

Abstract
E-cadherin loss is a key biological mechanism in tumour invasion. As a main regulator of epithelial-mesenchymal transition (EMT) mechanism-mediated invasion and metastasis, Twist1 plays an important role through its regulation of E-cadherin expression. However, whether or not Twist2 has the same function in tumour metastasis remains unclear. The purpose of this study is to investigate the expressions and different roles of Twist1 and Twist2 in human hepatocellular carcinoma (HCC). The expressions of Twist1 and Twist2 in HCC tissue were evaluated by immunohistochemical staining. The role of Twist1 and Twist2 in invasiveness was also evaluated in vitro by using HCC cell lines. Twist1 nuclear overexpression is found to be correlated with HCC metastasis, and its expression is negatively correlated with E-cadherin expression in human tissue. Twist2, a Twist1 homology protein, only expresses in the cytoplasm and shows no significant correlation with HCC metastasis. By ectopic transfection of Twist1 and Twist2 into the HCC cells, HepG2 and PLC, Twist1 is able to down-regulate E-cadherin expression and promote matrix metalloproteinase (MMP) activation, specifically in MMP2 and MMP9. In functional assays, Twist1 is found to promote invasion in HepG2 and PLC cells, but the invasion ability of the groups is not affected Twist2. Our findings indicate that Twist1 induces HCC invasion via increased activity in MMPs, leading to poor clinical prognoses. The results of this study also demonstrate a novel cogitation in Twist2, which has no effect on HCC invasion and metastasis. Twist1 may contribute to HCC invasion and metastasis and may be used as a novel therapeutic target for the inhibition of HCC metastasis.
Keywords: human hepatocellular carcinoma, metastasis, invasion, MMPs
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common malignancy worldwide and the third leading cause of cancer morbidities in China. As a rapidly growing tumour, HCC is associated with a high propensity for vascular invasion and metastasis, which leads to poor cancer prognoses [1, 2]. Metastasis occurs in more than 50% of patients after surgery therapy; intrahepatic metastasis is the type that occurs more frequently in these cases. The common extrahepatic metastasis sites are in the lungs, bones, peritoneum, spleen and lymph nodes [3, 4]. To understand the metastatic recurrent disease, cellular and molecular characterization of the metastatic tumour cells is essential.
The epithelial-mesenchymal transition (EMT) is a common example of the cellular plasticity of a tumour. It is the leading mechanism for cellular invasion. As the invasion process proceeds, epithelial cell layers lose polarity and cell–cell interaction, which finally leads to a complex remodelling of the cytoskeleton [5–7]. E-cadherin is an epithelial marker that disappears as a hallmark of EMT. It is a central component of cell–cell adhesion junctions, and it maintains cell polarity and environment [8, 9]. HCC is not a typical epithelial type of tumour. The mechanism of loss of E-cadherin expression and gain of mesenchymal markers is also associated with tumour invasiveness, metastasis and poor cancer prognoses, much like EMT [9, 10]. The regulatory mechanisms of E-cadherin are controlled by regulators that repress E-cadherin transcription via interactions with specific E-boxes of the proximal E-cadherin promoter. The Snail and Slug from zinc finger families are the most prominent EMT regulators [5–7, 12]. Recently, the basic helix-loop-helix (bHLH) transcription factor, Twist has been added to the list of EMT regulators. Twist1 and Twist2 are two homologous proteins identified in Drosophila melanogaster. They act as organizers of EMT during gastrulation and as regulators of mesoderm differentiation [13, 14]. They have also been included in the list of developmental genes with key roles in E-cadherin repression and in the promotion of tumour cell invasion.
Consistent with the known Twist functions, Twist1 has been found to be correlated with metastasis in various cancer cells, including those of the breast, prostate and HCC [5, 13, 15]. The same functions in metastasis have not been found for Twist2. Therefore, the difference between the roles of Twist1 and Twist2 in HCC metastasis is an interesting topic for study. To date, there have been no data on the difference in the Twist1 and Twist2 roles in HCC. This study aims to investigate the expressions and possible roles of Twist1 and Twist2 in HCC metastasis using human tissue samples and HCC cell lines. Twist1, Twist2 and their associated proteins were examined by immunohistochemical staining of the tissue specimens. In addition, functional tests were conducted based on the transfection of Twist1 or Twist2 cDNA into the HCC cell line. Our evidence suggests that Twist1 is correlated with HCC metastasis and cell invasion but not Twist2.
Materials and methods
Patient samples
Tissue specimens from 97 patients who underwent hepatectomy for HCC between 2001 and 2005 were obtained from the Tumor Tissue Bank of Tianjin Cancer Hospital. The diagnosis of these HCC samples was verified by pathologists. Detailed pathologic and clinical data were collected for all the samples, including Edmondson tumour grade, metastasis and survival duration. Paraffin-embedded tumour tissue samples were collected from patients in the surgical operation without any other therapy. The use of these tissues in this study was approved by the institutional research committee.
Immunostaining
Slides were deparaffinized in xylene. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide in 50% methanol for 10 min. at room temperature. Sections were rehydrated in alcohol, washed with phosphate-buffered saline (PBS), and then pretreated with citrate buffer (0.01 M citric acid, pH 6.0) for 20 min. at 95°C in a microwave oven. After the non-specific binding sites were blocked by exposing them to 10% normal goat serum in PBS for 20 min. at 37°C, sections were incubated with a series of antibodies (dilution 1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4°C overnight. Following incubation, the sections were rinsed with PBS and were incubated with biotinylated goat anti-mouse IgG for 20 min. at 37°C. The slides were then incubated with 3,3’-diaminobenzidine chromogen for 5–10 min. at room temperature and were washed with distilled water. Finally, sections were slightly counterstained with haematoxylin for 1 min., followed by dehydration and cover slip mounting. PBS was utilized in place of the primary antibodies for the negative control. The staining systems used in this study were PicTure PV6000 (Zhongshan Chemical Co., Beijing, China) and Elivision Plus (Zhongshan Chemical Co.).
Plasmids
Full-length cDNA of Twist was generated by reverse transcription using normal human embryo total RNA as template. This was followed by reverse transcription-polymerase chain reaction (RT-PCR) amplification with the primers derived from human Twist1 (primers: 5′-GCTCTTCTCCTCTGCCCCGG-3′ and 5′-CATCTAGGTCTCCGGCCCTG-3′) and Twist2 (5′-GCACAACGGCCGGAACTTTAG-3′ and 5′-TGTCCATGGCTGCGCGGACGTC-3′). The PCR products were digested with XhoI/EcoRI and were subcloned into pcDNA3.1 vectors. The resulting constructs were confirmed by DNA sequencing.
Cell lines
The HCC cell lines used in this study were HepG2, Bel7402, PLC and SMMC7221 (from the American Type Culture Collection, Rockville, MD, USA). The derivation and sources of these cell lines have been reported previously. These cells were maintained and propagated in vitro by serial passage in Dulecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum and 0.1% gentamicin sulphate (Invitrogen, Carlsbad, CA, USA). All the experiments were performed with 70–80% confluent cultures.
Cell transfection
Samples of pcDNA3-Negative, pcDNA3-Twist1 and pcDNA3-Twist2 were transfected into HepG2 or Bel7402 cells using polyethylenimine (PEI, Polysciences, Inc., PA, USA; Cat#23966). A pool transfectant (>150 colonies) was selected using G418 at a dose of 800 μg/ml.
Western blot
Cells were lysed with 1 × modified RIPA buffer (50 mM Tris, 150 mM NaCl, 1% Triton X-100 and 0.5% deoxycholate) containing 25 μg/ml leupeptin (Sigma Chemical Co., St. Louis, MO, USA), 10 μg/ml aprotinin (Sigma Chemical Co.) and 2 mM EDTA. Cells were removed from the dishes by cell scraping. The samples were then subjected to 30 min. cooling on ice and were centrifuged at 12,500 rpm for 30 min. The whole cell lysates were analysed by 8% SDS-PAGE and stained with Coomassie BBR-250 (Sigma) to ensure equal loading (about 60 μg whole protein in each lane). Samples were transferred to polyvinylidene difluoride membranes (PVDF, Millipore, Billerica, MA, USA). Blots were blocked with 5% fat-free milk for 1 hr at room temperature and then incubated with the monoclonal Twist1 and Twist2 antibodies (1:200 dilution; Abcam, Cambridge, MA, USA and Santa Cruz Biotechnology) for 2 hrs at room temperature with agitation. This was followed by incubation with a horseradish peroxidase-conjugated antimouse secondary antibody (1:2000; Santa Cruz). Blots were developed using an enhanced chemiluminescence detection kit (ECL) (Amersham Pharmacia Biotech, Piscataway, NJ, USA). For protein loading analysis, a monoclonal β-actin antibody (1:200; Santa Cruz) was used. The intensity of the protein bands for this analysis was determined by densitometry using the Gene Genius Super system (Gene Company Limited, EN).
Invasion assay
Cell migration assay was performed using Transwell cell culture inserts with 8-μm porosity polyethylene teraphthalate filters (Invitrogen). Briefly, confluent tumour cells were trypsinized, plated onto the upper Matrigel-coated insert and allowed to attach to the membrane for 1 hr. Cells were transfected for 48 hrs and then allowed to migrate for another 24 hrs. The upper surface of the membrane was wiped to remove non-migratory cells. The cells that invaded through the Matrigel and adhered to the bottom of the membrane were stained with a crystal violet solution. The cell-associated dye was eluted with 10% acetic acid, and its absorbance at 590 nm was determined. Each experiment was conducted in triplicate. The values were presented as mean ± SE.
Zymography assays
All media were collected and subjected to SDS-PAGE using 0.01% w/v gelatine containing 10% polyacrylamide gel. After electrophoresis, gels were equilibrated in 2.5% Triton X-100 and incubated in 50 mM Tris-HCl (pH 7.5), 10 mM CaCl2, 150 mM NaCl, 1 mM ZnCl2 and 0.02% NaN3 for 40 hrs at 37°C. These were stained with Coomassie R250 and then destained until the wash became clear, and the gels had apparent cleared zones associated with matrix metalloproteinase (MMP) activity.
Statistical analysis
All the data in the study were evaluated using SPSS13.5 (SPSS Inc., Chicago, IL, USA). Differences were considered significant at values of P < 0.05.
Results
Twist1 and Twist2 expression in human HCC tissue and cell lines
To determine the possible role of Twist1 and Twist2 in HCC metastasis, we evaluated their expressions in human tissue samples by immunostaining. Metastatic tumours numbered 49 from a total of 97 HCC samples (49/97). The corresponding clinical data are shown in Table 1. In the 97 HCC samples, Twist1 nuclear expression was detected in 33 (34.0%) cases, and Twist1 cytoplasm expression was detected in 51 (52.6%) cases. Twist2 expression in 97 HCC samples was only detected in the cytoplasm in 52 (53.6%) cases. Nuclei expression of Twist2 was not detected. Twist1 was expressed in both the cytoplasm and nuclei, but Twist2 was only detected in the cytoplasm (Fig. 1). To screen further for the expression levels of Twist1 and Twist2 in HCC cells, the mRNA and protein levels of Twist1 and Twist2 expression in various HCC cell lines were compared using RT-PCR and Western blotting results. HepG2 and PLC had a low level of Twist1 expression and a low level of Twist2 expression. Bel7402 exhibited a high level of Twist1 expression, while SMMC7221 exhibited a high level of Twist2 expression (Fig. 2).
Table 1.
Correlation between metastasis and clinicopathologic characteristics of patients with hepatocellular carcinoma
| Variant | Tissue samples | Chi-square test | P-value | |
|---|---|---|---|---|
| Metastasis(−) | Metastasis(+) | |||
| Age (years) | ||||
| <50 | 18 | 20 | 0.112 | 0.738 |
| ≥50 | 30 | 29 | ||
| Gender | ||||
| Male | 42 | 40 | 0.639 | 0.424 |
| Female | 6 | 9 | ||
| Tumour size (cm) | ||||
| >5 | 30 | 17 | 7.506 | 0.006* |
| ≤5 | 18 | 32 | ||
| Histological differentiation | ||||
| I/II | 20 | 11 | 4.118 | 0.042* |
| III/IV | 28 | 38 | ||
| Stage | ||||
| III | 42 | 11 | 41.399 | 0.000* |
| III IV | 6 | 38 | ||
Significant difference.
Fig 1.
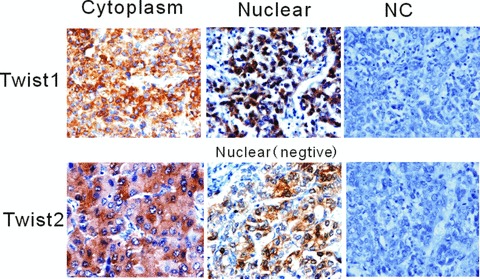
Positive Twist1 expression was detected in HCC cytoplasm and nucleus by immunostaining, and Twist2 expression was only detected in the cytoplasm. Nuclei expression for Twist2 was not detected. Twist1 was expressed in both the cytoplasm and nuclei, but Twist2 was only detected in the cytoplasm.
Fig 2.
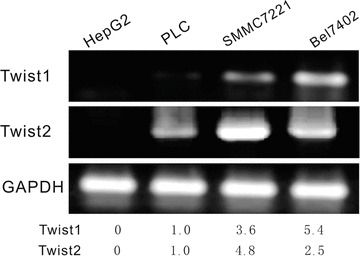
Twist1 and Twist2 expression level in HCC cell lines. RT-PCR was used to screen the level of Twist1 and Twist2 expression in the HCC cell lines HepG2, PLC, SMMC7221 and Bel7402. HepG2 and PLC had a low level of Twist1 expression and a low level of Twist2 expression. Bel7402 presented a high level of Twist1 expression, while SMMC7221 presented a high level of Twist2 expression.
Increased invasion ability in cells with Twist1 up-regulation
An up-regulated cell model using HepG2 and PLC cells transfected with Twist1 and Twist2 cDNA was determined. Based on Western blot analysis, Twist1 and Twist2 increased their expressions after ectopic transfection. To investigate the molecular changes in the HepG2-Twist1 and PLC-Twist1 transfectants, the expression of the epithelial marker E-cadherin was detected by Western blot analysis, and results show that it was down-regulated. For the Twist2 transfectant, E-cadherin showed no significant differences between the transfected and control groups for HepG2 and PLC cells. To investigate further the effects of Twist1 and Twist2 activation on cell invasion, a transwell invasion assay was used to analyse the different influences between Twist1 and Twist2. The invasion ability of HepG2 and PLC cells were studied after performing Twist1 and Twist2 ectopic transfection. As shown in Fig. 3, following a transwell invasion assay, the quantitative analysis suggests a significant difference between the Twist1 transfection groups and the control group (P < 0.01). Twist2, however, showed no significant effect on the invasion ability of HepG2 and PLC cells (P > 0.05). Based on zymographic assays, the activities of MMP2 and MMP9 in HepG2 and PLC cells were significantly higher in the Twist1 up-regulation group compared with the control group, but Twist2 did not present the same changes after transfection.
Fig 3.
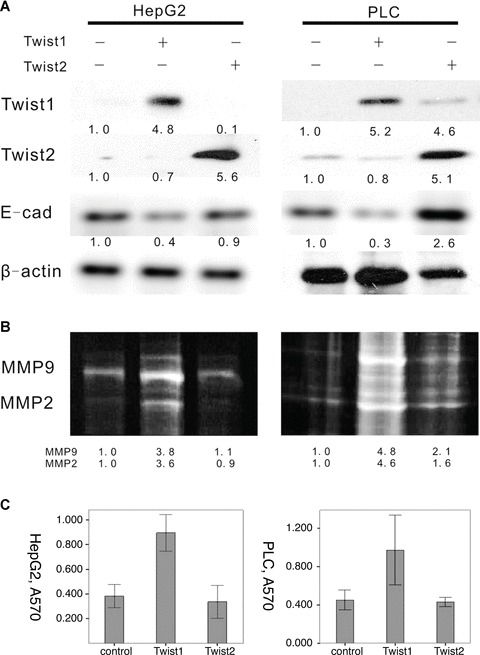
Effect of ectopic expression of Twist1 and Twist2 by pcDNA3.1 in the HCC cell lines HepG2 and PLC cells. (A) We determined that the expression of the epithelial marker E-cadherin was down-regulated after Twist1 transfectant, which was detected by Western blot analysis. For the Twist2 transfectant, E-cadherin showed no significant differences between the transfect and control groups for HepG2 and PLC cells. (B) Based on zymographic assays, the activities of MMP2 and MMP9 in HepG2 and PLC cells were significantly higher in the Twist1 up-regulation group than with the control group, but Twist2 did not present the same changes after transfection. (C) To investigate further the effects of Twist1 and Twist2 activation on cell invasion, we used transwell invasion assay to analyse the different influences between Twist1 and Twist2. We studied the invasion ability of HepG2 and PLC cells after performing Twist1 and Twist2 ectopic transfection. The quantitative analysis suggests a significant difference between the Twist1 transfection groups and the control group (P < 0.05). Twist2, however, showed no significant result in HepG2 and PLC cells (P > 0.05).
Significant correlation of Twist1 nuclear overexpression with E-cadherin and MMPs
Immunohistochemical analysis was performed to access the expression of Twist1, Twist1 (nuclear), Twist2, E-cadherin, MMP-2 and MMP-9 in the HCC samples (Fig. 4). The results show that for Twist1, expression was located both in the cytoplasm and nuclei of the HCC cells. For Twist2, expression was located only in the cytoplasm. E-cadherin expression was located in the membranes. MMP-2 and MMP-9 expressions were located in the cytoplasm of the HCC cells. The expressions of these proteins in the cells with and without metastasis were compared using the chi-square test (Table 2) and rank-sum test (Table 3). The positive rates of Twist1 (nuclear) and MMP-9 expression with metastasis were higher and E-cadherin was lower compared with cancers without metastasis. The differences in Twist1 (nuclear), E-cadherin, MMP-2 and MMP-9 were statistically significant (P < 0.01). Numeration data were collected using the positive cell rates, and analysis was conducted using the Pearson correlation test. Twist1 nuclear expression is correlated to Twist1 cytoplasm expression, E-cadherin and MMP9. Twist2 has no relationship with any other protein (Fig. 5). Kaplan–Meier survival analysis revealed that patients with metastasis and expression of Twist1, Twist1 (nuclear), MMP-2 and MMP-9 had shorter survival rates than those without expression (Fig. 6).
Fig 4.
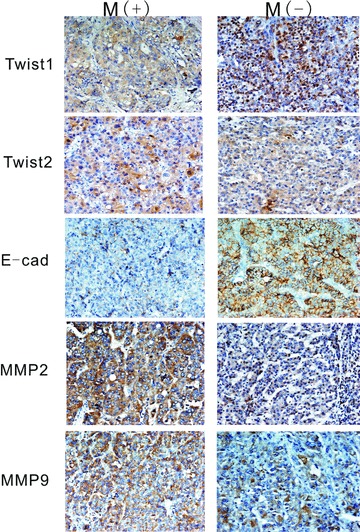
E-cadherin, MMP2 and MMP9 expression in HCC. Brown or yellow staining was observed in the membrane and cytoplasm. E-cadherin was located in membranes. MMP2 and MMP9 were located in the cytoplasm.
Table 2.
Relationship between metastasis and Twist1, Twist2, E-cad, MMP-2 and MMP-9 expression in HCC (chi-square test)
| Variant | Tissue samples | Chi-square test | P-value | ||
|---|---|---|---|---|---|
| Metastasis (−) | Metastasis (+) | ||||
| Twist1 (nuclear) | Negative | 39 | 25 | 9.871 | 0.002* |
| Positive | 9 | 24 | |||
| Twist1 (cytoplasm) | Negative | 27 | 19 | 2.970 | 0.085 |
| Positive | 21 | 30 | |||
| Twist2 | Negative | 24 | 21 | 0.497 | 0.481 |
| Positive | 24 | 28 | |||
| E-cadherin | Negative | 21 | 27 | 1.25 | 0.264 |
| Positive | 27 | 22 | |||
| MMP2 | Negative | 12 | 15 | 0.38 | 0.537 |
| Positive | 36 | 34 | |||
| MMP9 | Negative | 20 | 16 | 0.844 | 0.358 |
| Positive | 28 | 33 |
Significant difference.
Table 3.
Relationship between metastasis and Twist1, Twist2, E-cad, MMP-2 and MMP-9 expression (rank-sum test)
| Variant | Z | P-value |
|---|---|---|
| Twist1(c+n) | −3.619 | 0.000* |
| Twist2 | −0.239 | 0.811 |
| E-cadherin | −2.205 | 0.027* |
| Mmp2 | −2.623 | 0.009* |
| Mmp9 | −2.028 | 0.043* |
Significant difference, (c+n) cytoplasm and nuclear.
Fig 5.
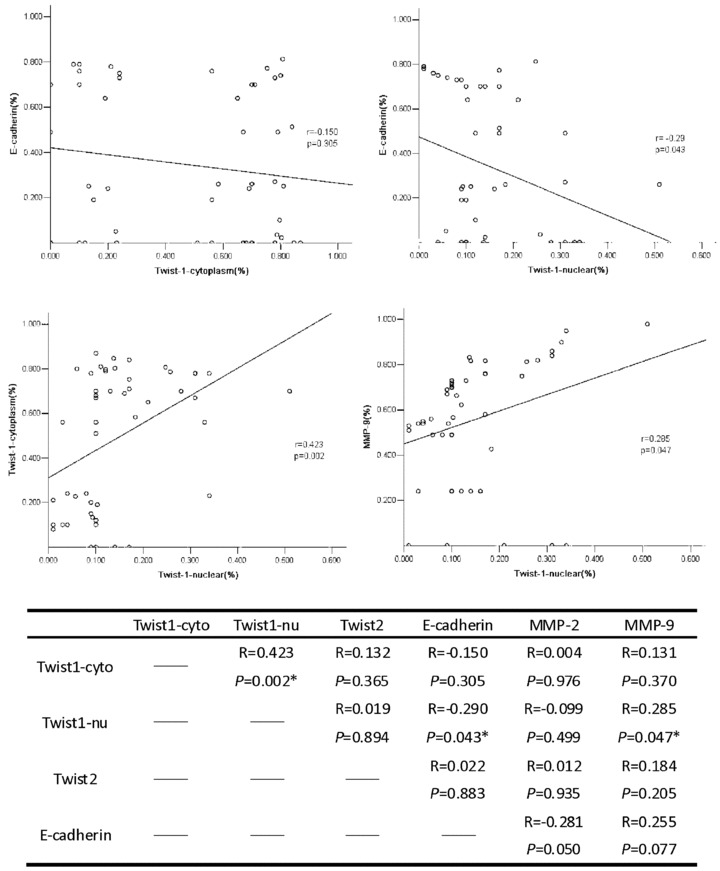
Correlation analysis between Twist1(cytoplasm), Twist1(nuclear), Twist2, E-cadherin, MMP2 and MMP9 expression was tested using the Pearson correlation test. Results show that the Twist1 (nuclear) is correlated with Twist1(cytoplasm), E-cadherin and MMP9 (*P < 0.05).
Fig 6.
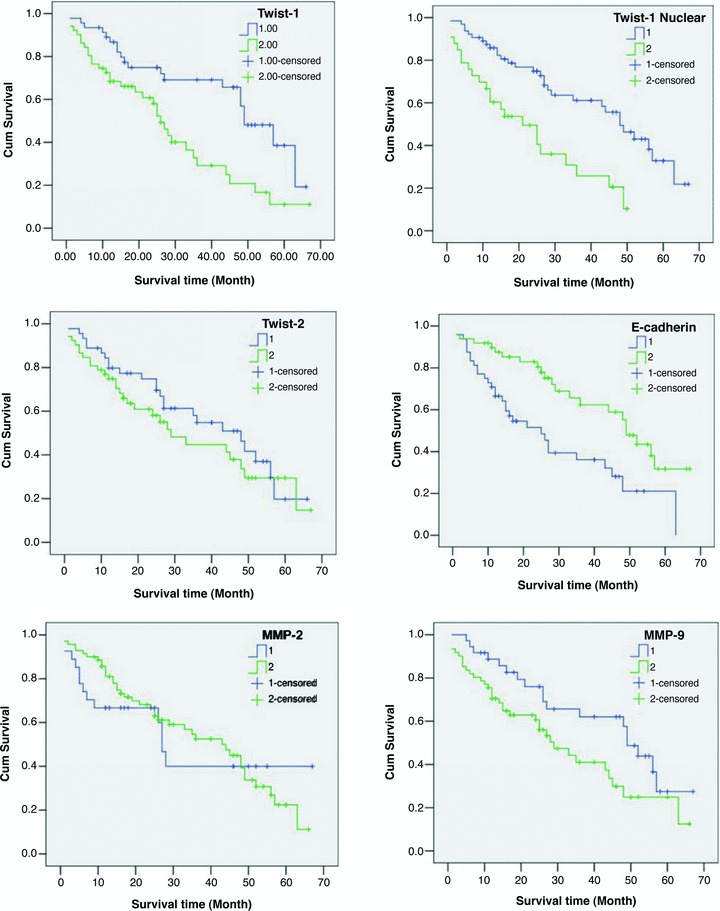
Kaplan–Meier survival analysis in HCCs. The P-value of Twist1 (cytoplasm) was 0.000, Twist1 (nuclear) was 0.002, Twist2 was 0.336, E-cadherin was 0.002, MMP2 was 0.805 and MMP9 was 0.039.
Discussion
The molecular mechanism of HCC invasiveness and metastasis is very important to research [1–4]. EMT plays a significant role in the development of tissues during embryogenesis. However, during pathological processes, as in the case of cancer, similar cell changes are recapitulated. Most of the research conducted on EMT focuses on the invasion and metastasis of cancer cells. In particular, several developmental genes that induce EMT have been shown to act as E-cadherin repressors. The first of these is a zinc finger protein, the Snail family, which is a DNA-binding factor family that recognizes E-box motifs in target promoters, such as E-cadherin [5–7, 11]. The new findings of Yang et al. have added Twist to this class of EMT inducers [12]. Twist was first considered an oncogene. Twist proteins are highly conserved bHLH transcription factors with important regulatory functions during embryogenesis. In Drosophila, the ancestral Twist protein is crucial for proper gastrulation and mesoderm formation [13, 14]. Recently, Twist overexpression has been found to correlate with the malignant transformation of melanoma, breast cancer and HCC [12, 15, 16]. This suggests that Twist induces E-cadherin loss, resulting in the promotion of tumour invasion. However, the role of Twist-mediated tumour metastasis remains controversial.
This study reported that Twist1 overexpression in HCC tumour tissues correlated with metastasis in human samples. Findings indicating that Twist2 does not behave in a similar manner as Twist1 were also reported for the first time. Instead, Twist2 plays a free role in HCC invasion and metastasis. The above results strongly suggest the role of Twist1 in HCC metastasis. Twist1, like other EMT-inducing transcription factors, such as Snail, Slug and SIP1, binds DNA through similar E-box sequence motifs and represses E-cadherin and other epithelial cell adhesion molecules. In mammals, two Twist-like proteins, Twist1 and Twist2, share high structural homology [17, 18]. Gene deletion experiments have shown that Twist1 and Twist2 have some functional redundancy [19, 20]. Twist1 and Twist2 are early drivers of tumour progression, and they provide a rationale for the high frequency of Twist1 and Twist2 overexpression in a large variety of human tumours. Twist2 knockout mice display an elevated expression of proinflammatory cytokines, which causes perinatal death [21]. However, interestingly, this phenotype is also found in individuals doubly heterozygous for Twist1 and Twist2 alleles, reflecting a functional redundancy [22]. As such, these findings shed new light on the role of Twist proteins in metastasis. Several studies have previously reported that high Twist1 expression promotes EMT and correlates with tumour invasion and metastasis in a variety of human cancers [12, 13, 23, 24].
In this study, Twist1 was found to be significant in HCC invasion and metastasis, but the same could not be said for Twist2. A similar phenomenon was present in HCC vasculogenic mimicry, which also confirms that Twist2 is different from Twist1. In human tissue samples, Twist1 nuclear-expression was negatively correlated with E-cadherin, but Twist2 expression showed no significant difference in the E-cadherin levels. Thus, we hypothesize that Twist1, but not Twist2, can induce EMT-mediated metastasis through E-cadherin repression. This result was further confirmed by comparing Twist1 and Twist2 expressions in various HCC cell lines with different metastatic potentials. To confirm this, we evaluated the ectopic transfection of Twist1 and Twist2 cDNA into HepG2 and PLC cells. After transfection, ectopic Twist1 and Twist2 were up-regulated in the cells, as detected by Western blot analysis. Ectopic Twist1 conferred with the morphological changes from the epithelial to the fibroblastic appearance and with the loss of epithelial markers, such as E-cadherin. Twist1 was overexpressed in the cell lines, and it increased the ability of invasion and up-regulated the activity of MMP2 and MMP9. These effects were not found in Twist2. Ectopic Twist2 did not confer with the HCC cell morphological changes. It could not down-regulate E-cadherin expression and could not up-regulate MMP activity. MMPs have been shown to be involved in cancer invasion and metastasis. Overexpression of MMP2 and especially MMP9 in HCC has been shown to be involved in increased tumour recurrence or metastasis after tumour resection. Twist1 expression and relocation into the nucleus and the up-regulation of MMP9 expression show that both have a significant effect on metastasis in patients. This results in patients presenting these expressions having a shorter survival period than those that do not present them.
During metastasis, tumour cells are involved in numerous interactions, such as with the extracellular matrix (ECM) itself; the proteins, growth factors and cytokines; and the microenvironment of the secondary site where tumour cells eventually displace normal tissue as they grow out and form metastatic foci [25–27]. Several regulatory pathways or networks are either altered or aberrantly expressed, providing the tumour cells with the ability to accomplish each and all of the steps of the metastatic process successfully [28, 29]. Experimental in vitro and in vivo findings indicate that interacting tumour and stromal cells can mutually induce MMPs, which in turn can contribute to overall tumour invasion [30]. Tumour-induced angiogenesis is regarded to be important in sustaining the growth of solid tumours until they reach a size at which they become invasive and capable of metastasizing [31]. The functional roles of MMPs in tumour angiogenesis are well confirmed; genetic evidence and reconstitution experiments have established MMP-9 as a critical pro-angiogenic molecule in the tumour invasion process [32]. In this study, we highlighted the role of MMP-9 in the downstream of induced HCC metastasis by Twist1. Recent reports have shown that MMP-9 promoted by Twist1 is also critical in tumour vasculogenic mimicry formation, which is considered a key metastasis process in a poorly differentiated tumour.
In conclusion, we showed for the first time that Twist1, but not Twist2, is correlated with HCC metastasis and that Twist1 induces HCC invasiveness through the suppression of E-cadherin expression and activated MMPs. Our findings not only provide a molecular basis for the different roles of Twist1 and Twist2 in HCC metastasis but also suggest a novel therapeutic target for the inhibition of HCC metastasis.
Acknowledgments
This work was partly supported by a grant from Key project of the National Natural Science Foundation of China (No. 30830049), the National Natural Science Foundation of China (No. 30770828), the Tianjin Natural Science Foundation (No. 09ZCZDSF04400, 08JCZDJC23500 and No. 09JCYBJC12100) and the 973 Program (No. 2009CB918903 to Z.Y.) from the Ministry of Science and Technology of China.
References
- 1.Kuper H, Ye W, Broome U, et al. The risk of liver and bile duct cancer in patients with chronic viral hepatitis, alcoholism, or cirrhosis. Hepatology. 2001;34:714–8. doi: 10.1053/jhep.2001.28233. [DOI] [PubMed] [Google Scholar]
- 2.Poon RT, Fan ST, Wong J. Risk factors, prevention and management of postoperative recurrence after resection of hepatocellular carcinoma. Ann Surg. 2000;232:10–24. doi: 10.1097/00000658-200007000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.El-Assal ON, Yamanoi A, SodaY, et al. Proposal of invasiveness score to predict recurrence and survival after curative hepatic resection for hepatocellular carcinoma. Surgery. 1997;122:571–7. doi: 10.1016/s0039-6060(97)90130-6. [DOI] [PubMed] [Google Scholar]
- 4.Toyosaka A, Okamoto E, Mitsunobu M, et al. Intrahepatic metastases in hepatocellular carcinoma: evidence for spread via the portal vein as an efferent vessel. Am J Gastroenterol. 1996;91:1610–5. [PubMed] [Google Scholar]
- 5.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–42. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 6.Kang Y, Massagué J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell. 2004;118:277–9. doi: 10.1016/j.cell.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 7.Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–6. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 8.Hirohashi S. Inactivation of the E-cadherin-mediated cell adhesion system in human cancers. Am J Pathol. 1998;153:333–9. doi: 10.1016/S0002-9440(10)65575-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sugimachi K, Taguchi K, Aishima S, et al. Altered expression of β-catenin without genetic mutation in intrahepatic cholangiocarcinoma. Mod Pathol. 2001;14:900–5. doi: 10.1038/modpathol.3880409. [DOI] [PubMed] [Google Scholar]
- 10.Kanai Y, Ushijima S, Hui AM, et al. The E-cadherin gene is silenced by CpGme thylation in human hepatocellular carcinomas. Int J Cancer. 1997;71:355–9. doi: 10.1002/(sici)1097-0215(19970502)71:3<355::aid-ijc8>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 11.Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002;3:155–66. doi: 10.1038/nrm757. [DOI] [PubMed] [Google Scholar]
- 12.Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–39. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 13.Simpson P. Maternal-zygotic gene interactions during formation of the dorsoventral pattern in Drosophila embryos. Genetics. 1983;105:615–32. doi: 10.1093/genetics/105.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thisse B, Messal M, Perrin-Schmitt F. The twist gene: isolation of a Drosophila zygotic gene necessary for the establishment of dorsoventral pattern. Nucleic Acids Res. 1987;15:3439–53. doi: 10.1093/nar/15.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li L, Cserjesi P, Olson EN, et al. Dermo-1: a novel twist-related bHLH protein expressed in the developing dermis. Dev Biol. 1995;172:280–92. doi: 10.1006/dbio.1995.0023. [DOI] [PubMed] [Google Scholar]
- 16.Lee TK, Poon RT, Yuen AP, et al. Twist overexpression correlates with hepatocellular carcinoma metastasis through induction of epithelial-mesenchymal transition. Clin Cancer Res. 2006;12:5369–76. doi: 10.1158/1078-0432.CCR-05-2722. [DOI] [PubMed] [Google Scholar]
- 17.Kang Y, Massagué J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell. 2004;118:277–9. doi: 10.1016/j.cell.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 18.Wolf C, Thisse C, Stoetzel C, et al. The M-twist gene of Mus is expressed in subsets of mesodermal cells and is closely related to the Xenopus X-twi and the Drosophila twist genes. Dev Biol. 1991;143:363–73. doi: 10.1016/0012-1606(91)90086-i. [DOI] [PubMed] [Google Scholar]
- 19.Tsuji T, Ibaragi S, Shima K, et al. Epithelial-mesenchymal transition induced by growth suppressor p12CDK2-AP1 promotes tumor cell local invasion but suppresses distant colony growth. Cancer Res. 2008;68:10377–86. doi: 10.1158/0008-5472.CAN-08-1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bialek P, Kern B, Yang X, et al. A twist code determines the onset of osteoblast differentiation. Dev Cell. 2004;6:423–35. doi: 10.1016/s1534-5807(04)00058-9. [DOI] [PubMed] [Google Scholar]
- 21.Sosic D, Richardson JA, Yu K, et al. Twist regulates cytokine gene expression through a negative feedback loop that represses NF-kappaB activity. Cell. 2003;112:169–80. doi: 10.1016/s0092-8674(03)00002-3. [DOI] [PubMed] [Google Scholar]
- 22.Bialek P, Kern B, Yang X, et al. A twist code determines the onset of osteoblast differentiation. Dev Cell. 2004;6:423–35. doi: 10.1016/s1534-5807(04)00058-9. [DOI] [PubMed] [Google Scholar]
- 23.Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17:548–58. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 24.Kwok WK, Ling MT, Lee TK, et al. Up-regulation of Twist in prostate cancer and its implication as a therapeutic target. Cancer Res. 2005;65:5153–62. doi: 10.1158/0008-5472.CAN-04-3785. [DOI] [PubMed] [Google Scholar]
- 25.Lee SL, Rouhi P, Dahl Jensen L, et al. Hypoxia-induced pathological angiogenesis mediates tumor cell dissemination, invasion, and metastasis in a zebrafish tumor model. Proc Natl Acad Sci. 2009;106:19485–90. doi: 10.1073/pnas.0909228106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao Y, Cao R, Hedlund EM. R Regulation of tumor angiogenesis and metastasis by FGF and PDGF signaling pathways. J Mol Med. 2008;86:785–9. doi: 10.1007/s00109-008-0337-z. [DOI] [PubMed] [Google Scholar]
- 27.Cao Y, Liu Q. Therapeutic targets of multiple angiogenic factors for the treatment of cancer and metastasis. Adv Cancer Res. 2007;97:203–24. doi: 10.1016/S0065-230X(06)97009-2. [DOI] [PubMed] [Google Scholar]
- 28.Cao Y. Opinion: emerging mechanisms of tumour lymphangiogenesis and lymphatic metastasis. Nat Rev Cancer. 2005;5:735–43. doi: 10.1038/nrc1693. [DOI] [PubMed] [Google Scholar]
- 29.Björndahl MA, Cao R, Burton JB, et al. Vascular endothelial growth factor-a promotes peritumoral lymphangiogenesis and lymphatic metastasis. Cancer Res. 2005;65:9261–8. doi: 10.1158/0008-5472.CAN-04-2345. [DOI] [PubMed] [Google Scholar]
- 30.Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25:9–34. doi: 10.1007/s10555-006-7886-9. [DOI] [PubMed] [Google Scholar]
- 31.Cao R, Björndahl MA, Religa P, et al. PDGF-BB induces intratumoral lymphangiogenesis and promotes lymphatic metastasis. Cancer Cell. 2004;6:333–45. doi: 10.1016/j.ccr.2004.08.034. [DOI] [PubMed] [Google Scholar]
- 32.Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Cell. 2005;7:513–20. doi: 10.1016/j.ccr.2005.05.024. [DOI] [PubMed] [Google Scholar]