

Abstract
Macrophage migration inhibitory factor (MIF) is a pleiotropic inflammatory cytokine that was recently identified as a non-cognate ligand of the CXC-family chemokine receptors 2 and 4 (CXCR2 and CXCR4). MIF is expressed and secreted from endothelial cells (ECs) following atherogenic stimulation, exhibits chemokine-like properties and promotes the recruitment of leucocytes to atherogenic endothelium. CXCR4 expressed on endothelial progenitor cells (EPCs) and EC-derived CXCL12, the cognate ligand of CXCR4, have been demonstrated to be critical when EPCs are recruited to ischemic tissues. Here we studied whether hypoxic stimulation triggers MIF secretion from ECs and whether the MIF/CXCR4 axis contributes to EPC recruitment. Exposure of human umbilical vein endothelial cells (HUVECs) and human aortic endothelial cells (HAoECs) to 1% hypoxia led to the specific release of substantial amounts of MIF. Hypoxia-induced MIF release followed a biphasic behaviour. MIF secretion in the first phase peaked at 60 min. and was inhibited by glyburide, indicating that this MIF pool was secreted by a non-classical mechanism and originated from pre-formed MIF stores. Early hypoxia-triggered MIF secretion was not inhibited by cycloheximide and echinomycin, inhibitors of general and hypoxia-inducible factor (HIF)-1α-induced protein synthesis, respectively. A second phase of MIF secretion peaked around 8 hrs and was likely due to HIF-1α-induced de novo synthesis of MIF. To functionally investigate the role of hypoxia-inducible secreted MIF on the recruitment of EPCs, we subjected human AcLDL+ KDR+ CD31+ EPCs to a chemotactic MIF gradient. MIF potently promoted EPC chemotaxis in a dose-dependent bell-shaped manner (peak: 10 ng/ml MIF). Importantly, EPC migration was induced by supernatants of hypoxia-conditioned HUVECs, an effect that was completely abrogated by anti-MIF- or anti-CXCR4-antibodies. Thus, hypoxia-induced MIF secretion from ECs might play an important role in the recruitment and migration of EPCs to hypoxic tissues such as after ischemia-induced myocardial damage.
Keywords: endothelial cell (EC), endothelial progenitor cells (EPCs), macrophage migration inhibitory factor (MIF), stromal cell-derived factor-1α (SDF-1α/CXCL12)
Introduction
Macrophage migration inhibitory factor (MIF) is a pleiotropic inflammatory cytokine that serves as an upstream regulator of innate and adaptive immune responses. When dysregulated, MIF plays a pivotal role in a broad spectrum of inflammatory and immune diseases such as septic shock, rheumatoid arthritis, glomerulonephritis, inflammatory bowel disease and atherosclerosis [1–4].
MIF is widely expressed in various tissues, an observation that is in line with its additional intracellular function in cell homeostasis and cell cycle regulation. Yet, secretion of MIF from T lymphocytes and monocytes/macrophages is specifically triggered by inflammatory stimuli such as endotoxin (LPS) or tumour necrosis factor-α (TNF-α). Endothelial cells (ECs) secrete MIF upon atherogenic stimulation by oxidized low-density lipoprotein [2, 4]. Of note, MIF was recently demonstrated to exhibit chemokine-like functions and to play a prominent role in inflammatory and atherogenic leucocyte recruitment [5]. At the molecular level, this activity is based on a high affinity non-cognate interaction between MIF and the CXC chemokine receptors CXCR2 and CXCR4 [5, 6]. CXCR4 has previously been known as the receptor for stromal cell-derived factor-1α (SDF-1α)/CXCL12. The CXCL12/CXCR4 axis is critically involved in various homeostatic and inflammatory cell migration processes, including inflammatory and atherogenic T-cell recruitment, stem cell homing and cancer cell metastasis [7–9]. Stem cell recruitment by CXCL12/CXCR4 includes the recruitment of circulating endothelial progenitor cells (EPCs) into ischemic tissues [10, 11].
EPCs have been shown to amply contribute to the revascularization of ischemic areas. While the underlying molecular pathways are only partly understood, the discovery of EPCs has substantively altered our view of adult tissue angiogenesis, indicating that circulating bone marrow-derived cells can contribute to new blood vessel formation. This has opened up potential new avenues to repair damaged ischemic tissues by using EPCs to improve neo-angiogenesis or re-endothelialization [12–17]. However, clinical studies applying EPCs in myocardial infarction (MI) have only been partially encouraging [18]. Moreover, the identity of EPCs as characterized by their surface markers has been a matter of debate [19, 20]. The underlying molecular pathways of the angiogenic and vasculogenic functions of EPCs are only partly understood and are likely to be diverse. There is evidence for two principle mechanisms: (i) EPCs recruited to ischemic tissues differentiate into ECs to participate in re-endothelialization and re-vascularization; (ii) EPCs promote angiogenesis and vasculogenesis by virtue of the release of paracrine angiogenic factors which they carry with them as cargo [13, 16]. For example, mouse embryonic EPCs (eEPCs) have been demonstrated to express and release a number of factors that could be correlated with eEPC-mediated beneficial angiogenic effects, i.e. inducing blood vessel growth and cardioprotection in severe ischemic conditions [21]. In addition to various growth factors and prominent angiogenic factors such as vascular endothelial growth factor (VEGF), EPCs also strongly express MIF, suggesting that MIF may contribute to the angiogenic potential of these cells [21].
The CXCL12/CXCR4 chemokine/chemokine receptor axis has been proposed to play a pivotal role in the recruitment of EPCs into ischemic tissues. CXCL12 gene expression is regulated by the transcription factor hypoxia-inducible factor-1 (HIF-1) in ECs, resulting in expression and secretion of CXCL12 in ischemic tissue in direct proportion to reduced oxygen tension. In turn, HIF-1-induced CXCL12 secretion increases the adhesion, migration and homing of circulating CXCR4-positive progenitor cells to ischemic tissue, whereas blockade of CXCL12 in ischemic tissue or CXCR4 on circulating cells prevents EPC recruitment to such sites of injury [10, 22].
The expression of MIF is also subject to induction by HIF-1α[23] and in line with the lack of an N-terminal signal sequence, the secretion of MIF follows a non-classical, ER-Golgi-independent pathway [24, 25]. MIF secretion resembles that of other leaderless mediators such as IL-1β, FGF2 or HMGB1 [26] and occurs from pre-formed intracellular stores. Thus, secretion of MIF encompasses a rapid early-phase (secretion from pre-formed stocks) and a late-phase (involves de novo synthesis of MIF protein).
Here, we have studied the hypoxia-induced secretion of MIF from human umbilical vascular endothelial (HUVECs) and human heart aortic endothelial (HAoECs) cells. Release of MIF following stimulation with 1% hypoxia was compared with that of normoxic cells by MIF ELISA from conditioned cell supernatants. The phases, kinetics and mechanism of secretion were probed by analysing various time intervals and treatment with secretion, protein biosynthesis and HIF-1α inhibitors. Finally, a potential role of MIF in the hypoxic recruitment of EPCs was investigated by exposing EPCs to chemotactic gradients of recombinant human MIF, CXCL12 or hypoxia-conditioned culture supernatants of HUVECs in combination with blocking monoclonal antibodies against MIF and CXCR4.
Methods
Endothelial cells and cell culture
Human umbilical vein endothelial cells (HUVECs) were isolated from human umbilical cord veins obtained from the Department of Gynaecology and Obstetrics at the RWTH Aachen University Hospital according to the protocol of Jaffe et al.[27] and was approved by informed consent of the patients of the Department of Gynaecology and Obstetrics. Umbilical cord sampling was according to local ethics regulations. A sterile technique was used in all manipulations of the cord. The cord was separated from the placenta soon after birth, placed in a sterile container filled with 15 mM HEPES buffer, pH 7.4 (Sigma, Taufkirchen, Germany).
Generally, cell isolation started immediately after cord sampling, in some instances the cord was stored at 4°C for up to 1 hr. The cord was cleaned and all areas with clamp marks were cut off with a scalpel (Feather, Osaka, Japan). The umbilical vein was cannulated on both ends with olive canules, and the canules were secured. The vein was perfused with 50 ml of PBS to remove blood clots. Ten ml of 0.1% collagenase (Worthington, Lakewood, NJ, USA) in PBS were infused and the vein closed on both ends. The cord was placed in a water bath and incubated at 37°C for 20 min. One end was opened and the collagenase solution, containing the ECs was collected. The cord was flushed with 20 ml of endothelial growth medium (PromoCell MV2, Heidelberg, Germany), cell-containing solutions pooled and HUVECs centrifuged at 300 ×g for 5 min., resuspended in 10 ml of fresh medium and incubated at 37°C. HUVECs were plated, cultured for one week and their identity verified by morphologic and immunologic criteria. Passages 2–5 were used for the experiments.
HAoECs were purchased from Promocell and were cultured in EGM MV1Media (PromoCell).
Isolation and characterization of endothelial progenitor cells
EPCs were isolated from the mononuclear cell fraction obtained by density gradient centrifugation from human blood as previously described [28]. ‘Buffy coats’ were obtained from healthy volunteers after informed consent in accordance with the local ethics committee. Mononuclear cells were separated by Biocoll density gradient centrifugation (Biochrom, Berlin, Germany) and CD34+ cells were enriched to >90% by magnetic separation applying a human CD34 selection kit (StemCell Technologies, Cologne, Germany) in accordance with the manufacturer’s protocol. CD34+ cells were plated on fibronectin (Biochrom)-coated 6-well plates and cultured in MV2 endothelial growth medium (PromoCell). Media were changed on day 4 and cells were harvested on day 7. EPCs were characterized by FACS Analysis (FACS Canto, Becton Dickinson, Heidelberg, Germany). Preparations of cells that co-stained for lectin-FITC (Sigma) and DiI-conjugated acLDL (Cell Systems, St. Katharinen, Germany), and co-expressed CD31 (Chemicon Europe, Hofheim, Germany; anti human CD31, CBL468F/anti-PECAM-1, clone HC1/6, FITC-conjugated) and VEGFR-2 (KDR, anti-VEGFR-2 mAb, Clone KDR-1, Sigma V9134) at a rate of >90% were considered as EPCs and were used for the experiments.
Hypoxic cell treatment
Hypoxic conditions (1% O2) were achieved in a hypoxic incubator (In vivo 200, Ruskinn) by continuous infusion of a pre-analysed gas mixture (94% N2, 5% CO2, 1% O2). Serum-reduced endothelial growth medium, containing 0.5% foetal calf serum, was used in all experiments for the cells that were subjected to hypoxic conditions. Media were rendered hypoxic by equilibration in the hypoxic incubator for at least 14 hrs prior to the experiment.
MIF and CXCL12 release experiments
MIF and CXCL12 secretion experiments were performed as follows: HUVECs (15,000 cells per well) were added to 24-well plates and allowed to adhere overnight in an incubator under normoxic conditions. For hypoxic experiments, plates were transferred into the hypoxic incubator. At t= 0 hr, medium was replaced with pre-conditioned hypoxic medium (1% O2, 5% CO2 and 37°C and cells incubated for 0, 1, 2, 8, 24 hrs or 0, 20, 40, 60, 80, 100 and 120 min. as indicated under Results. For normoxic control experiments, cells were treated identically except that plates remained in the regular normoxic incubator. Cell supernatants collected at indicated time intervals were frozen at –80°C and used to determine MIF and CXCL12 concentrations by ELISA technique. In some experiments, certain inhibitors were added to the media. Inhibitors were added 1 hr prior to hypoxia treatment. Glyburide (inhibitor of the non-classical ABC-A1 transporter-mediated secretion pathway) was added at a concentration of 5 μM, cycloheximide (inhibitor of protein biosynthesis) was added at 2 μg/mL and echinomycin (inhibitor of HIF-1 transcriptional activity) was used at 10 nM.
Enzyme-linked immunosorbent assays (ELISAs) for MIF and CXCL12
The human MIF ELISA was performed essentially as described previously [24] applying capture antibody MAB289 and detection antibody BAF289 from R&D Systems (Wiesbaden, Germany). Concentrations of CXCL12 (SDF-1α) were determined by a human CXCL12 Quantikine kit (R&D Systems) according to the manufacturer’s protocol.
Cell integrity and cell viability assays
Cell integrity was verified by determining lactate dehydrogenase (LDH) activities in the supernatants of the examined cells. Specimens were sampled before and after hypoxia. LDH activities were determined in the Laboratory of Clinical Chemistry of the RWTH Aachen University Hospital by routine clinical chemistry procedures. Cell viability was confirmed by Trypan blue staining 1 hr and 24 hrs after normoxia versus hypoxia and was expressed in percentage of viable cells.
EPC chemotaxis measurements
Chemotactic assays were performed using Transwell cell migration chambers in combination with FluoroBlok inserts (Becton Dickinson; 8 μm pore size) in a 24-well plate format. Lower chambers contained increasing concentrations of recombinant human MIF (0, 5, 10, 50, 100 and 200 ng/ml) or recombinant human CXCL12 (200 ng/ml) in MV2 basal endothelial growth medium containing 0.5% BSA. Chemotaxis towards CXCL12 was examined for comparison. Calcein-stained (Calbiochem/Merck, Darmstadt, Germany) EPCs in MV2 basal endothelial growth medium containing 0.5% BSA were placed into upper chambers (50,000 cells per insert). Fluorescence signals representing the migration of calcein-stained cells into the bottom chambers were determined 3 hrs later using a fluorescent microplate reader at 485/535 nm (Victor, Wallac/Perkin Elmer, Waltham, MA, USA). The chemotactic indices were obtained by dividing the fluorescence observed in the lower migration chambers loaded with various chemokine concentrations by the fluorescence of the bottom chamber containing the control medium without chemokine.
Chemotaxis of EPCs was also studied using hypoxically versus normoxically conditioned HUVEC media obtained 1 hr after hypoxic or normoxic treatment, respectively. Conditioned media were added to the bottom chamber as chemotactic stimulus. For blockade experiments, neutralizing anti-MIF mAbs (NIH/III.D9; described previously in [5]) or anti-CXCR4 mAbs (MAB171, R&D Systems) were added as indicated. The percentage of cells migrated to the lower chamber was determined after 3 hrs.
Statistics
Statistical significance of data sets was evaluated by Student’s t-test. P-values < 0.05 were considered to be statistically significant. Asterisks represent different levels of statistical power: *, P < 0.05; **, P < 0.01 and ***, P < 0.005.
Results
Hypoxia-induced secretion of MIF by HUVECs
We initially investigated whether HUVECs release MIF when exposed to hypoxia. HUVECs were exposed to 1% hypoxia and cell supernatants analysed for MIF by ELISA at various time intervals after hypoxic exposure. Figure 1A shows that HUVECs exposed to hypoxia released substantial amounts of MIF over time. Hypoxic MIF release was significantly higher than that observed under normoxic conditions at all time-points analysed (0 min. to 24 hrs). MIF release reached levels of up to 30 ng/ml and was thus comparable to secreted MIF levels observed previously following cell stimulation with endotoxin or inflammatory cytokines [29, 30]. It should be noted that the time-point t= 0 hr, due to handling delays and cell stress related to hypoxic medium change, was not a true 0 min. time-point but represented an interval of 2–3 min., indicating that hypoxia and/or handling stress triggered the release of MIF very rapidly. Hypoxia-stimulated MIF release was specific as 1% hypoxia did not cause any appreciable cell damage. Table 1 shows that hypoxic treatment did not lead to the release of cytoplasmic LDH nor was any measurable Trypan blue staining of HUVECs detected. We next calculated relative cumulative release rates at the time intervals analysed (Fig. 1B). It became apparent that hypoxia-induced MIF secretion from HUVECs occurred in two phases. Substantial amounts of MIF were secreted by HUVECs within the first hour of hypoxic treatment, likely reflecting MIF secretion from pre-formed stores. Secretion then dropped as indicated by the low MIF pool secreted between 1 and 8 hrs. Between 8 and 24 hrs, MIF secretion then increased again, with appreciable amounts released within this time period. Late secretion of MIF likely derived from renewed MIF pools generated following induction of de novo protein synthesis of MIF. The observed marked secretion of MIF in the early post-hypoxic phase prompted us to investigate this secretion phase in more detail. Analysis of short time intervals between 0 and 120 min. revealed that early hypoxia-induced MIF secretion already peaked 60 min. after exposure to hypoxia and declined thereafter towards 120 min. (Fig. 1C). The observed decrease between 60 and 120 min. did not reach statistical significance, but it may be speculated that it is due to MIF degradation or re-internalization. While CXCL12 is known to be secreted from ECs following hypoxic challenge within 24 hrs [10], we were unable to detect any CXCL12 in our HUVEC supernatants 1 hr after treatment with 1% hypoxia (data not shown).
Fig 1.
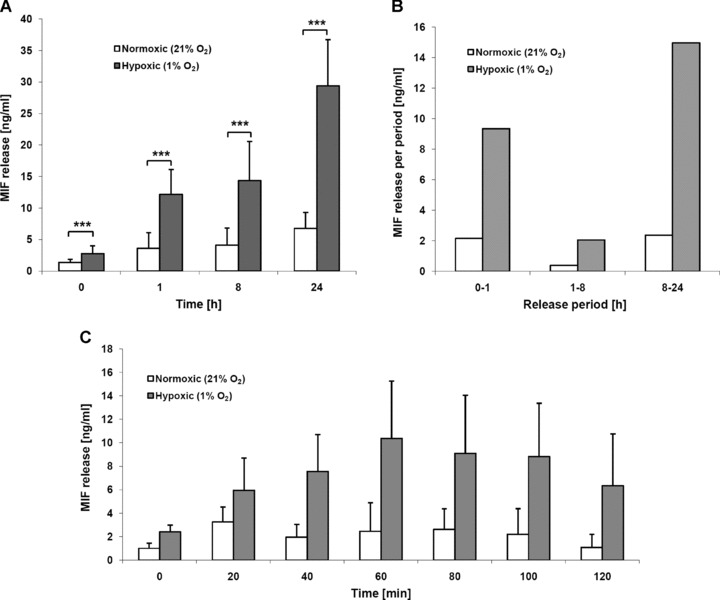
Biphasic hypoxia-induced MIF secretion from ECs. (A) Cumulative MIF release from HUVEC cells under hypoxic and normoxic conditions between 0 and 24 hrs. (B) Period-specific MIF release under hypoxic and normoxic conditions (data deduced from Fig. 1A). (C) Short-term cumulative MIF release between 0 and 120 min. 15,000 cells were incubated overnight in 24-well plates in a normoxic incubator, one plate for each time-point. Half of the plates were transferred into a hypoxic incubator, the other half remained in the normoxic incubator. At t= 0, media were replaced by media that were conditioned under either normoxic or 1% hypoxia conditions (>14 hrs). Supernatants were collected at 0, 1, 8 and 24 hrs or 0, 20, 40, 60, 80, 100 and 120 min. after medium change and MIF concentrations in the cell supernatants were determined by ELISA. Bars represent mean values, error bars refer to the corresponding standard deviations (n= 9). ***, indicates statistical significance with a power of P < 0.005.
Table 1.
Cell integrity and viability
| Sample | LDH | Trypan blue | ||
|---|---|---|---|---|
| Activity [units/ml] | Vitality [%] | |||
| Medium | 1 | |||
| 1% O2 1 hr | <1 | 91 | ||
| 1% O2 24 hrs | <1 | 91 | ||
| 21% O2 1 hr | 1 | 94 | ||
| 21% O2 24 hrs | <1 | 92 | ||
| 1% O2+ echinomycin 8 hrs | 2 | n.d. | ||
Hypoxia-induced secretion of MIF by HAoECs
EC-derived MIF has been demonstrated to play an important role in early atherogenic processes [5, 31–33]. Thus, we next asked whether hypoxia-triggered MIF release may also occur from HAoECs, a process that may be of relevance in myocardial ischemia and infarction. In fact, a significant increase in MIF secretion was noted in these cells within the first 60 min. following exposure to 1% hypoxia and was similar in extent to that measured in the HUVEC supernatants (Fig. 2). However, unlike the MIF release seen in HUVECs, MIF levels secreted from hypoxia-treated HAoECs peaked at 8 hrs and declined thereafter, suggesting that the second phase of MIF release, i.e. the one representing de novo MIF protein synthesis, followed different kinetics in these cells.
Fig 2.
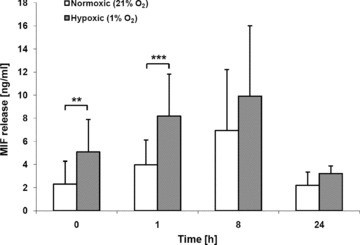
Hypoxia-induced MIF secretion from HAoECs. Experiments were performed essentially as described in the legend to Figure 1. Bars represent mean values (n= 3) ± S.D. Asterisks, * and ***, indicate statistical significance with P < 0.05 and P < 0.005, respectively.
Blockade of non-classical protein secretion pathways but not protein synthesis or HIF-1 inhibits hypoxia-triggered rapid secretion of MIF
Secretion of MIF follows an ER/Golgi-independent non-classical pathway [24, 25]. MIF release by monocytes is not inhibited by monensin or brefeldin A, but can be strongly blocked by glyburide, a pharmacological inhibitor of ABC transporter proteins [24]. We tested whether glyburide would interfere with hypoxia-triggered MIF secretion in HUVECs as well. Hypoxia-stimulated MIF release by HUVECs following pre-treatment of cells with glyburide was compared with that of cells treated with solvent control solution. Figure 3 demonstrates that early-phase hypoxic MIF release (t= 1 hr) was markedly and significantly blocked by glyburide. In contrast, glyburide exhibited no inhibitory effect on late-phase MIF secretion (t= 24 hrs), in line with the notion that inhibitors of non-classical secretion pathways predominantly target rapid protein secretion processes from preformed stores. MIF secretion within an initial 8-hour time interval at first sight seemed to be affected by glyburide (Fig. 3A), but period-specific analysis (Fig. 3B) revealed that this effect was due to the glyburide effect in the first hour after hypoxic stimulation.
Fig 3.
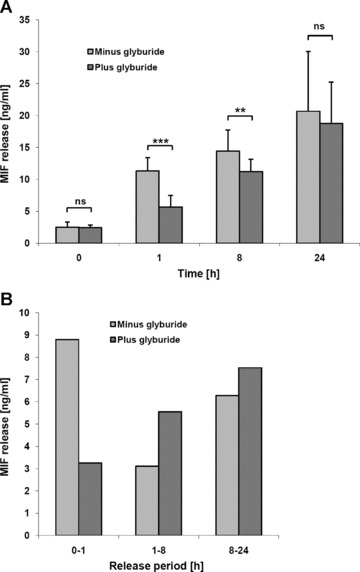
Inhibition of hypoxia-induced rapid MIF release by the ABCA1 transporter inhibitor glyburide. HUVECs (15,000 cells) were incubated overnight in 24-well plates in a normoxic incubator. At t= 0, media were replaced by hypoxic media containing 5 μM glyburide. (A) Cumulative hypoxia-induced MIF release in the presence or absence of 5 μM glyburide at the indicated time intervals as determined by ELISA. (B) Period-specific MIF release from HUVECs under hypoxic conditions in the absence versus presence of glyburide. Data were derived from the experiment of Figure 4A. Bars represent mean values (n= 6) ± S.D. Asterisks, ** and ***, indicate statistical significance with P < 0.01 and P < 0.005, respectively.
Together, these data suggested that hypoxia-induced early MIF release from HUVECs mainly occurred through specific non-classical secretion from pre-formed cytosolic MIF stores. To further confirm this notion, cells were pre-treated with cycloheximide or echinomycin, inhibitors of protein synthesis and HIF-1α-mediated transcriptional processes, respectively. In fact, neither inhibitor affected hypoxia-stimulated MIF secretion within the first hour (Fig. 4).
Fig 4.
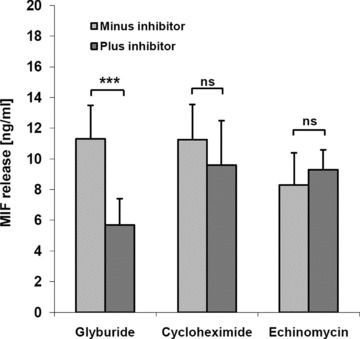
Hypoxia-induced rapid MIF release is not affected by cycloheximide or echinomycin. HUVECs (15,000 cells) were incubated overnight in 24-well plates in a normoxic incubator. At t= 0, media were replaced by hypoxic media either containing 5 μM glyburide, 2 μg/ml cycloheximide or 10 nM echinomycin. Solvent was used as a control. MIF concentrations were determined from 1 hr supernatants by ELISA. Bars represent mean values (n= 3) ± S.D. Asterisks, ***, indicate statistical significance with P < 0.005.
Enhancement of chemotactic EPC migration by recombinant MIF
The CXCL12/CXCR4 chemokine/chemokine receptor axis plays a critical role in the recruitment of EPCs into hypoxic areas. EPCs abundantly express CXCR4 and are thus amenable to CXCL12 gradients as they are generated by a hypoxically exposed endothelium [10]. Because MIF acts as a non-cognate chemokine-like ligand of CXCR4 and triggers T-cell arrest onto atherogenic endothelium [5] and because we had observed substantial MIF secretion following hypoxia, we suggested that it may share with CXCL12 important functions in EPC recruitment processes.
To address this notion, we isolated EPCs from blood of healthy human volunteers following the method described in Grieb et al.[28]. The EPC population which was harvested after a 7-day culture period on fibronectin-coated plates was characterized by the uptake of DiI-labelled acLDL and expression of CD31 and VEGFR-2 (data not shown) to be appreciably homogeneous (Fig. 5). Enrichment of EPCs was in line with previously obtained rates ranging from 28% in a day-2 culture (Fig. 5A) to >90% in the day-7 culture (Fig. 5B).
Fig 5.
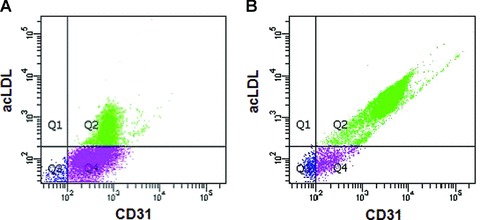
EPCs take up DiI-labelled acLDL and co-express CD31 and VEGFR2. Characterization of EPCs was performed by flow cytometric analysis. (A) Cell population obtained after 2 days on fibronectin. (B) Cell population harvested after 7 days culture on fibronectin and removal of non-adhering cells.
After verifying the identity, homogeneity and purity of our human EPC population, calcein-stained EPCs were loaded into the upper chamber of a Transwell chemotaxis device and their chemotactic migration studied. As potential chemotactic stimulus MIF or CXCL12 were added to the bottom chamber. Figure 6 shows that MIF strongly supported migration of EPCs. The observed chemotactic effect of MIF was concentration-dependent and followed the typical bell-shaped curve seen for chemokine-induced chemotactic migration processes, with a peak pro-chemotactic activity seen at 10 ng/ml of MIF. Also, MIF-mediated EPC chemotaxis was specific as it was fully blocked by a neutralizing anti-MIF antibody. For comparison, EPC chemotactic movement towards a dose of 200 ng/ml CXCL12 was analysed, a concentration at which CXCL12 has been known to exhibit optimal chemotactic responses [10]. Of note, the chemotactic index (CTX) as achieved in response to 200 ng/ml CXCL12 was smaller (CTX = 2.5) than that obtained at the peak MIF concentration of 10 ng/ml (CTX > 3).
Fig 6.
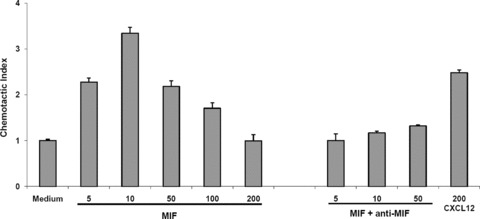
MIF specifically induces EPC chemotaxis. Dose-dependent enhancement of EPC chemotaxis by recombinant human MIF and blockade by neutralizing anti-MIF mAbs. Chemotaxis was evaluated in Transwell chambers. The upper chamber contained 50,000 calcein-stained EPCs. The lower chamber contained varying concentrations of recombinant human MIF (ng/ml) or recombinant human CXCL12 (ng/ml). MIF and anti-MIF antibodies or recombinant human CXCL12 were added as indicated. Bars represent mean values ± S.D. (n= 5).
MIF/CXCR4-mediated chemotaxis of EPCs following hypoxia-induced secretion of MIF from endothelial cells
We next sought to address the question whether MIF secreted from ECs upon hypoxic challenge would also act to chemotactically attract EPCs. Hypoxia-conditioned (1 hr hypoxic treatment at 1% O2) HUVEC supernatants were collected and transferred to the lower chamber of a Transwell device and chemotactic migration of calcein-stained EPCs evaluated. For comparison, normoxia-conditioned (1 hr normoxic treatment at 21% O2) HUVEC supernatants were studied. Hypoxia-conditioned EC supernatants markedly and significantly promoted EPC chemotaxis when compared with normoxia-conditioned media. The chemotactic effect was fully mediated by MIF, because it could be completely blocked by a neutralizing anti-MIF mAb (Fig. 7A), suggesting that EC-derived MIF secreted upon hypoxic stimulation supports EPC recruitment.
Fig 7.
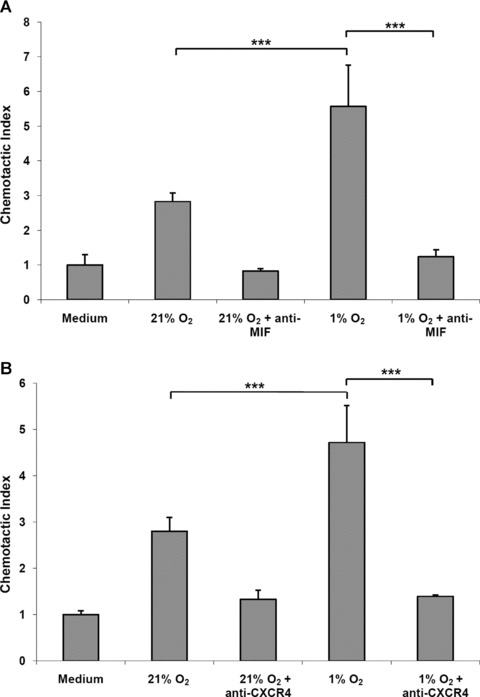
Stimulation of EPC chemotaxis by hypoxia-conditioned HUVEC media and blockade by anti-MIF and anti-CXCR4 antibodies. Hypoxia-conditioned HUVEC supernatants but not normoxia-conditioned control media promotes EPC chemotaxis and this effect is abolished by anti-MIF antibodies (A). (B) as in (A), but blockade by anti-CXCR4 antibodies. The hypoxia-conditioned HUVEC media were added to the lower chamber of a Transwell device. Bars represent mean values ± S.D. (n= 3). Asterisks, ***, indicates statistical significance with P < 0.005.
In line with this notion, the stimulatory effect of hypoxia-conditioned EC medium on EPC chemotactic migration was fully reversed by a neutralizing anti-CXCR4 antibody (Fig. 7B). As EPCs markedly express surface CXCR4 (data not shown) and because HUVECs do not release any CXCL12 within 1 hr of hypoxic stimulation, this finding indicated that EC-derived MIF promoted EPC chemotaxis through non-cognate interaction with the chemokine receptor CXCR4 expressed on EPCs.
Discussion
In the present study, we have demonstrated that hypoxia specifically triggered the secretion of the chemokine-like function chemokine MIF from ECs and that MIF, once secreted, enhanced the chemotactic migration of EPCs in a CXCR4-dependent manner. Hypoxia-induced MIF secretion by EPCs followed two timely separated phases: a rapid and a late-phase. Using pharmacological inhibitors, we determined that early-phase MIF release occurred from pre-formed intracellular stores of MIF, which is known to be released by a so-called non-conventional secretion pathway. In contrast, late-phase secretion of MIF was most likely coupled to de novo protein synthesis. Early-phase secretion of MIF from hypoxia-treated ECs strikingly differed from the secretion pattern of CXCL12, for which secretion has not been observed within the first 4 hrs after stimulation [10] and confirmed herein. Thus, CXCL12 and MIF, representing high affinity, cognate and non-cognate ligands of CXCR4, respectively, are both secreted from ECs following hypoxic challenge, but differ regarding their kinetic production profile. They might thus account for different phases of the EPC recruitment process following ischemic stress in vivo. Hypoxic MIF release from ECs not only occurred from HUVECs, but also from aortic ECs, indicating that the above mechanism may be operable in situations of arterial ischemia, such as myocardial infarction.
Hypoxia-induced expression and secretion of MIF has been observed before. Baugh and colleagues observed elevated MIF mRNA levels in HeLa cells upon hypoxic treatment, an effect that was dependent on HIF-1α[23]. Similarly, adipocytes increase their MIF mRNA and protein levels under hypoxia [34]. Moreover, MIF was found to be released from MCF-7 breast cancer cells during hypoxia; however, this occurred in a HIF-1α-independent manner, as siRNA treatment against HIF-1α and HIF-2 α did not interfere with the effect [35]. Importantly, recent reports indicate that hypoxic release of MIF may be relevant in pathogenic ischemic and atherosclerotic events in vivo[32, 36]. For example, Miller et al. demonstrated MIF induction and secretion in a murine model of myocardial infarction after ischemia/reperfusion in conjunction with a subsequent protective paracrine activity of MIF [36]. A correlation between cardiac MIF expression and local tissue hypoxia was confirmed by Jian et al. who compared cardiac MIF expression levels in infants with cyanotic cardiac defects [37], while Wang et al. found that MIF levels were increased in stroke patients and in focal ischemic rat brains [38].
Applying comparable conditions, in our study, hypoxia-induced MIF release from ECs was triggered by 1% hypoxia and was monitored over a wide time range from 0 min. to 24 hrs. Experimental hypoxia is frequently performed at hypoxic conditions between 0.5% and 2–4% O2. We elected 1% O2 hypoxia, because this oxygen level corresponded to the most ischemic tissue area in the mouse model as studied by Ceradini et al.[10]. The experiments performed in our study thus simulated situations of pathophysiological ischemia, further confirming that hypoxia-induced secretion of MIF may be of relevance in vivo. The rate of hypoxia-triggered MIF release under such conditions was compared to that seen under normoxic but otherwise identical conditions. The observed hypoxia-induced MIF release occurred in two timely separated phases: a rapid and a late release, whereas no significant increase of MIF secretion was observed under normoxic conditions. The rapid release phase showed an increase in the cumulative concentration of MIF between 0 and 1 hr followed by a subsequent stagnant phase, resulting in a drop of cumulative increase, before cumulative MIF concentrations increased again in the late-phase. This release profile was similar, yet not identical, in aortic ECs and was confirmed by a detailed kinetic analysis of hypoxic MIF secretion between 0 and 120 min., in which MIF secretion increased up to 60 min. and then began to decrease again towards 120 min. While, as discussed above and below, the rapid early-phase release apparently occurs from pre-formed MIF stores, the notable stagnation of post-hypoxic MIF secretion between 1 and 8 hrs could be due to the following effects: (i) a gap of MIF secretion between the rapid emptying of pre-formed intracellular MIF stores and their re-filling by de novo protein synthesis; (ii) an increased re-internalization rate of MIF coupled to a maintained release rate or (iii) maintained MIF secretion together with an increased degradation rate of MIF. MIF has been shown to be rapidly internalized by various cell types [39–41] and ECs express appreciable concentrations of the MIF receptor CD74, which itself undergoes rapid internalization [42]. Similarly, ECs also express some surface levels of CXCR4 and CXCL12-induced internalization and lysosomal degradation of CXCR4 has been shown to increase in lymph node metastases upon exposure to hypoxia [43], suggesting that a portion of the released MIF may be re-internalized through CXCR4. Because MIF has been shown to be relatively proteolysis resistant, we thus speculate that the gap phase between storage emptying and resumption of protein synthesis in conjunction with some re-internalization of MIF into the ECs could explain the apparent mid-phase drop in hypoxia-triggered MIF secretion.
Rapid hypoxia-induced MIF release was significantly inhibited by glyburide, a known pharmacological inhibitor of ATP-binding cassette transporter, family A, member 1 (ABCA1) transporters. Of note, glyburide has previously been shown to interfere with the non-classical secretion track of both MIF and interleukin-1β following inflammatory stimulation by endotoxin [24, 44]. This notion was confirmed by our data showing that rapid MIF release was not inhibited by the protein synthesis inhibitor cycloheximide and the HIF-1 blocker echinomycin. Thus, in line with the prior observations, we postulate that hypoxia-induced rapid MIF secretion occurs through exocytosis of pre-formed MIF from endosome-like vesicles involving the action of an ABCA1 transporter.
It is widely accepted that the increase of the hypoxia-inducible transcription factor HIF-1α level results from inhibition of its oxygen-dependent degradation by the von Hippel-Lindau protein E3 ligase pathway. At low oxygen concentrations, HIF-1α will no longer be hydroxylated by prolylhydroxylase, which will lead to the stabilization of HIF-1α and hence to its dimerization with HIF-1β resulting in the formation of the active HIF1 heterodimer, which then drives the biosynthesis of a large number of proteins. Among them are chemokines like CXCL12 and, as discussed, MIF [22, 23]. Baugh et al. propose a model according to which hypoxia-induced MIF expression is driven by HIF-1, but amplified by the hypoxia-induced degradation of CREB [23], indicating that hypoxia-triggered yet HIF-1-independent pathways of MIF induction exist. Interestingly, hypoxia-induced release of MIF by ECs as demonstrated in our current study was HIF-1-independent within the rapid phase.
We next investigated whether hypoxia-triggered MIF release from ECs might play a direct role in the recruitment of EPCs to hypoxic or ischemic tissues. Indications that supported such a scenario encompassed MIF’s recently unravelled function in atherogenic T-cell recruitment, which is mediated through non-cognate binding of CXCR4 by MIF [5], as well as the well-documented CXCR4-dependent role of CXCL12 in ischemic EPC recruitment [10, 22]. We used primary human EPCs for these experiments which we isolated by the procedure of [28] and which were reasonably pure as judged by a number of EPC-specific markers. Specifically, our EPCs were found to express CD31 and VEGFR2, and efficiently took up DiI-acLDL, indicating that we had prepared a >90% EPC population, containing only neglectible portions of monocytes and other blood-borne precursor cells. Various experimental approaches applying endotoxin-free, purified recombinant MIF and MIF-containing hypoxia-conditioned EC cell supernatants in combination with blocking antibodies against MIF and CXCR4 unanimously demonstrated that MIF at physiologically relevant concentrations of 5–50 ng/ml and subsequent to rapid secretion from hypoxia-stimulated ECs is able to potently promote the chemotactic migration of EPCs. This MIF effect followed a bell-shaped concentration curve, typically observed for chemotactic effects of chemokines, i.e. for CXCL12/CXCR4-mediated activities [43]. The observation that anti-CXCR4 mAbs completely inhibited the effect of hypoxia-conditioned EC media collected after 1 hr of hypoxia, during which no CXCL12 is secreted, indicated that MIF is the predominant EPC chemoattractant during the early post-hypoxic phase and that it acts through CXCR4. In contrast, earlier work by Gurtner and colleagues clearly suggests that it is likely that the CXCL12/CXCR4 axis is the major player governing EPC recruitment into ischemic tissues in mid-to-late post-hypoxic phases [10]. For example, these authors showed that a hypoxia-induced increase of CXCL12 in HUVEC supernatants was not observed until 6 hrs of hypoxic treatment. We also investigated the hypoxia-induced CXCL12 release by HUVECs; initial results suggested that CXCL12 release is not substantial within the first 2 hrs after hypoxia. Of note, CXCL12, in contrast to MIF, bears an N-terminal leader sequence and is released through the classical ER/Golgi-dependent secretion pathway.
In summary, we have shown that MIF is secreted from ECs upon hypoxia and can serve to promote EPC recruitment into ischemic tissue, reminiscent of the recruitment function of SDF-1α/CXCL12. Ongoing efforts aim at elucidating the role of MIF in wound healing and vascularization and may open further avenues for applying MIF-based molecular strategies to improve the regeneration of ischemic tissues.
Acknowledgments
We thank L. Rink and P. Ucziekowski (Department of Immunology, RWTH Aachen University), for making their hypoxic incubator accessible. We also thank O. Gressner, (Laboratory of Clinical Chemistry, RWTH Aachen University), for assistance with the LDH activity measurements. The excellent technical assistance of Andrea Fritz and Barbara Lippok is acknowledged. This study was supported by grants of the Deutsche Forschungsgemeinschaft, DFG, (TP1-FOR809/BE1977/4–1, J.B.; TP4-FOR809/WE1913/11–1, C.W.; TP-A7/SFB542, J.B.) and the Faculty of Medicine at RWTH Aachen University (Interdisciplinary Centre for Clinical Research, ‘IZKF-Aachen’ VV 113-f, J.B and C.W. and in part START-Program, G.G.).
References
- 1.Bernhagen J, Calandra T, Bucala R. Regulation of the immune response by macrophage migration inhibitory factor: biological and structural features. J Mol Med. 1998;76:151–61. doi: 10.1007/s001090050204. [DOI] [PubMed] [Google Scholar]
- 2.Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3:791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morand EF, Leech M, Bernhagen J. MIF: a new cytokine link between rheumatoid arthritis and atherosclerosis. Nat Rev Drug Discov. 2006;5:399–410. doi: 10.1038/nrd2029. [DOI] [PubMed] [Google Scholar]
- 4.Zernecke A, Bernhagen J, Weber C. Macrophage migration inhibitory factor in cardiovascular disease. Circulation. 2008;117:1594–602. doi: 10.1161/CIRCULATIONAHA.107.729125. [DOI] [PubMed] [Google Scholar]
- 5.Bernhagen J, Krohn R, Lue H, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13:587–96. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- 6.Weber C, Kraemer S, Drechsler M, et al. Structural determinants of MIF functions in CXCR2-mediated inflammatory and atherogenic leukocyte recruitment. Proc Natl Acad Sci USA. 2008;105:16278–83. doi: 10.1073/pnas.0804017105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beider K, Abraham M, Peled A. Chemokines and chemokine receptors in stem cell circulation. Front Biosci. 2008;13:6820–33. doi: 10.2741/3190. [DOI] [PubMed] [Google Scholar]
- 8.Luster AD. Chemokines – chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338:436–45. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 9.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–21. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 10.Ceradini DJ, Kulkarni AR, Callaghan MJ, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–64. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 11.Yu JX, Huang XF, Lv WM, et al. Combination of stromal-derived factor-1alpha and vascular endothelial growth factor gene-modified endothelial progenitor cells is more effective for ischemic neovascularization. J Vasc Surg. 2009;50:608–16. doi: 10.1016/j.jvs.2009.05.049. [DOI] [PubMed] [Google Scholar]
- 12.Hristov M, Zernecke A, Liehn EA, et al. Regulation of endothelial progenitor cell homing after arterial injury. Thromb Haemost. 2007;98:274–7. [PubMed] [Google Scholar]
- 13.Jujo K, Ii M, Losordo DW. Endothelial progenitor cells in neovascularization of infarcted myocardium. J Mol Cell Cardiol. 2008;45:530–44. doi: 10.1016/j.yjmcc.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Real C, Caiado F, Dias S. Endothelial progenitors in vascular repair and angiogenesis: how many are needed and what to do. Cardiovasc Hematol Disord Drug Targets. 2008;8:185–93. doi: 10.2174/187152908785849071. [DOI] [PubMed] [Google Scholar]
- 15.Hristov M, Weber C. Endothelial progenitor cells: characterization, pathophysiology, and possible clinical relevance. J Cell Mol Med. 2004;8:498–508. doi: 10.1111/j.1582-4934.2004.tb00474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hristov M, Weber C. Endothelial progenitor cells in vascular repair and remodeling. Pharmacol Res. 2008;58:148–51. doi: 10.1016/j.phrs.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 17.Hristov M, Weber C. Progenitor cell trafficking in the vascular wall. J Thromb Haemost. 2009;7:31–4. doi: 10.1111/j.1538-7836.2009.03406.x. [DOI] [PubMed] [Google Scholar]
- 18.Pearson JD. Endothelial progenitor cells – hype or hope. J Thromb Haemost. 2009;7:255–62. doi: 10.1111/j.1538-7836.2008.03214.x. [DOI] [PubMed] [Google Scholar]
- 19.Timmermans F, Plum J, Yoder MC, et al. Endothelial progenitor cells: identity defined. J Cell Mol Med. 2009;13:87–102. doi: 10.1111/j.1582-4934.2008.00598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sieveking DP, Buckle A, Celermajer DS, et al. Strikingly different angiogenic properties of endothelial progenitor cell subpopulations: insights from a novel human angiogenesis assay. J Am Coll Cardiol. 2008;51:660–8. doi: 10.1016/j.jacc.2007.09.059. [DOI] [PubMed] [Google Scholar]
- 21.Kupatt C, Horstkotte J, Vlastos GA, et al. Embryonic endothelial progenitor cells expressing a broad range of proangiogenic and remodeling factors enhance vascularization and tissue recovery in acute and chronic ischemia. FASEB J. 2005;19:1576–8. doi: 10.1096/fj.04-3282fje. [DOI] [PubMed] [Google Scholar]
- 22.Ceradini DJ, Gurtner GC. Homing to hypoxia: HIF-1 as a mediator of progenitor cell recruitment to injured tissue. Trends Cardiovasc Med. 2005;15:57–63. doi: 10.1016/j.tcm.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 23.Baugh JA, Gantier M, Li L, et al. Dual regulation of macrophage migration inhibitory factor (MIF) expression in hypoxia by CREB and HIF-1. Biochem Biophys Res Commun. 2006;347:895–903. doi: 10.1016/j.bbrc.2006.06.148. [DOI] [PubMed] [Google Scholar]
- 24.Flieger O, Engling A, Bucala R, et al. Regulated secretion of macrophage migration inhibitory factor is mediated by a non-classical pathway involving an ABC transporter. FEBS Lett. 2003;551:78–86. doi: 10.1016/s0014-5793(03)00900-1. [DOI] [PubMed] [Google Scholar]
- 25.Merk M, Baugh J, Zierow S, et al. The Golgi-associated protein p115 mediates the secretion of macrophage migration inhibitory factor. J Immunol. 2009;182:6896–906. doi: 10.4049/jimmunol.0803710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nickel W, Rabouille C. Mechanisms of regulated unconventional protein secretion. Nat Rev Mol Cell Biol. 2009;10:148–55. doi: 10.1038/nrm2617. [DOI] [PubMed] [Google Scholar]
- 27.Jaffe EA, Nachman RL, Becker CG, et al. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest. 1973;52:2745–56. doi: 10.1172/JCI107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grieb G, Groger A, Piatkowski A, et al. Tissue substitutes with improved angiogenic capabilities: an in vitro investigation with endothelial cells and endothelial progenitor cells. Cells Tissues Organs. 2010;191:96–104. doi: 10.1159/000231473. [DOI] [PubMed] [Google Scholar]
- 29.Nishihira J, Koyama Y, Mizue Y. Identification of macrophage migration inhibitory factor (MIF) in human vascular endothelial cells and its induction by lipopolysaccharide. Cytokine. 1998;10:199–205. doi: 10.1006/cyto.1997.0276. [DOI] [PubMed] [Google Scholar]
- 30.Okamoto T, Atsumi T, Shimizu C, et al. The potential role of macrophage migration inhibitory factor on the migration of vascular smooth muscle cells. J Atheroscler Thromb. 2008;15:13–9. doi: 10.5551/jat.e495. [DOI] [PubMed] [Google Scholar]
- 31.Burger-Kentischer A, Goebel H, Seiler R, et al. Expression of macrophage migration inhibitory factor in different stages of human atherosclerosis. Circulation. 2002;105:1561–6. doi: 10.1161/01.cir.0000012942.49244.82. [DOI] [PubMed] [Google Scholar]
- 32.Schmeisser A, Marquetant R, Illmer T, et al. The expression of macrophage migration inhibitory factor 1alpha (MIF 1alpha) in human atherosclerotic plaques is induced by different proatherogenic stimuli and associated with plaque instability. Atherosclerosis. 2005;178:83–94. doi: 10.1016/j.atherosclerosis.2004.08.038. [DOI] [PubMed] [Google Scholar]
- 33.Schober A, Bernhagen J, Thiele M, et al. Stabilization of atherosclerotic plaques by blockade of macrophage migration inhibitory factor after vascular injury in apolipoprotein E-deficient mice. Circulation. 2004;109:380–5. doi: 10.1161/01.CIR.0000109201.72441.09. [DOI] [PubMed] [Google Scholar]
- 34.Wang B, Wood IS, Trayhurn P. Dysregulation of the expression and secretion of inflammation-related adipokines by hypoxia in human adipocytes. Pflugers Arch. 2007;455:479–92. doi: 10.1007/s00424-007-0301-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larsen M, Tazzyman S, Lund EL, et al. Hypoxia-induced secretion of macrophage migration-inhibitory factor from MCF-7 breast cancer cells is regulated in a hypoxia-inducible factor-independent manner. Cancer Lett. 2008;265:239–49. doi: 10.1016/j.canlet.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 36.Miller EJ, Li J, Leng L, et al. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature. 2008;451:578–82. doi: 10.1038/nature06504. [DOI] [PubMed] [Google Scholar]
- 37.Jian Z, Li JB, Ma RY, et al. Increase of macrophage migration inhibitory factor (MIF) expression in cardiomyocytes during chronic hypoxia. Clin Chim Acta. 2009;405:132–8. doi: 10.1016/j.cca.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 38.Wang L, Zis O, Ma G, et al. Upregulation of macrophage migration inhibitory factor gene expression in stroke. Stroke. 2009;40:973–6. doi: 10.1161/STROKEAHA.108.530535. [DOI] [PubMed] [Google Scholar]
- 39.Kleemann R, Hausser A, Geiger G, et al. Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature. 2000;408:211–6. doi: 10.1038/35041591. [DOI] [PubMed] [Google Scholar]
- 40.Lue H, Kapurniotu A, Fingerle-Rowson G, et al. Rapid and transient activation of the ERK MAPK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on JAB1/CSN5 and Src kinase activity. Cell Signal. 2006;18:688–703. doi: 10.1016/j.cellsig.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 41.Berndt K, Kim M, Meinhardt A, et al. Macrophage migration inhibitory factor does not modulate co-activation of androgen receptor by Jab1/CSN5. Mol Cell Biochem. 2008;307:265–71. doi: 10.1007/s11010-007-9578-3. [DOI] [PubMed] [Google Scholar]
- 42.Koch N, Moldenhauer G, Hofmann WJ, et al. Rapid intracellular pathway gives rise to cell surface expression of the MHC class II-associated invariant chain (CD74) J Immunol. 1991;147:2643–51. [PubMed] [Google Scholar]
- 43.Shim H, Lau SK, Devi S, et al. Lower expression of CXCR4 in lymph node metastases than in primary breast cancers: potential regulation by ligand-dependent degradation and HIF-1alpha. Biochem Biophys Res Commun. 2006;346:252–8. doi: 10.1016/j.bbrc.2006.05.110. [DOI] [PubMed] [Google Scholar]
- 44.Rubartelli A, Cozzolino F, Talio M, et al. A novel secretory pathway for interleukin-1β, a protein lacking a signal sequence. EMBO J. 1990;9:1503–10. doi: 10.1002/j.1460-2075.1990.tb08268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]