

Abstract
Although angiogenesis is viewed as a fundamental component of inflammatory bowel disease (IBD) pathogenesis, we presently lack a thorough knowledge of the cell type(s) involved in its induction and maintenance in the inflamed intestinal mucosa. This study aimed to determine whether platelet (PLT) adhesion to inflamed intestinal endothelial cells of human origin may favour angiogenesis. Unstimulated or thrombin-activated human PLT were overlaid on resting or tumour necrosis factor (TNF)-α-treated human intestinal microvascular endothelial cells (HIMEC), in the presence or absence of blocking antibodies to either vascular cell adhesion molecule (VCAM)-1, intercellular adhesion molecule (ICAM)-1, integrin αvβ3, tissue factor (TF) or fractalkine (FKN). PLT adhesion to HIMEC was evaluated by fluorescence microscopy, and release of angiogenic factors (VEGF and soluble CD40L) was measured by ELISA. A matrigel tubule formation assay was used to estimate PLT capacity to induce angiogenesis after co-culturing with HIMEC. TNF-α up-regulated ICAM-1, αvβ3 and FKN expression on HIMEC. When thrombin-activated PLT were co-cultured with unstimulated HIMEC, PLT adhesion increased significantly, and this response was further enhanced by HIMEC activation with TNF-α. PLT adhesion to HIMEC was VCAM-1 and TF independent but ICAM-1, FKN and integrin αvβ3 dependent. VEGF and sCD40L were undetectable in HIMEC cultures either before or after TNF-α stimulation. By contrast, VEGF and sCD40L release significantly increased when resting or activated PLT were co-cultured with TNF-α-pre-treated HIMEC. These effects were much more pronounced when PLT were derived from IBD patients. Importantly, thrombin-activated PLT promoted tubule formation in HIMEC, a functional estimate of their angiogenic potential. In conclusion, PLT adhesion to TNF-α-pre-treated HIMEC is mediated by ICAM-1, FKN and αvβ3, and is associated with VEGF and sCD40L release. These findings suggest that inflamed HIMEC may recruit PLT which, upon release of pro-angiogenic factors, actively contribute to inflammation-induced angiogenesis.
Keywords: angiogenesis, tumour necrosis factor, inflammation, inflammatory bowel disease
Introduction
Angiogenesis is an intricate process resulting in the generation of novel vessels from pre-existing ones, through the promotion of endothelial cell (EC) division, degradation of vascular basement membrane and surrounding extracellular matrix, and EC migration [1]. Angiogenesis is tightly regulated by a balance between expression and function of pro-angiogenic and anti-angiogenic mediators. Vascular endothelial growth factor (VEGF) is recognized as a potent pro-angiogenic cytokine and is secreted by monocytes/macrophages, EC and a variety of other cell types [2]. Interleukin (IL)-8 exerts direct angiogenic effects on EC in vitro and in vivo. Colonic mucosal IL-8 is up-regulated in Crohn’s disease in direct correlation with the degree of inflammation [3]. In addition to pro-inflammatory actions, IL-8 has been implicated in tumour angiogenesis and may be secreted by colonic epithelial cells and microvessel EC [4]. Similarly, basic fibroblast growth factor (bFGF) may mediate tumour necrosis factor (TNF)-α-induced angiogenesis both in vitro and in vivo[5]. In this respect, we have recently shown that angiogenesis may be implicated in the pathogenesis of inflammatory bowel disease (IBD) [6]. In line with this assumption, microvessel density is increased in IBD mucosal tissues, a phenomenon that is mostly dependent upon heightened IL-8, bFGF and VEGF release [6]. In addition, angiogenesis blockade may represent a new and promising therapeutic target in experimental models of IBD [7, 8].
The CD40-CD40 ligand (CD40L) axis is a regulator and amplifier of immune reactivity and contributes to leucocyte and platelet (PLT) adhesion to inflamed intestinal EC [8–10]. CD40 expression is prominent in pathological conditions associated with angiogenesis and inflammation, and ligation of CD40 on EC and monocytes leads to production of bFGF and VEGF [11]. For instance, CD40 engagement on synovial fibroblasts by CD40L-expressing activated T cells up-regulates the production of VEGF, providing a potential explanation for the occurrence of neovascularization in rheumatoid arthritis [12]. CD40L is over-represented in patients with IBD, mainly reflecting enhanced expression on the PLT surface and spontaneous release into the circulation [10, 13]. Plasma sCD40L is particularly elevated in patients with active Crohn’s disease and ulcerative colitis compared with patients with inactive disease and with healthy controls [10, 13].
IBD has been associated with PLT dysfunction, and increased PLT aggregation to epinephrine, collagen and/or ADP [14]. Increased PLT expression of P-selectin [15] and elevation of PLT-derived microparticles have been previously reported in patients with IBD. In addition to qualitative abnormalities, PLT are numerically elevated in patients with IBD in relation with increased thrombopoietin and IL-6 serum levels [16, 17]. Interestingly, an increased tendency to form PLT-leucocyte aggregates has been described both in the affected colonic mucosa and in the peripheral blood of patients with active IBD, but not in other chronic inflammatory disorders [18, 19]. PLT-derived inflammatory and pro-angiogenic mediators such as sCD40L and VEGF may be instrumental in the in vitro migration and vessel-like organization of EC, pointing to a role for PLT in inflammatory neoangiogenesis [20].
The present study was designed and conducted to determine whether activated PLT may contribute to angiogenesis through an enhanced adhesiveness to inflamed EC with subsequent release of pro-angiogenic growth factors. We also addressed the potential molecular determinants of PLT–EC interactions that may contribute to angiogenesis and inflammation in the IBD microvasculature [21]. We show herein that PLT adhesion to inflamed microvascular EC translates into an enhanced release of pro-angiogenic mediators, providing clues on the potential role of activated PLT in the promotion of inflammation-driven angiogenesis in the gut.
Materials and methods
Patient population
Patients with active IBD were studied after their informed consent. The investigations were reviewed and approved by the local Ethical Committee. All diagnoses were confirmed by clinical, radiological, endoscopic and histological criteria, as previously detailed [10, 13]. Anatomical disease extension was assessed by radiological and endoscopic examination. Peripheral blood samples were also obtained from consented healthy blood donors and were used to isolate PLT for control experiments, as reported [13, 22]. Patients’ characteristics were summarized in Table 1.
Table 1.
Patients’ characteristics
| Number of patients | 17 |
|---|---|
| UC | 8 |
| CD | 9 |
| Sex | |
| Male | 9 |
| Female | 8 |
| Disease duration (years) | 8 (2–18) |
| Location | |
| Ileal | 6 |
| Colonic | 9 |
| Ileo-colonic | 2 |
| Proximal | 0 |
| Concomitant medications | |
| Steroids | 4 |
| Azathioprine | 7 |
| Mesalamine | 12 |
| Antibiotics | 2 |
Procurement and culture of HIMEC
Surgical specimens of colonic origin were used to isolate human intestinal microvascular endothelial cells (HIMEC), as reported elsewhere [23, 24]. Briefly, after enzymatic digestion of intestinal mucosal strips, samples were gently compressed to extrude EC clumps, which adhered to fibronectin-coated plates, and were subsequently cultured in MCDB131 medium (Sigma Aldrich, St. Louis, MO, USA) supplemented with 20% FBS, antibiotics, heparin, and EC growth factor. HIMEC were routinely plated on fibronectin-coated wells of a 24-well cluster plate at a density of 5 × 104/ml/well. For HIMEC activation, cells were supplemented with 100 IU/ml TNF-α (R&D Systems, Oxon, UK). Cultures of HIMEC were maintained at 37°C in 5% CO2 and cells were used between passages 3 and 10 [23].
Isolation of PLT and PLT-HIMEC co-culture
PLT from normal donors were obtained by gel filtration of PLT-rich plasma (PRP) onto Sepharose 2B columns (25 × 1 cm) equilibrated with a buffer containing 20 mM Hepes, 135 mM NaCl, 5 mM KCl, 5 mM glucose, 0.2% albumin (pH 7.4). Ethylenediaminetetraacetic acid (EDTA) disodium salt (1 mM final concentration) was added to the PRP prior to gel filtration to minimize PLT activation during washing procedures. The resulting PLT population was essentially free of contaminating erythrocytes (<0.1%) and peripheral blood mononuclear cells. In order to rule out PLT activation due to the isolation procedure, PLT activation state was assessed before and after isolation by measuring P-selectin expression levels, as previously detailed [10]. The PLT count was adjusted to 1 × 105/μl with washing buffer in all the experiments [13, 25].
Confluent monolayers of HIMEC were incubated with either 2% bovine serum albumin alone or 0.5 U/ml thrombin alone, or they were overlaid with 100 × 106 resting or thrombin-activated PLT. Plates were immediately centrifuged at 700×g for 2 min. to bring PLT and HIMEC in close apposition. Each experiment was performed in duplicate. After 4 hrs at 37°C, supernatants were harvested, transferred to polypropylene tubes, centrifuged at 1300×g at 4°C for 10 min. to remove cell debris, and stored at −70°C until analysis. In preliminary experiments, we ensured, by visual microscopy and flow cytometry with PLT-specific CD42b antibodies, that washings had removed virtually all PLT from the HIMEC monolayers. At the end of the co-culture, HIMEC were washed five times in cold phospate buffer saline (PBS) and a single cell suspension obtained using a detaching buffer (PBS, 20 mmol/l HEPES, pH 7.4, 10 mmol/l EDTA and 0.5% bovine serum albumin) for 10 min. each on ice and at 37°C, followed by vigorous pipetting.
Release of pro-angiogenic factors
VEGF-A and sCD40L levels in culture supernatants were measured in triplicate with commercially available ELISA, following the manufacturer’s instructions. The limits of detection were as follows: <9 pg/ml VEGF-A and <10.1 pg/ml sCD40L.
In vitro tube formation assay
EC tube formation was assessed using Matrigel™, a solubilized extracellular basement membrane matrix extracted from the Engelbreth-Holm-Swarm mouse sarcoma, as detailed elsewhere [26]. Briefly, multi-well dishes were coated with 250 μl of complete medium containing 5 mg/ml Matrigel™ and HIMEC re-suspended in complete growth medium were seeded at a density of 5 × 104. Cells were cultured on Matrigel™ for 16 hrs and inverted phase-contrast microscopy was used to assess formation of endothelial tube-like structures. Five high-power fields per condition were examined and experiments were performed in duplicate.
Statistical analysis
The approximation of data distribution to normality was preliminarily tested with statistics for kurtosis and symmetry. Results were presented as mean and S.D. All comparisons were performed with the Student’s t-test for paired or unpaired determinations or with the ANOVA, as appropriate. The criterion for statistical significance was defined as P≤ 0.05.
Results
The pro-inflammatory cytokine TNF-α enhances the expression of cell adhesion molecules (CAM) on HIMEC
In a first set of experiments, we determined how the exposure of HIMEC to TNF-α would affect the expression pattern of a spectrum of adhesion molecules deemed relevant for HIMEC interaction with PLT. To this end, vascular cell adhesion molecule (VCAM)-1, intercellular adhesion molecule (ICAM)-1, aVβ3 integrin, tissue factor (TF) and fractalkine (FKN) expression was investigated by flow cytometry on HIMEC that were exposed to TNF-αin vitro for 16 hrs. As shown in Fig. 1A, TNF-α-treated HIMEC up-regulated ICAM-1, aVβ3 integrin and FKN, and expressed de novo VCAM-1 and TF when compared to CAM levels on untreated HIMEC. The analysis of the mean fluorescence intensity ratios indicated that the magnitude of adhesion molecule induction by TNF-α differed significantly, with aVβ3 integrin and FKN being particularly responsive to the pro-inflammatory stimulus applied to HIMEC (Fig. 1B).
Fig 1.
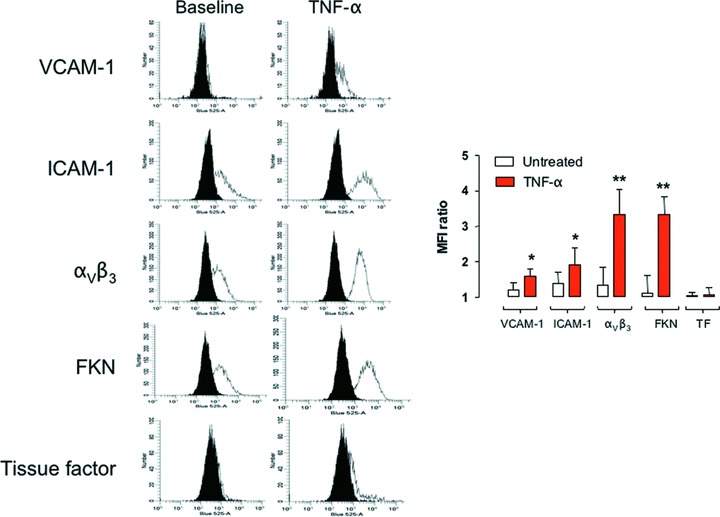
Expression of adhesion molecules after exposure to TNF-α. HIMEC were treated with TNF-α, and then labelled with monoclonal antibodies directed against VCAM-I, ICAM-I, integrin αVβ3, TF and FKN prior to flow cytometry analysis. A representative experiment out of six with similar results is shown in (A). Black histograms depict isotypic controls. The mean fluorescence intensity ratios of histogram distributions recorded in these experiments have been pooled and are shown as mean and S.D. in (B). *P < 0.05 and **P < 0.01 compared with HIMEC that were not challenged with TNF-α.
Inflammation promotes PLT adhesion to HIMEC
To ascertain whether a prototypical inflammatory cytokine such as TNF-α was endowed with the ability to promote PLT adhesion to HIMEC, 10 × 106 PLT from healthy control patients were co-cultured with 50 × 103 HIMEC that were activated with TNF-α, as above detailed. To dissect the role of PLT activation status, if any, in the interaction with HIMEC, PLT were treated with thrombin prior to HIMEC co-culture.
The baseline adhesion of resting PLT to untreated HIMEC is shown in Fig. 2, left part. The stimulation of HIMEC with TNF-α enhanced PLT adhesion (Fig. 2). Interestingly, activation with thrombin further increased the number of adhering PLT to resting HIMEC and, to an even greater extent, the number of PLT that adhered to TNF-α-activated HIMEC (Fig. 2). It should be pointed out that provision of TNF-α to HIMEC per se was capable of enhancing the adhesion of resting PLT compared to that recorded with unstimulated HIMEC, as shown in Fig. 2. Collectively, these studies suggest that a pro-inflammatory cytokine stimulus promotes the adhesion of resting PLT to HIMEC and that this effect is significantly enhanced by PLT activation prior to the co-culture.
Fig 2.
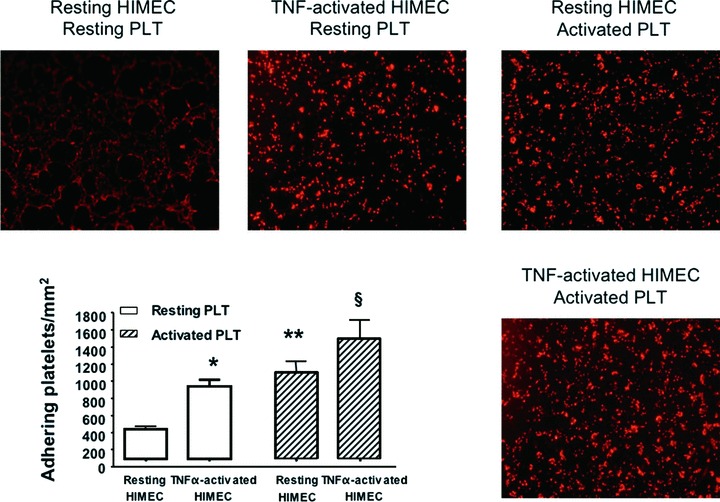
Assays of PLT adhesion to HIMEC. Prior to the adhesion assay, PLT and HIMEC were either left untouched or activated with thrombin and TNF-α, respectively, as detailed in ‘Materials and methods’. The bar graph (mean ± S.D.) summarizes the results obtained in six independent experiments. *P < 0.05 compared with PLT adhesion to resting HIMEC; **P < 0.01 compared with adhesion of resting PLT to HIMEC; §P < 0.01 compared with adhesion of resting PLT to TNF-α-activated HIMEC.
PLT adhesion to inflamed HIMEC is mediated through ICAM-I, aVβ3 and FKN
We next attempted to dissect the mechanism(s) that govern PLT adhesion to HIMEC under the inflammatory conditions that we established herein. Because provision of TNF-α to HIMEC translates into the up-regulation of selected adhesion molecules, we maintained PLT-HIMEC co-cultures either in the presence or absence of blocking antibodies directed against VCAM-1, ICAM-1, aVβ3 integrin, TF and FKN. Co-cultures were also performed with resting HIMEC and resting PLT (negative control for PLT adhesion) and with TNF-α-stimulated HIMEC and thrombin-activated PLT (positive control for PLT adhesion). As shown in Fig. 3, neutralization of either surface ICAM-1, aVβ3 integrin, or FKN significantly, albeit not completely, inhibited PLT adhesion to HIMEC. Conversely, no changes in the number of adhering PLT were recorded in co-cultures established in the presence of blocking antibodies to either VCAM-1 or TF compared with the control co-cultures containing TNF-α-activated HIMEC and thrombin-activated PLT. Collectively, neutralization studies suggest that a restricted panel of adhesion molecules is involved in the enhanced PLT adhesion to inflamed endothelial surfaces in our system model.
Fig 3.
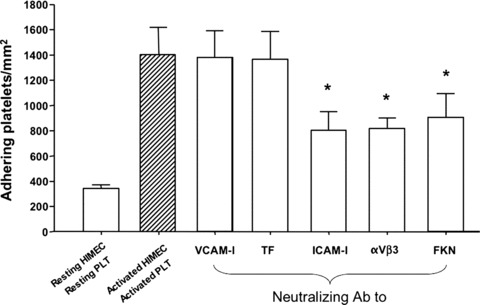
PLT adhesion to HIMEC after antibody-mediated neutralization of adhesion molecules. Neutralising antibodies to VCAM-I, TF, ICAM-1, integrin αVβ3 and FKN were provided to the HIMEC-PLT co-cultures. The number of adhering PLT was counted and plotted in the y-axis. The adhesion of thrombin-activated PLT to TNF-α-activated HIMEC was measured as positive control. The addition of neutralising antibodies to ICAM-I, integrin αVβ3 and FKN translated into a statistically significant reduction of the number of adhering PLT. The bar graph (mean ± S.D.) summarizes the results obtained in six independent experiments. *P < 0.01 compared with adhesion of activated PLT to TNF-α-activated HIMEC.
PLT from patients with active IBD are particularly effective at releasing pro-angiogenic growth factors upon co-culture with HIMEC
We also aimed to ascertain whether PLT interaction with inflamed HIMEC may promote the release of pro-angiogenic growth factors. To accomplish this goal, we initially measured the release of VEGF-A and sCD40L in the supernatant of co-cultures established with HIMEC and PLT from healthy donors. VEGF-A was undetectable in the supernatant of unstimulated HIMEC, but was readily measured in supernatants of resting PLT (Fig. 4). PLT activation with thrombin translated into a significant up-regulation of VEGF-A levels compared with those detected in the supernatant of resting PLT. Of interest, pre-treatment of HIMEC with the pro-inflammatory cytokine TNF-α enhanced the release of VEGF-A in the co-culture with both resting and thrombin-activated PLT (Fig. 4).
Fig 4.
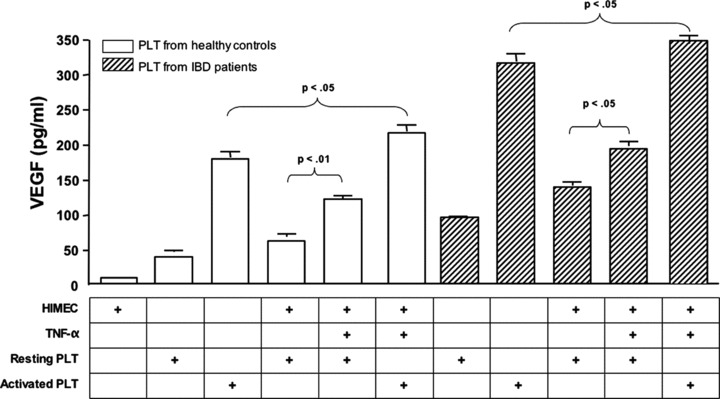
Release of VEGF upon co-culture of HIMEC with PLT from healthy controls and patients with active IBD. PLT from either healthy controls (n= 8; empty columns) or patients with active IBD (8 UC and 9 CD; shaded columns) were co-cultured with HIMEC that were either left untouched or activated with TNF-α. VEGF release was measured with conventional ELISA in culture supernatants. The bar graph (mean ± S.D.) summarizes the results obtained in six independent experiments.
We subsequently reproduced the above co-culture experiments using PLT from patients with active IBD, either Crohn’s disease or ulcerative colitis. PLT from patients with IBD released greater amounts of VEGF-A both spontaneously and following activation with thrombin compared with equal numbers of PLT from healthy controls (Fig. 4). Notably, VEGF-A production by resting PLT was further enhanced in the co-cultures containing TNF-α-activated HIMEC. Not unexpectedly, the highest release of VEGF-A was detected in the co-cultures established with thrombin-activated PLT and TNF-α-activated HIMEC.
The same culture conditions were applied in further experiments aimed at quantifying sCD40L production. As depicted in Fig. 5, PLT from healthy controls released sCD40L and this phenomenon was significantly enhanced by PLT activation with thrombin. Whereas TNF-α-activated HIMEC promoted sCD40L release from unstimulated PLT, no such increase in sCD40L production occurred in the co-cultures established with activated PLT and TNF-α-stimulated HIMEC compared with those containing activated PLT alone (Fig. 5). Interestingly, PLT from patients with active IBD released significantly greater quantities of sCD40L compared with PLT from healthy controls, both spontaneously and following thrombin activation. Finally, the co-culture of PLT from IBD patients with TNF-α-activated HIMEC further promoted sCD40L secretion, as depicted in Fig. 5. Collectively, PLT isolated from patients with active IBD were endowed with a greater capacity to secrete both VEGF-A and sCD40L either untouched or following their in vitro activation with thrombin. No differences were found between PLTs derived from Crohn’s disease or ulcerative colitis patients (data not shown).
Fig 5.
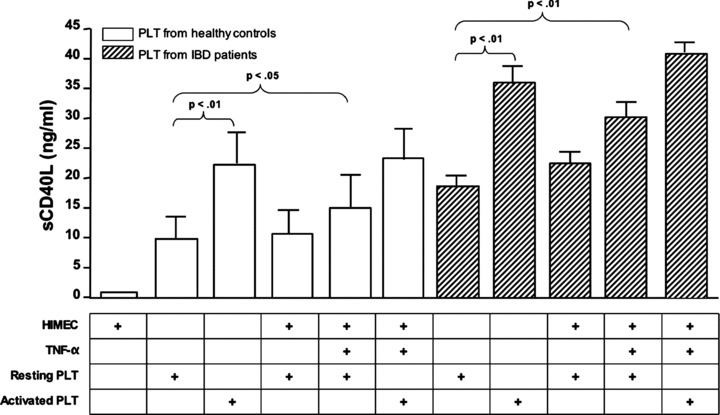
Release of sCD40L upon co-culture of HIMEC with PLT from healthy controls and patients with active IBD. PLT from either healthy controls (n= 8; empty columns) or patients with active IBD (8 UC and 9 CD; shaded columns) were co-cultured with HIMEC that were either left untouched or activated with TNF-α. The release of sCD40L was measured with conventional ELISA in culture supernatants. The bar graph (mean ± S.D.) summarizes the results obtained in six independent experiments.
Activated PLT favour tubule formation in HIMEC cultures
In a further set of functional studies, we wanted to determine whether thrombin-activated PLT may induce tubule formation in HIMEC. To answer this, HIMEC were cultured on a solubilized extracellular basement membrane extract and used to assess the formation of endothelial-like structures by inverted phase-contrast microscopy [26]. As clearly shown in Fig. 6A, tube-like structures could be seen after co-culturing fluorescently labelled resting HIMEC with thrombin-activated PLT derived from healthy controls but not in cultures established with HIMEC alone. Thrombin alone failed to induce tubule formation by HIMEC (data not shown). Fluorescent microscopy experiments with PLT-HIMEC co-cultures stained with different fluorochromes indicated that activated PLT preferentially localized at endothelial luminal surfaces, as shown in a 3-dimensional adhesion assay on matrigel (Fig. 6B).
Fig 6.
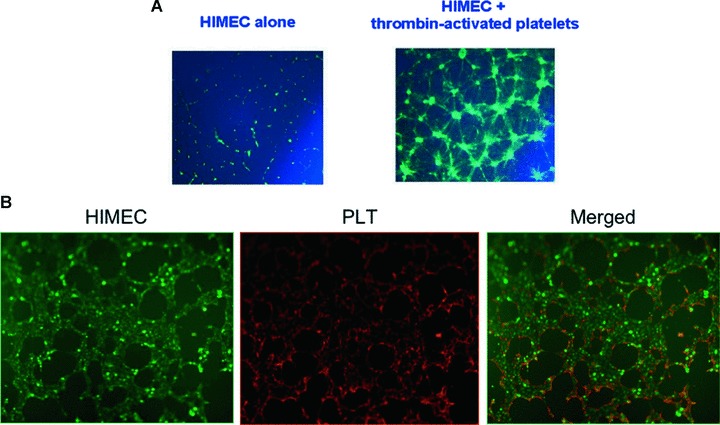
Tubule formation assay and visualization of PLT-HIMEC interaction. (A) PLT from healthy controls were either left untouched or activated with thrombin. Tubule formation was evaluated as detailed in ‘Materials and methods’. One representative experiment out of six with similar results is shown. (B) PLT from healthy controls were either left untouched or activated with thrombin. The interaction of HIMEC (green) and PLT (red) was visualized by confocal microscopy. One representative experiment out of four with similar results is shown.
Discussion
It is presently believed that IBD results from the interaction of genetic, environmental, microbial and immune factors. Accumulating evidence suggests that non immune cells such as mucosal EC, fibroblasts, neurons and PLT actively contribute to IBD pathogenesis [27]. In particular, we and others have previously provided novel and substantial evidence that PLT are dynamic participants in the multi-component system responsible for mucosal inflammation and injury [28]. Since the number and activation state of PLT are markedly increased in IBD patients, PLT contained within the systemic circulation represent a potential risk factor for triggering an inflammatory response at the intestinal level. For these considerations, PLT are currently viewed as an attractive target for therapeutic intervention [13, 22, 29]. In a different disease context such as multiple sclerosis, PLT-derived VEGF reportedly sustains angiogenesis, a process that may be exacerbated as a result of PLT interaction with the injured ECs [30].
CD40L, also referred to as gp39, is a cell surface molecule largely restricted to activated CD4+ T cells, and is also expressed by human vascular EC, smooth muscle cells and macrophages. The CD40-CD40L axis has been implicated in the pathogenesis of several human diseases, including atherosclerosis [31], rheumatoid arthritis [12] and IBD [8]. PLT have been reported to trigger CD40-dependent inflammatory responses in the IBD microvasculature and to induce endothelial CAM up-regulation, chemokine secretion and leucocyte recruitment [10]. However, the role of activated PLT in inducing mucosal angiogenesis has not been explored yet, although extensive data on PLT-microvasculature interactions have been reported in mice [32]. Since PLT aggregation and microthrombi are frequent findings in the IBD microvasculature [28, 33, 34], it would be particularly important to gain insights into the molecular determinants of such interaction in human beings.
Our data show that human PLT adhesion to HIMEC mainly occurs when PLT, HIMEC or both are activated, and that PLT-HIMEC interactions are mediated by ICAM-1, aVβ3 integrin, and FKN as suggested by antibody blockade experiments. Not unexpectedly, TNF-α, a potent pro-inflammatory stimulus, induced adhesion molecule expression on HIMEC, leading to the promotion of PLT adhesion. FKN and aVβ3 integrin manifested the highest degree of induction in response to TNF-α stimulation, suggesting that these CAM may be exquisitely sensitive to a pro-inflammatory milieu. Remarkably, FKN has been shown to mediate the adhesion of FKN receptor-expressing T cells to HIMEC, pointing to this molecule as a determinant of HIMEC interaction with both immune and non-immune cells [35, 36].
In addition to adhering to the activated endothelium, PLT are known to secrete a wide array of pro-inflammatory mediators [28]. In the present study, we focused mainly on angiogenic molecules such as VEGF-A and sCD40L. As shown by the co-culture experiments, PLT were the main source of both VEGF-A and sCD40L, and PLT secretion was higher when PLT were activated with thrombin, or maintained in co-culture with TNF-α-treated HIMEC, a finding compatible with higher PLT adhesion and with enhanced PLT activation. Notably, PLT from patients with active IBD were particularly prone to release pro-angiogenic VEGF-A and sCD40L, either spontaneously or in response to thrombin, when compared with PLT from healthy volunteers. In addition, VEGF-A and sCD40L production by PLT from patients with IBD were maximal upon co-culture with TNF-α-activated HIMEC. Indeed, these in vitro experiments might recapitulate an in vivo scenario, where PLT circulate in the inflamed IBD microvasculature, encounter an activated endothelium, adhere and get activated themselves, contributing to foster intestinal inflammation [28]. When translating our findings to an in vivo context, it is conceivable that neither PLT adhesion to EC nor PLT release of pro-angiogenic mediators perturb intestinal homeostasis under physiological conditions. This is backed by our observation that both the number of resting PLT adhering to HIMEC and the release of VEGF and sCD40L by resting PLT are significantly lower compared with the data recorded with thrombin-activated PLT, suggesting that in vivo PLT activation occurring in patients with IBD is required to trigger PLT recruitment and adhesion [9], and to foster the release of pro-angiogenic cytokines upon interaction with the inflamed EC. Unequivocal evidence is now available in favour of PLT activation in patients with IBD, including an increased tendency to form PLT-leucocyte aggregates [37], an increased PLT aggregation response to epinephrine, collagen and ADP [14], and the ability of activated PLT to induce the formation of reactive oxygen species by polymorphonuclear leucocytes [38]. It is thus tempting to speculate that PLT activation status will dictate the outcome of PLT–EC interaction in vivo.
In addition, we explored the role of PLT as a novel cell type involved in mediating angiogenesis. EC tube formation on basement membranes replicates many of the steps in angiogenesis, encompassing adhesion, migration, protease activity, alignment and tube formation [39]. In our tubule formation assays with a solubilized extracellular membrane matrix, the presence of thrombin-activated PLT prompted robust angiogenesis, suggesting a potential active role of PLT in mediating inflammation-induced angiogenesis. We have recently shown that CD40L and VEGF-A are crucial molecules that act at the cross-road between angiogenesis and inflammation [7]. In support of this, preclinical models of experimental IBD have shown that blockade of either CD40L or VEGF-A may induce significant amelioration of experimental colitis, associated with dramatic inhibition of inflammation-induced angiogenesis. Therefore, it is tempting to speculate that such therapeutic effect could also result from the inhibition of PLT-derived pro-angiogenic and pro-inflammatory sCD40L and VEGF-A.
Collectively, our study indicates that PLT may serve as partners for the promotion of angiogenesis and sheds some light into the molecular determinants that drive inflammation-induced angiogenesis. Whether PLT may be successfully targeted to limit pro-angiogenic factor release and dampen inflammation in clinical IBD will have to be tested by future clinical trials.
Acknowledgments
This study was supported by grants from the Broad Medical Research Program, the Italian Ministery of Health (Ricerca Finalizzata 2006, n.72 and Bando Giovani Ricercatori), Fondazione Cariplo, and Italian Association for Cancer Research (My first AIRC Grant) to S.D.
Conflict of interest
The authors declare no competing financial interests.
References
- 1.Folkman J. Seminars in Medicine of the Beth Israel Hospital, Boston. Clinical applications of research on angiogenesis. N Engl J Med. 1995;333:1757–63. doi: 10.1056/NEJM199512283332608. [DOI] [PubMed] [Google Scholar]
- 2.Leung DW, Cachianes G, Kuang WJ, et al. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–9. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- 3.Mazzucchelli L, Hauser C, Zgraggen K, et al. Expression of interleukin-8 gene in inflammatory bowel disease is related to the histological grade of active inflammation. Am J Pathol. 1994;144:997–1007. [PMC free article] [PubMed] [Google Scholar]
- 4.Lugering N, Kucharzik T, Gockel H, et al. Human intestinal epithelial cells down-regulate IL-8 expression in human intestinal microvascular endothelial cells; role of transforming growth factor-β 1 (TGF-β1) Clin Exp Immunol. 1998;114:377–84. doi: 10.1046/j.1365-2249.1998.00718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida S, Ono M, Shono T, et al. Involvement of interleukin-8, vascular endothelial growth factor, and basic fibroblast growth factor in tumor necrosis factor α-dependent angiogenesis. Mol Cell Biol. 1997;17:4015–23. doi: 10.1128/mcb.17.7.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Danese S, Sans M, de la Motte C, et al. Angiogenesis as a novel component of inflammatory bowel disease pathogenesis. Gastroenterology. 2006;130:2060–73. doi: 10.1053/j.gastro.2006.03.054. [DOI] [PubMed] [Google Scholar]
- 7.Scaldaferri F, Vetrano S, Sans M, et al. VEGF-A links angiogenesis and inflammation in inflammatory bowel disease pathogenesis. Gastroenterology. 2009;136:585–95. doi: 10.1053/j.gastro.2008.09.064. [DOI] [PubMed] [Google Scholar]
- 8.Danese S, Scaldaferri F, Vetrano S, et al. Critical role of the CD40 CD40-ligand pathway in regulating mucosal inflammation-driven angiogenesis in inflammatory bowel disease. Gut. 2007;56:1248–56. doi: 10.1136/gut.2006.111989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vowinkel T, Anthoni C, Wood KC, et al. CD40-CD40 ligand mediates the recruitment of leukocytes and platelets in the inflamed murine colon. Gastroenterology. 2007;132:955–65. doi: 10.1053/j.gastro.2006.12.027. [DOI] [PubMed] [Google Scholar]
- 10.Danese S, Katz JA, Saibeni S, et al. Activated platelets are the source of elevated levels of soluble CD40 ligand in the circulation of inflammatory bowel disease patients. Gut. 2003;52:1435–41. doi: 10.1136/gut.52.10.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melter M, Reinders ME, Sho M, et al. Ligation of CD40 induces the expression of vascular endothelial growth factor by endothelial cells and monocytes and promotes angiogenesis in vivo. Blood. 2000;96:3801–8. [PubMed] [Google Scholar]
- 12.Cho CS, Cho ML, Min SY, et al. CD40 engagement on synovial fibroblast up-regulates production of vascular endothelial growth factor. J Immunol. 2000;164:5055–61. doi: 10.4049/jimmunol.164.10.5055. [DOI] [PubMed] [Google Scholar]
- 13.Danese S, de la Motte C, Sturm A, et al. Platelets trigger a CD40-dependent inflammatory response in the microvasculature of inflammatory bowel disease patients. Gastroenterology. 2003;124:1249–64. doi: 10.1016/s0016-5085(03)00289-0. [DOI] [PubMed] [Google Scholar]
- 14.Andoh A, Yoshida T, Yagi Y, et al. Increased aggregation response of platelets in patients with inflammatory bowel disease. J Gastroenterol. 2006;41:47–54. doi: 10.1007/s00535-005-1721-x. [DOI] [PubMed] [Google Scholar]
- 15.Fagerstam JP, Whiss PA, Strom M, et al. Expression of platelet P-selectin and detection of soluble P-selectin, NPY and RANTES in patients with inflammatory bowel disease. Inflamm Res. 2000;49:466–72. doi: 10.1007/s000110050618. [DOI] [PubMed] [Google Scholar]
- 16.Kapsoritakis AN, Potamianos SP, Sfiridaki AI, et al. Elevated thrombopoietin serum levels in patients with inflammatory bowel disease. Am J Gastroenterol. 2000;95:3478–81. doi: 10.1111/j.1572-0241.2000.03364.x. [DOI] [PubMed] [Google Scholar]
- 17.Heits F, Stahl M, Ludwig D, et al. Elevated serum thrombopoietin and interleukin-6 concentrations in thrombocytosis associated with inflammatory bowel disease. J Interferon Cytokine Res. 1999;19:757–60. doi: 10.1089/107999099313604. [DOI] [PubMed] [Google Scholar]
- 18.Irving PM, Macey MG, Shah U, et al. Formation of platelet-leukocyte aggregates in inflammatory bowel disease. Inflamm Bowel Dis. 2004;10:361–72. doi: 10.1097/00054725-200407000-00007. [DOI] [PubMed] [Google Scholar]
- 19.Pamuk GE, Vural O, Turgut B, et al. Increased circulating platelet-neutrophil, platelet-monocyte complexes, and platelet activation in patients with ulcerative colitis: a comparative study. Am J Hematol. 2006;81:753–9. doi: 10.1002/ajh.20655. [DOI] [PubMed] [Google Scholar]
- 20.Russo S, Bussolati B, Deambrosis I, et al. Platelet-activating factor mediates CD40-dependent angiogenesis and endothelial-smooth muscle cell interaction. J Immunol. 2003;171:5489–97. doi: 10.4049/jimmunol.171.10.5489. [DOI] [PubMed] [Google Scholar]
- 21.Macdonald TT, Monteleone G. Immunity, inflammation, and allergy in the gut. Science. 2005;307:1920–5. doi: 10.1126/science.1106442. [DOI] [PubMed] [Google Scholar]
- 22.Danese S, de la Motte C, Reyes BM, et al. Cutting edge: t cells trigger CD40-dependent platelet activation and granular RANTES release: a novel pathway for immune response amplification. J Immunol. 2004;172:2011–5. doi: 10.4049/jimmunol.172.4.2011. [DOI] [PubMed] [Google Scholar]
- 23.Vogel JD, West GA, Danese S, et al. CD40-mediated immune-nonimmune cell interactions induce mucosal fibroblast chemokines leading to T-cell transmigration. Gastroenterology. 2004;126:63–80. doi: 10.1053/j.gastro.2003.10.046. [DOI] [PubMed] [Google Scholar]
- 24.Binion DG, West GA, Ina K, et al. Enhanced leukocyte binding by intestinal microvascular endothelial cells in inflammatory bowel disease. Gastroenterology. 1997;112:1895–907. doi: 10.1053/gast.1997.v112.pm9178682. [DOI] [PubMed] [Google Scholar]
- 25.De Candia E, Hall SW, Rutella S, et al. Binding of thrombin to glycoprotein Ib accelerates the hydrolysis of Par-1 on intact platelets. J Biol Chem. 2001;276:4692–8. doi: 10.1074/jbc.M008160200. [DOI] [PubMed] [Google Scholar]
- 26.Heidemann J, Ogawa H, Dwinell MB, et al. Angiogenic effects of interleukin 8 (CXCL8) in human intestinal microvascular endothelial cells are mediated by CXCR2. J Biol Chem. 2003;278:8508–15. doi: 10.1074/jbc.M208231200. [DOI] [PubMed] [Google Scholar]
- 27.Danese S. Nonimmune cells in inflammatory bowel disease: from victim to villain. Trends Immunol. 2008;29:555–64. doi: 10.1016/j.it.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 28.Danese S, de la Motte C, Fiocchi C. Platelets in inflammatory bowel disease: clinical, pathogenic, and therapeutic implications. Am J Gastroenterol. 2004;99:938–45. doi: 10.1111/j.1572-0241.2004.04129.x. [DOI] [PubMed] [Google Scholar]
- 29.Ziche M, Morbidelli L, Choudhuri R, et al. Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not basic fibroblast growth factor-induced angiogenesis. J Clin Invest. 1997;99:2625–34. doi: 10.1172/JCI119451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Solanilla A, Villeneuve J, Auguste P, et al. The transport of high amounts of vascular endothelial growth factor by blood platelets underlines their potential contribution in systemic sclerosis angiogenesis. Rheumatology. 2009;48:1036–44. doi: 10.1093/rheumatology/kep154. [DOI] [PubMed] [Google Scholar]
- 31.Mach F, Schonbeck U, Sukhova GK, et al. Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for CD40-CD40 ligand signaling in atherosclerosis. Proc Natl Acad Sci USA. 1997;94:1931–6. doi: 10.1073/pnas.94.5.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshida H, Granger DN. Inflammatory bowel disease: a paradigm for the link between coagulation and inflammation. Inflamm Bowel Dis. 2009;15:1245–55. doi: 10.1002/ibd.20896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Danese S, Papa A, Saibeni S, et al. Inflammation and coagulation in inflammatory bowel disease: the clot thickens. Am J Gastroenterol. 2007;102:174–86. doi: 10.1111/j.1572-0241.2006.00943.x. [DOI] [PubMed] [Google Scholar]
- 34.Yoshida H, Russell J, Stokes KY, et al. Role of the protein C pathway in the extraintestinal thrombosis associated with murine colitis. Gastroenterology. 2008;135:882–8. doi: 10.1053/j.gastro.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scaldaferri F, Sans M, Vetrano S, et al. The role of MAPK in governing lymphocyte adhesion to and migration across the microvasculature in inflammatory bowel disease. Eur J Immunol. 2009;39:290–300. doi: 10.1002/eji.200838316. [DOI] [PubMed] [Google Scholar]
- 36.Kobayashi T, Okamoto S, Iwakami Y, et al. Exclusive increase of CX3CR1+CD28−CD4+ T cells in inflammatory bowel disease and their recruitment as intraepithelial lymphocytes. Inflamm Bowel Dis. 2007;13:837–46. doi: 10.1002/ibd.20113. [DOI] [PubMed] [Google Scholar]
- 37.Kayo S, Ikura Y, Suekane T, et al. Close association between activated platelets and neutrophils in the active phase of ulcerative colitis in humans. Inflamm Bowel Dis. 2006;12:727–35. doi: 10.1097/00054725-200608000-00009. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki K, Sugimura K, Hasegawa K, et al. Activated platelets in ulcerative colitis enhance the production of reactive oxygen species by polymorphonuclear leukocytes. Scand J Gastroenterol. 2001;36:1301–6. doi: 10.1080/003655201317097164. [DOI] [PubMed] [Google Scholar]
- 39.Arnaoutova I, George J, Kleinman HK, et al. The endothelial cell tube formation assay on basement membrane turns 20: state of the science and the art. Angiogenesis. 2009;12:267–74. doi: 10.1007/s10456-009-9146-4. [DOI] [PubMed] [Google Scholar]