and 10.9%, respectively, and was higher in TLR4i and TLR2 + 4i subgroups compared with control subgroup (both P < 0.05), although no significant difference was noted in α-SMC content among the control, mock and TLR2i subgroups. In comparison with the control group, the relative increase of the α-SMCs content in plaques of the TLR2i, TLR4i and TLR2 + 4i subgroups was 5.7%, 20.7% and 17.2%, respectively (Fig. 4F).
The relative content of collagen in plaques of control, mock, TLR2i, TLR4i and TLR2 + 4i subgroups was 22.4%, 23.8%, 29.7%, 29.4% and 40.9%, respectively, and was significantly higher in the three gene-knockdown subgroups than the control or mock subgroups (all P < 0.05). Compared with the control group, the relative increase of the collagen content in plaques of the TLR2i, TLR4i and TLR2 + 4i subgroups was 33.3%, 32.4% and 82.6%, respectively (Fig. 4G).
The relative content of lipids in plaques of control, mock, TLR2i, TLR4i and TLR2 + 4i subgroups was 30.0%, 29.8%, 25.5%, 25.6% and 21.0%, respectively, and was significantly lower in the three gene-knockdown subgroups than the control or mock subgroups (all P < 0.05). In comparison with the control group, the relative reduction of the relative content of lipids in plaques of the TLR2i, TLR4i and TLR2 + 4i subgroups was 17.7%, 17.0% and 30.3%, respectively (Fig. 4H).
Thus, gene-knockdown subgroups showed higher content of collagen and lower content of lipid and macrophages than the control or mock subgroups and factorial ANOVA revealed that TLR2i and TLR4i had a synergistic effect on reducing the macrophage content and increasing the collagen content in plaques (P < 0.05).
The plaque vulnerability index in control, mock, TLR2i, TLR4i and TLR2 + 4i subgroups was 2.11 ± 0.18, 2.08 ± 0.14, 1.50 ± 0.26, 1.40 ± 0.20 and 1.02 ± 0.26, respectively, and was significantly lower in three gene-knockdown subgroups than the control or mock subgroups (all P < 0.05). In comparison with the control group, the relative reduction of the plaque vulnerability index in TLR2i, TLR4i and TLR2 + 4i subgroups was 33.0%, 34.0% and 52.2%, respectively. Moreover, TLR2i and TLR4i had a significant synergistic effect on decreasing the plaque vulnerability index (P < 0.05, Fig. 4I).
Plaque inflammation
The mRNA expression level of IL-6, MCP-1, MMP-2 and MMP-9 in the TLR2i subgroup and that of IL-1β, IL-6, TNF-α, MCP-1, MMP-2 and MMP-9 in the TLR4i subgroup were lower than the corresponding measurements in the control or mock subgroups (all P < 0.05). The TLR2 + 4i subgroup showed a significantly lower mRNA expression level of IL-1β, IL-6, TNF-α, MCP-1, MMP-2 and MMP-9 than the other four subgroups (P < 0.05, Fig. 6). Furthermore, the synergistic effect of TLR2i and TLR4i in the reduction of the mRNA expression level of these cytokines was confirmed by factorial ANOVA analysis (P < 0.05, Fig. 6).
Fig 6.
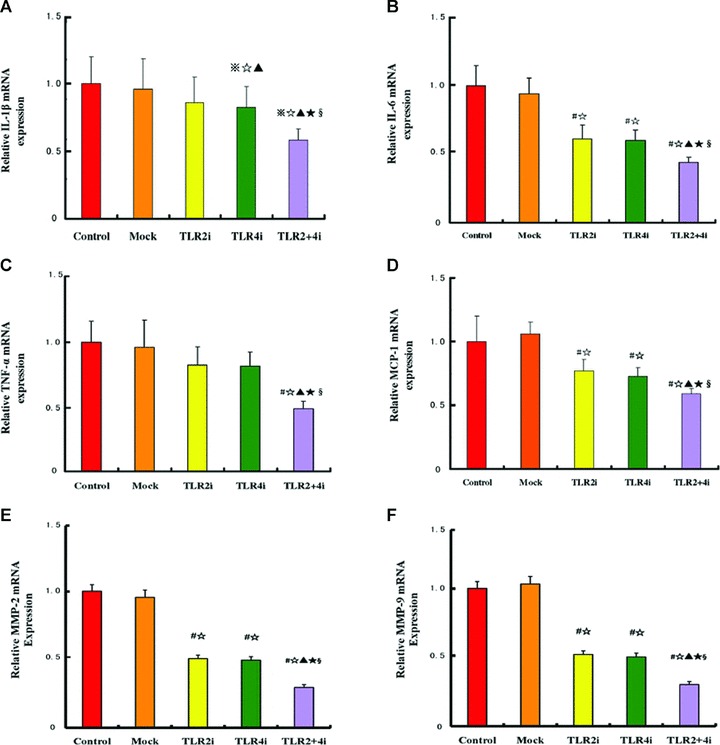
mRNA expression of inflammatory cytokines in carotid plaques of 5 subgroups of mice. (A) mRNA expression of IL-1β in the control, mock and three gene interference subgroups; (B) mRNA expression of IL-6 in the control, mock and three gene interference subgroups; (C) mRNA expression of TNF-α in the control, mock and three gene interference subgroups; (D) mRNA expression of MCP-1 in the control, mock and three gene interference subgroups; (E) mRNA expression of MMP-2 in the control, mock and three gene interference subgroups; (F) mRNA expression of MMP-9 in the control, mock and three gene interference subgroups (n= 12 per group). #P < 0.05 versus control, ⋆P < 0.05 versus mock, ▴P < 0.05 versus TLR2i, ⋆P < 0.05 versus TLR4i; §P < 0.05, interaction between TLR2i and TLR4i.
Discussion
The major finding of the present study was that using lentivirus-mediated RNA interference, mRNA and protein expression of TLR2 or TLR4 can be effectively knocked down in carotid plaques of ApoE−/− mice, leading to reduced local inflammatory cytokine expression and plaque content of lipid and macrophages, increased plaque content of collagen and lowered plaque vulnerability index. More importantly, there is a synergistic effect between TLR4i and TLR2i and combinatorial interference of TLR2 and TLR4 reduces local inflammation and stabilizes plaques more effectively than interference of TLR2 or TLR4 alone. To the best of our knowledge, this is the first study to show a synergistic effect between TLR2i and TLR4i in stabilizing atherosclerotic plaques.
Atherosclerosis is a chronic inflammatory disease of the vascular wall, characterized by accumulation of lipids and macrophage-derived foam cells in the sub-endothelial space [19]. TLR2 and TLR4 genes represent attractive therapeutic targets in atherosclerosis because they are important in recognizing evolutionary highly conserved molecular motifs in pathogens. Experimental studies have identified that activation of TLR2 and TLR4 plays a proatherogenic role [4, 20]. In the present study, therefore, the TLR2 and TLR4 genes were selected as a therapeutic target for vulnerable atherosclerotic plaques.
RNA interference is an effective method for selectively silencing mRNA for a wide range of proteins [21], and has been applied in several diseases for gene intervention [22, 23]. Although a number of delivering approaches are available, significant challenges remain, such as a low success rate, a low safety and off-target effects of RNAi in vivo. A major problem for synthetic siRNAs and plasmid-based siRNAs as a delivering approach is their transient expression and poor transfection efficiency in vivo. To overcome this limitation, a lentivirus expression cassette was used in this study to increase the transfection efficiency and such an approach has proven effective in silencing gene function in primary mammalian cells, stem cells and transgenic mice. By instilling a high-titre lentivirus suspension into the carotid artery, targeted gene interference was achieved. The efficacy of this method was confirmed not only by our in vitro observation that the protein expression levels of TLR2, TLR4, IL-1β, IL-6, TNF-α and MCP-1 in RAW264.7 cells were substantially lowered by TLR2 and TLR4 interference, but also by our in vivo observation of GFP fluorescence persisting for 4 weeks. The fact that no adverse effects were observed in our mice during the experiment and no significant difference was found in body weight between the treatment and non-treatment groups indicates that virus transfection was safe in these animals. The possibility that the therapeutic effects in the three treatment subgroups might be caused by non-specific immune stimulation was denied by the absence of significant difference in plaque inflammation and vulnerability between the control and the mock subgroup. Thus, lentiviral vectors expressing siRNA provide a safe, efficient and specific tool to knock down TLR genes.
In the present study, the effects of isolated and combinatorial interference of TLR2 and TLR4 genes on advanced atherosclerotic lesions were investigated. Our results demonstrated that both TLR2 and TLR4 were critical in the progression of atherosclerosis, as the three gene-knockdown subgroups of mice consistently exhibited a lower content of macrophages and lipids, a higher content of collagen, a higher cap/core area ratio and a thicker fibrous cap in atherosclerotic plaques than the control or mock subgroups of mice. All these morphological changes led to a decreased plaque vulnerability index in the three gene-knockdown subgroups compared to the control or mock subgroups. Plaque vulnerability index, by incorporating the cumulative effects of protective and destructive morphological factors, has been recognized as a histological marker for vulnerable plaques. Inhibited metabolism of active macrophages in plaques may explain the anti-atherosclerotic effects induced by TLR gene interference. Interestingly, combinatorial silencing of TLR2 and TLR4 genes had a synergistic effect on reducing the macrophage content, increasing the collagen content, thickening the fibrous cap and decreasing the plaque vulnerability index, which suggests that combinatorial interference of TLR2 and TLR4 is superior to isolated interference of TLR 2 or TLR 4 for stabilization of vulnerable plaques. In a recent published study, Shinohara et al. transfected cDNA encoding human TLR2 and TLR4 into the carotid arterial wall of rabbits fed high-cholesterol diets and found that overexpression of both TLR2 and TLR4 in the vessel wall synergistically accelerated local inflammation and atherosclerosis [24], which lends support to our results. It should be noted that at the end of the experiment, although plaque area and the ratio of plaque area/internal elastic lamina area tended to be lower in the TLR2i, TLR4i and TLR2i + TLR4i subgroups than in the control and mock subgroups, the difference was not significant, indicating that the 4 weeks gene therapy only tended to reduce plaque burden using the current animal model. These results demonstrated that the major effects of TLR interference were improvement of plaque components rather than plaque size, which led to a stabilized rather than regressed atherosclerotic plaque.
Recent studies have demonstrated that enhanced expression of TLR2 and TLR4 in atherosclerotic lesions is associated with production and activation of cytokines [5, 25]. TLRs recognize exogenous and endogenous ligands and initiate a signal cascade that leads to macrophage activation and secretion of inflammatory cytokines. MCP-1 expression has been detected in experimental and human atherosclerotic lesions, suggesting an active role of MCP-1 in monocyte recruitment [26]. In contrast, inactivation of MCP-1, or its receptor, CCR2, resulted in markedly reduced macrophage accumulation and attenuated atherosclerosis in mice [27]. Similarly, IL-6, IL-1β and TNF-α as the major pro-inflammatory cytokines are abundantly expressed in atherosclerotic lesions [28]. An elevated level of IL-6 and IL-1β has been detected in the coronary circulation in patients with acute coronary syndrome. TNF-α plays a major role in the recruitment and activation of inflammatory cells, stimulation of MMP production and inhibition of extracellular matrix synthesis [29, 30] whereas increased MMPs in atherosclerotic plaques may further induce monocyte and SMC migration and proliferation, and degrade extracellular matrix, therefore contributing to thinning and fissuring of the fibrous cap of plaques [31]. In the present study, the mRNA expression levels of IL-6, MCP-1, MMP-2 and MMP-9 were consistently reduced in the TLR2i, TLR4i and TLR2 + 4i subgroups compared with that in the control or mock subgroups and a synergistic effect of TLR2i and TLR4i in the reduction of the expression levels of IL-6, MCP-1, MMP-2 and MMP-9 was confirmed by factorial ANOVA analysis. On the other hand, the mRNA expression levels of IL-1β and TNF-α were reduced only in the TLR4i and TLR2 + 4i subgroups, suggesting that production of these two cytokines is more closely related to TLR4 than to TLR2 function. These results support a critical role for inflammatory cytokines such as IL-6, MCP-1, MMP-2, MMP-9, IL-1β and TNF-α in the progression of atherosclerosis and suggest that a reduced level of these cytokines induced by isolated and combinatorial interference of TLR2 and TLR4 contributed to the stabilization of atherosclerotic plaques.
Several limitations in this study should be mentioned. First, collar-induced carotid atherosclerosis is different from natural aortic atherosclerosis in the ApoE−/− mice. The carotid lesions occur in a region proximal to the constrictive collar with a prominent plaque burden and expansive arterial remodelling, whereas the aortic lesions develop in the ascending aorta with scattered plaques of small size and no expansive arterial remodelling. These phenotype differences may explain why the carotid plaque area did not differ among our five groups of mice, whereas the aortic lesions may actually regress in TLR2 and TLR4 knockout mice [12, 13]. Nonetheless, the carotid lesions in ApoE−/− mice resemble advanced human atherosclerosis and represent the most reproducible and reliable model for studies of vulnerable plaques [32]. Second, different amounts of TLR2 or TLR4 siRNA lentiviral vectors were given to different groups: 10 μl in the solo treated group and 5 μl of each vector in the combined treatment group. A more potent TLR interference effect would have been achieved if 10 μl of each vector had been given to the combined treatment group. The dose design of our study was based on the results of our preliminary experiment in which we tested the effectiveness of 5 titres of the lentivirus carrying TLR2 or TLR4 gene and found that the transduction effectiveness was enhanced with increased titres but reached a complete plateau when the titre went up to 109 TU/ml. Thus, we chose the titre of 2 × 109 TU/ml for all gene treatment groups to eliminate any possible dose-related effects. For the concern that an overdose of vectors might cause negative side effects of lentiviral infections, 10 μl vector was given to each of the four treatment groups. The fact that a combination of 5 μl TLR2i lentivirus and 5 μl TLR4i lentivirus was more potent than 10 μl of the individual vectors lent support to the presence of a synergistic effect between TLR2i and TLRi4. Third, adventitia may play an important role in atherogenesis but was not examined in this study. Placement of a constrictive collar may induce adventitial inflammation and adhesion to the collar and during collar removal, the carotid adventitia may become incomplete. Further studies using animal models with intact adventitia are required to elucidate the possible changes of adventitia induced by TLR2 and TLR4 interference.
In summary, our study demonstrated that lentivirus-mediated RNA interference was effective in knocking down TLR2 and TLR4 genes in ApoE−/− mice, which resulted in reduced inflammatory cytokine expression and plaque content of lipid and macrophages, increased plaque content of collagen and lowered plaque vulnerability index. A synergistic effect exists between TLR2i and TLR4i in stabilizing atherosclerotic plaques and thus, combinatorial interference is superior to isolated interference of TLR2 and TLR4 genes for this purpose which provides a novel approach to the treatment of vulnerable atherosclerotic plaques.
Acknowledgments
We thank Dr. Jin Bo Feng for his excellent technical assistance. This study was supported by the National 973 Basic Research Program of China (No. 2009CB521904), the National High-tech Research and Development Program of China (No. 2006AA02A406), the Program of Introducing Talents of Discipline to Universities (No. B07035), the State Key Program of National Natural Science of China (No. 60831003), Cultivation Fund of the Key Scientific and Technical Innovation Project, Ministry of Education of China (No.704030), grants from the National Natural Science Foundation of China (No. 30871037, No. 30900607, No. 30900523, No. 30470702 and No. 30971096), grants from the Specialized Research Fund for the Doctoral Program of Higher Education of China (No. 20090131120066) and grants from the Natural Science Foundation of Shandong Province, China (No. ZR2009CM021).
Conflict of interest
None declared.
References
- 1.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 2.Yamamoto M, Takeda K, Akira S. TIR domain-containing adaptors define the specificity of TLR signaling. Mol Immunol. 2004;40:861–8. doi: 10.1016/j.molimm.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 3.Tobias P, Curtiss LK. Thematic review series: the immune system and atherogenesis. Paying the price for pathogen protection: toll receptors in atherogenesis. J Lipid Res. 2005;46:404–11. doi: 10.1194/jlr.R400015-JLR200. [DOI] [PubMed] [Google Scholar]
- 4.Hollestelle SC, De Vries MR, Van Keulen JK, et al. Toll-like receptor 4 is involved in outward arterial remodeling. Circulation. 2004;109:393–8. doi: 10.1161/01.CIR.0000109140.51366.72. [DOI] [PubMed] [Google Scholar]
- 5.Schoneveld AH, Hoefer I, Sluijter JP, et al. Atherosclerotic lesion development and Toll like receptor 2 and 4 responsiveness. Atherosclerosis. 2008;197:95–104. doi: 10.1016/j.atherosclerosis.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 6.Faure E, Thomas L, Xu H, et al. Bacterial lipopolysaccharide and IFN-gamma induce Toll-like receptor 2 and Toll-like receptor 4 expression in human endothelial cells: role of NF-kappa B activation. J Immunol. 2001;166:2018–24. doi: 10.4049/jimmunol.166.3.2018. [DOI] [PubMed] [Google Scholar]
- 7.Jimenez R, Belcher E, Sriskandan S, et al. Role of Toll-like receptors 2 and 4 in the induction of cyclooxygenase-2 in vascular smooth muscle. Proc Natl Acad Sci USA. 2005;102:4637–42. doi: 10.1073/pnas.0407655101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu XH, Shah PK, Faure E, et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. 2001;104:3103–8. doi: 10.1161/hc5001.100631. [DOI] [PubMed] [Google Scholar]
- 9.Vink A, Schoneveld AH, der Meer JJv, et al. In vivo evidence for a role of toll-like receptor 4 in the development of intimal lesions. Circulation. 2002;106:1985–90. doi: 10.1161/01.cir.0000032146.75113.ee. [DOI] [PubMed] [Google Scholar]
- 10.Bulut Y, Faure E, Thomas L, et al. Chlamydial heat shock protein 60 activates macrophages and endothelial cells through Toll-like receptor 4 and MD2 in a MyD88-dependent pathway. J Immunol. 2002;168:1435–40. doi: 10.4049/jimmunol.168.3.1435. [DOI] [PubMed] [Google Scholar]
- 11.Zanin-Zhorov A, Nussbaum G, Franitza S, et al. T cells respond to heat shock protein 60 via TLR2: activation of adhesion and inhibition of chemokine receptors. FASEB J. 2003;17:1567–9. doi: 10.1096/fj.02-1139fje. [DOI] [PubMed] [Google Scholar]
- 12.Schoneveld AH, Oude NMM, van MB, et al. Toll-like receptor 2 stimulation induces intimal hyperplasia and atherosclerotic lesion development. Cardiovasc Res. 2005;66:162–9. doi: 10.1016/j.cardiores.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 13.Michelsen KS, Wong MH, Shah PK, et al. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci USA. 2004;101:10679–84. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qi LH, Wang Y, Gao F, et al. Enhanced stabilization of atherosclerotic plaques in apolipoprotein E-knockout mice by combinatorial Toll-like receptor-1 and -2 gene silencing. Hum Gene Ther. 2009;20:739–50. doi: 10.1089/hum.2008.203. [DOI] [PubMed] [Google Scholar]
- 15.Morris KV, Rossi JJ. Lentiviral-mediated delivery of siRNAs for antiviral therapy. Gene Ther. 2006;13:553–8. doi: 10.1038/sj.gt.3302688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.der Thusen JHv, van BTJ, Biessen EA. Induction of rapid atherogenesis by perivascular carotid collar placement in apolipoprotein E-deficient and low-density lipoprotein receptor-deficient mice. Circulation. 2001;103:1164–70. doi: 10.1161/01.cir.103.8.1164. [DOI] [PubMed] [Google Scholar]
- 17.der Thusen JHv, van VBJ, Hoeben RC, et al. Induction of atherosclerotic plaque rupture in apolipoprotein E−/− mice after adenovirus-mediated transfer of p53. Circulation. 2002;105:2064–70. doi: 10.1161/01.cir.0000015502.97828.93. [DOI] [PubMed] [Google Scholar]
- 18.Shiomi M, Ito T, Hirouchi Y, et al. Fibromuscular cap composition is important for the stability of established atherosclerotic plaques in mature WHHL rabbits treated with statins. Atherosclerosis. 2001;157:75–84. doi: 10.1016/s0021-9150(00)00708-5. [DOI] [PubMed] [Google Scholar]
- 19.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–43. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 20.Liu X, Ukai T, Yumoto H, et al. Toll-like receptor 2 plays a critical role in the progression of atherosclerosis that is independent of dietary lipids. Atherosclerosis. 2008;196:146–54. doi: 10.1016/j.atherosclerosis.2007.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elbashir SM, Harborth J, Lendeckel W, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–8. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 22.Song E, Lee SK, Wang J, et al. RNA interference targeting Fas protects mice from fulminant hepatitis. Nat Med. 2003;9:347–51. doi: 10.1038/nm828. [DOI] [PubMed] [Google Scholar]
- 23.Zender L, Hutker S, Liedtke C, et al. Caspase 8 small interfering RNA prevents acute liver failure in mice. Proc Natl Acad Sci USA. 2003;100:7797–802. doi: 10.1073/pnas.1330920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shinohara M, Hirata K, Yamashita T, et al. Local overexpression of toll-like receptors at the vessel wall induces atherosclerotic lesion formation: synergism of TLR2 and TLR4. Arterioscler Thromb Vasc Biol. 2007;27:2384–91. doi: 10.1161/ATVBAHA.106.139253. [DOI] [PubMed] [Google Scholar]
- 25.Markus HS, Labrum R, Bevan S, et al. Genetic and acquired inflammatory conditions are synergistically associated with early carotid atherosclerosis. Stroke. 2006;37:2253–9. doi: 10.1161/01.STR.0000236637.72124.3f. [DOI] [PubMed] [Google Scholar]
- 26.Martinovic I, Abegunewardene N, Seul M, et al. Elevated monocyte chemoattractant protein-1 serum levels in patients at risk for coronary artery disease. Circ J. 2005;69:1484–9. doi: 10.1253/circj.69.1484. [DOI] [PubMed] [Google Scholar]
- 27.Yla-Herttuala S, Lipton BA, Rosenfeld ME, et al. Expression of monocyte chemoattractant protein 1 in macrophage-rich areas of human and rabbit atherosclerotic lesions. Proc Natl Acad Sci USA. 1991;88:5252–6. doi: 10.1073/pnas.88.12.5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boring L, Gosling J, Cleary M, et al. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–7. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 29.Rajavashisth TB, Xu XP, Jovinge S, et al. Membrane type 1 matrix metalloproteinase expression in human atherosclerotic plaques: evidence for activation by proinflammatory mediators. Circulation. 1999;99:3103–9. doi: 10.1161/01.cir.99.24.3103. [DOI] [PubMed] [Google Scholar]
- 30.Zhang C, Zhang MX, Shen YH, et al. TNF-alpha suppresses prolyl-4-hydroxylase alpha1 expression via the ASK1-JNK-NonO pathway. Arterioscler Thromb Vasc Biol. 2007;27:1760–7. doi: 10.1161/ATVBAHA.107.144881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 2002;90:251–62. [PubMed] [Google Scholar]
- 32.Rosenfeld ME, Schwartz SM. Murine models of advanced atherosclerosis. In: Virmani R, Narula J, Leon MB, Willerson JT, editors. The vulnerable atherosclerotic plaque: strategies for diagnosis and management. Malden: Blackwell Publishing, Inc; 2007. pp. 105–27. [Google Scholar]