

Abstract
Traumatic brain injury (TBI) is a leading cause of acquired neurologic disability in children. Erythropoietin (EPO), an anti-apoptotic cytokine, improved cognitive outcome in adult rats after TBI. To our knowledge, EPO has not been studied in a developmental TBI model. Hypothesis: We hypothesized that EPO would improve cognitive outcome and increase neuron fraction in the hippocampus in 17-day-old (P17) rat pups after controlled cortical impact (CCI). Methods: EPO or vehicle was given at 1, 24, and 48 h after CCI and at post injury day (PID) 7. Cognitive outcome at PID14 was assessed using Novel Object Recognition (NOR). Hippocampal EPO levels, caspase activity, and mRNA levels of the apoptosis factors Bcl2, Bax, Bcl-xL, and Bad were measured during the first 14 days after injury. Neuron fraction and caspase activation in CA1, CA3, and DG were studied at PID2. Results: EPO normalized recognition memory after CCI. EPO blunted the increased hippocampal caspase activity induced by CCI at PID1, but not at PID2. EPO increased neuron fraction in CA3 at PID2. Brain levels of exogenous EPO appeared low relative to endogenous. Timing of EPO administration was associated with temporal changes in hippocampal mRNA levels of EPO and pro-apoptotic factors. Conclusion/Speculation: EPO improved recognition memory, increased regional hippocampal neuron fraction, and decreased caspase activity in P17 rats after CCI. We speculate that EPO improved cognitive outcome in rat pups after CCI as a result of improved neuronal survival via inhibition of caspase-dependent apoptosis early after injury.
Key words: : apoptosis, controlled cortical impect, developmental, EPO, memory
Introduction
Traumatic brain injury (TBI) is the leading cause of traumatic death and disability in children 1–14 years of age1,2 in the United States, affecting nearly half a million children each year. TBI causes cognitive disabilities, such as impaired learning and memory,3–7 that impose a heavy strain on society.8,9 Despite these facts, no treatments in clinical use today specifically decrease the cognitive sequelae of TBI.
Erythropoietin (EPO) administration may blunt TBI-induced cognitive dysfunction. EPO is an anti-apoptotic neuroprotective cytokine10,11 that is used to treat anemia in adults and children. In pre-clinical studies using adult rats, EPO administration decreased neuronal death and improved cognitive outcome after TBI.12–15 The effects of EPO on immature rats after TBI are unknown.
The immature rat may respond differently than the adult to EPO treatment after TBI. In the immature brain, given its increased susceptibility to pathologic apoptosis after injury,16 the anti-apoptotic effects of EPO may lead to greater neuronal survival after TBI than in the adult. However, should EPO interfere with developmentally appropriate apoptosis,17 then EPO could worsen cognitive dysfunction after TBI in immature rats. A preclinical study of EPO in a developmental model of TBI is warranted, particularly in light of ongoing clinical trials of EPO in adult TBI (NCT00987454 and NCT00313716) and newborn hypoxic-ischemic brain injury (NCT01471015).
Endogenous EPO is largely produced in the kidney and liver.10 Until recently, EPO was known only for its hematopoietic effects. In the bone marrow, EPO stimulates hematopoiesis by inhibiting apoptosis of the erythroblast while promoting proliferation and differentiation of erythroblast progenitor cells. Mounting evidence now supports an additional neuroprotective role of EPO.
Both EPO and its receptor are produced by neuronal, glial, and endothelial cells in the rodent18 and human brain19 in a developmentally regulated fashion.19,20 In animal models, endogenous EPO improves brain tolerance to ischemic and traumatic insults.21,22 In vivo, administration of recombinant human EPO (Rh EPO) decreased neuronal apoptosis23,24 and increased the expression of the anti-apoptotic factors Bcl2 and Bcl-xL25 relative to the pro-apoptotic factors Bad and Bax26,27 in the rodent brain. In vitro, Rh EPO increased neuronal proliferation and differentiation of neuronal progenitors.28 Penetration of Rh EPO into the brain, however, is limited. Therefore, neuroprotective doses of Rh EPO are much larger than those used to stimulate hematopoiesis.20,29,30
We hypothesized that, in a rat model of pediatric TBI, Rh EPO would improve cognitive outcome and increase neuron fraction in the hippocampus, a brain region critical for learning and memory. To test this hypothesis, we used controlled cortical impact (CCI) in 17-day-old rat pups to model pediatric TBI, as previously described.31 The Novel Object Recognition test at post-injury day (PID) 14 was used to assess cognitive outcome.32 CCI rats were treated with Rh EPO or vehicle (VEH), and SHAM rats with VEH. Histologic analyses of hippocampal neuron fraction and neuronal caspase activation were done at PID2, based on the expected peak in apoptotic cell death in the immature brain after trauma.33,34 Hippocampal caspase 3 activity, and mRNA levels of Bcl2, Bax, Bad and Bcl-xL in the first 14 days after CCI was used to evaluate the impact of Rh EPO on the intrinsic pathway of apoptosis, considered to be the predominant form of apoptotic death in neuronal cells.35
Methods
Animals
All experimental protocols were approved by the Animal Care and Use Committees at the University of Utah, in accordance with US NIH guidelines and carried out at the University of Utah. All surgical procedures were performed using aseptic technique.
Briefly, male Sprague-Dawley rats were obtained from Charles Rivers Laboratories (Raleigh, NC) on post-natal day (P) 7–10. We studied only males to eliminate any potential confounding effects of gender. Rats were housed in litters of 10 with the lactating dam until weaning on P 21–23. After weaning, rats were housed 3–5 per cage and allowed free access to food and water. All cages were kept in a temperature- and light-controlled (12 h on/12 h off ) environment.
In order to control for maternal rearing characteristics, randomization was distributed evenly within litters. Rats were randomized to experimental group at age P17, at which time rats underwent CCI or SHAM craniotomy followed by Rh EPO or vehicle (VEH) injection. After weaning, rats were placed in cages without any segregation based on experimental group.
CCI procedure
CCI was carried out as previously described.31 At least two litters of ten rats each were used to generate the numbers needed per group (6–8 rats per group for molecular studies and 6–7 rats per group for functional testing) for each time point in our study. On P17, rats undergoing CCI (n=6–7/ litter) were anesthetized with 3% isoflurane for induction, followed by 2–2.5% isoflurane for the duration of surgical preparation, using a VetEquip Bench Top Isoflurane Anesthesia System (Pleasanton, CA). Core temperature was monitored via a rectal probe and continuously controlled at 37±0.5°C using a servo-controlled heating pad. Oxygenation, heart rate, and respiratory rate were monitored using femoral probe pulse oximetry (Mousox ®, Starr Life Sciences, Oakmont, PA).
The rat was placed into a stereotaxic frame (David Kopf, Tujunga, CA). After shaving, prepping with povidone-iodine and incising the scalp, a craniotomy (6 mm x 6 mm) was performed over the left parietal cortex (centered at the point 4 mm posterior and 4 mm lateral to bregma). Care was taken not to perforate the dura. Once the craniotomy was complete, anesthesia was reduced to 1% isoflurane for a 5 min equilibration period. CCI was then delivered (Pittsburgh Precision Instruments, Pittsburgh, PA) to the left parietal cortex using a 5 mm rounded tip to deliver a 1.5 mm deformation at 4 m/s velocity and 100 ms duration. Immediately after CCI, isoflurane was increased to 2–2.5%, and the bone flap was replaced and secured with dental cement (Patterson Dental, Salt Lake City, UT). The scalp incision was sutured, and triple antibiotic ointment and bupivacaine 0.5% were applied topically. Isoflurane was stopped, and rats were allowed to recover in a temperature-controlled chamber. Once fully awake, rats were returned to their dams and littermates. SHAM rats (n=3–4/litter) underwent identical surgical craniotomy, equilibration, and closure procedures without CCI.
EPO dosing and rationale for dosing paradigm
Rats were dosed by intraperitoneal (IP) injection of Rh EPO (Procrit®) manufactured by Amgen Inc, CA. Rh EPO exerts neuroprotective and erythropoietic functions in rodents.36 Rat EPO has a predicted size of 36 kDa, while human EPO has a predicted size as high as 40kDa, given its one additional glycosylation site.
In the 7-day-old37 and adult rat,38 5000 U/kg Rh EPO given IP reliably produces measurable brain levels of EPO. Since Rh EPO takes between 3 and 9 h to peak in the brain,20,37 we chose to give the first dose 1 h after injury so it would be both clinically relevant and likely to produce an effect on early cell death. We chose to give two more doses, one at 24 and the other at 48 h after injury, to coincide with the expected peak in apoptotic cell death in the immature brain.33,34 Finally, based on data that an additional dose of Rh EPO at post injury day (PID) 7 improved long-term outcome in a rat model of neonatal stroke,39 we added a fourth dose. Thus, EPOCCI rats received 5000 U/kg Rh EPO IP at 1, 24, and 48 h after CCI and again at PID 7. At these time points, VEHCCI and SHAMVEH rats received an equal volume of IP vehicle (0.25 g BSA, 0.58 g sodium citrate, 0.58 g sodium chloride, and 6 mg citric acid in 100 mL dH2O (pH 6.9±0.3) as described in the Procrit® package insert).
Tissues
Rats were anesthetized with IP xylazine (8 mg/kg) and ketamine (40 mg/kg) for tissue collection. Rats were killed by swift decapitation either at 6 h after Rh EPO or VEH (on PID1, 2, and 7) or at an equivalent time of the day (PID3 and 14). The left (or ipsilateral to injury) hippocampus from CCI and SHAM animals was labeled either CCIipsi or SHAMipsi, respectively. The right (or contralateral to injury) hippocampus from CCI and Sham animals was labeled CCIcontra or SHAMcontra, respectively. Tissue was dissected on ice, snap frozen in liquid nitrogen, and stored at −80°C. For histology, rats underwent transcardiac perfusion-fixation at PID 2. Brains were removed, cryoprotected via placement in graded sucrose solutions (5, 15, and 30%), embedded in 2% gelatin medium, and frozen for immunofluorescence (IF) studies.
Hematocrit
Hematocrit determination was made using the I-Stat® system (Abbott Laboratories, Abbott Park, Il) on samples obtained by left ventricular puncture from a separate cohort of anesthetized EPOCCI (n=4–6), VEHCCI (n=4–6), and SHAMVEH (n=4) rats upon completion of CCI or SHAM surgery (on PID 0) and on PID 3, 7, and 14.
RNA isolation and real-time RT-PCR
Total RNA was extracted with Nucleospin II (Macherey-Nagel Inc. Bethlehem, PA), treated with DNase I (Ambion, Austin, TX), and quantified by spectrophotometry (Nano-Drop ND-1000, NanoDrop Technologies, DE). Samples were run on the 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA) using gene-specific primers and probes. Primers and probes used from Applied Biosystems were Rn01481376_m1 (EPO); Rn 00575519_m1 (Bad), and Rn 00437783_m1 (Bcl-xL). Primer pairs were designed for the remaining genes, as follows: Bcl-2: Forward primer, 5′- CAG GAT AAC GGA GGC TGG G-3′. Reverse primer, 5′- GAA ATC AAA CAG AGG TCG CAT G-3′. Probe, 5′- TGC CTT TGT GGA ACT ATA TGG CCC CA-3′; Bax: Forward primer, 5′- TGG TTG CCC TCT TCT ACT TTG C-3′. Reverse primer, 5′- TGA TCA GCT CGG GCA CTT TA-3′, Probe, 5′- AAC TGG TGC TCA AGG CCC TGT GCA-3′. Relative quantification of PCR products was based on value differences between the target and GAPDH control by the comparative threshold cycle method (Taqman Gold RT-PCR manual, PE Applied Biosystems).
Determination of total (Immunoblot) and Rh EPO (ELISA) levels in rat brain
We measured total EPO (endogenous rat EPO and exogenous Rh EPO) levels in rat brain by immunoblot using an antibody derived from an epitope common to both rat and human EPO. Protein samples were extracted by homogenizing ipsilateral PID1 hippocampi in RIPA buffer (150 mM NaCl, 50 mM Tris-HCl pH 8.0, 0.5% DOC, 1% NP-40, 1% SDS) with protease inhibitors (Roche Applied Science, Indianapolis, IN). Protein concentration determined by the BCA method (Pierce Protein Research Products, Rockford, IL) was used to calculate volume for equal protein loading (100 mcg/sample). Proteins were separated by SDS PAGE using Criterion XT Precast 8% Bis-Tris Gels (Bio-Rad, Hercules, CA), followed by transfer to PVDF membranes in MOPS buffer. After blocking with 5% milk in Tris-buffered saline and 0.1% Tween 20 (TBST), membranes were exposed to specific antibodies against EPO (rabbit polyclonal 1:500, sc-7956, Santa Cruz Biotechnology, Santa Cruz, CA) followed by goat anti-rabbit HRP secondary antibody (1:5,000, Cell Signaling Technology MA). Signals were detected with Western Lightning ECL (PerkinElmer Life Sciences) and quantified relative to GAPDH (rabbit monoclonal 1:4000, 14C10, Cell Signaling Technology, Danvers, MA) by densitometry on a Kodak Image Station 2000R (Eastman Kodak/SIS, Rochester, NY). The same methodology and antibody (sc-7956, Santa Cruz Biotechnology) were used to run varying dilutions of Procrit® in vehicle to determine the sensitivity of immunoblot to detect Procrit®.
Based on pharmacokinetic studies in neonatal and adult rats,37,38 we anticipated that Rh EPO would reach near-maximal levels in the brain at 6 h after the second Rh EPO injection (PID1). Therefore, PID1 tissue was used to measure rat brain Rh EPO levels using the Human EPO Immunoassay (DEP00 Quantikine® IVD®, R & D Systems, Minneapolis, MN) used in both these publications. Half the forebrain per rat was loaded because hippocampus alone did not yield detectable levels of Rh EPO using this assay. Briefly, 200 mg tissue were homogenized in 400 mcL of ice-cold lysis buffer (0.32 M sucrose, 1 mM EDTA, 5 mM Tris, 0.1 mM phenyl-methyl-sulfonyl-fluoride, 1 mM β-mercaptoethanol) using a 22G needle followed by manual homogenization with a sterile pestle. Samples were incubated for 40 min at 4°C and then centrifuged for 30 min at 13,000 g to get 300 mcL supernatant. ELISA was performed according to manufacturer's instructions. The optical density of each well was measured using the GENiosPro microplate reader (Tecan, Research Triangle Park, NC). Protein concentration determined by the BCA method was used to normalize ELISA Rh EPO results.
Caspase activity
Frozen hippocampal samples were sonicated in five times the volume of ice-cold lysis buffer (0.03 M Tris, 0.15 M NaCl) on ice and then spun at 10,000 g for 5 min at 4°C. Aliquots of 30 micrograms/50mcL were prepared, based on the protein concentration as determined by BCA, and loaded into the Caspase-Glo® 3/7 assay (Promega Corporation, Madison WI) plate. The plate was then gently shaken for 30 sec in a plate shaker, followed by incubation at RT for 1 h. Luminescence of each sample was read using GENiosPro microplate reader (Tecan). Because each plate used only samples from a given time point, results are only comparable between groups for each time point.
Novel Object Recognition testing
We used a black Plexiglas® NOR chamber (52×52×30 cm) as described for juvenile rats (P29–40)40 to test PID14 rat pups. The box was housed in a small room with soft lighting. Objects used for testing were small, plastic, and easily cleaned objects secured to the box using Velcro.® Interaction of the rat with an object was measured using automated video tracking and data analysis equipment from EthoVision® v 7.1 (Noldus Information Technology, Wageningen, NL) and confirmed by dual observer analysis of the videotaped session when needed. Exploration was timed for the duration that the rat's nose was within 2 cm of the object, as determined by a digitally drawn arena. Time spent rearing, if present, was not considered exploration. The box and objects were cleaned between rat pups with 70% alcohol to remove odor traces. During the habituation phase, each rat pup was allowed to explore the empty box individually for 15 min per day over 2 days to ensure acclimation to the testing environment. During the 5 min familiarization phase, a single rat was placed in the arena containing two objects. The rat was returned to the arena 30 min later. During the 5 min testing phase, the rat was exposed to one object from the familiarization trial and to one novel object. Performance on the testing phase was measured by the percent novel exploration time (100* (N/ (N+F)) where N is the time spent on the new object and F on the familiar. A higher exploration time is considered to reflect greater memory ability, while an exploration time of 50% is not better than chance.
Histology
Frozen brains (n=4 per group) were sliced into coronal sections from −2.52 to −3.24 Bregma.
For each animal, three serial 20 micron-thick sections from dorsal hippocampus taken 120 microns apart were placed on a single slide. Each of the three sections was taken from a comparable anatomical level as determined by The Rat Brain Atlas in Stereotaxic Coordinates.41 All slides were processed on the same day, and exposed to freshly made reagents at the same times to minimize variation in conditions between groups. Antigen retrieval was performed by equilibrating slides in antigen retrieval solution (H-3300, Vector Laboratories, UK). Nonspecific binding sites were blocked using 3% normal goat serum and 0.1% Triton X-100. Sections were incubated overnight at 4°C in a humidified chamber with two primary antibodies: anti-NeuN 1:500 (mouse monoclonal MAB377, Millipore, MA) and anti-cleaved caspase3 antibody (activated caspase 3) 1:250 (rabbit polyclonal AB3623, Millipore), diluted in 0.1% Triton with PBS. After washing, slides were then incubated with a secondary goat anti-mouse 1:250 (AlexaFluor633®, Molecular Probes, OR) and a secondary goat anti-rabbit 1:500 (AlexaFluor488®, Molecular Probes) 1:500 followed by Orange Nucleic Acid Stain 1:30,000 (SYTOX® Molecular Probes).
Hippocampal subregions were imaged using a Nikon A1 confocal microscope (Melville, NY) and 10x PLANAPO NA 0.45 Objective. Automated XY scanning and stitching of multiple fields was done using the Nikon Elements software v4.1 Scan large Image module with 10% overlap. The hippocampal subregions CA1, CA3, DG regions of interest (ROIs) were then drawn manually (as shown schematically in Fig. 8A) after thresholding the SYTOX channel to separate dense nuclear areas from the surrounding tissue. The volume of interest was thus limited to the granule cell body layer (DG) and stratum pyramidale (CA1 and CA3). Each ROI was then imported into Imaris v4.5 (Bitplane, South Windsor, CT) where confocal z-stack images were 3D-reconstructed.42,43 The Imaris software spots analysis was performed to identify individual nuclei. Spots analysis places a colored sphere at the centroid of the regions of interest from thresholding SYTOX or NeuN signal. The region target size used for spots was based on measurements of the average size of SYTOX positive labeling in NeuN positive cells (2 μM). Spots were generated using local background correction and displayed as spheres with smaller diameter than the maximum (1 μM rather than 2 μM) for clarity as shown in Figure 8B. Spheres from the NeuN signal which overlapped SYTOX were counted as NeuN-positive cells (red spheres), co-localization of activated caspase-3 and SYTOX was used to determine the number of activated caspase3-positive cells (blue spheres) and co-localization for NeuN, and activated caspase 3 (purple spheres) was used to determine the number of activated caspase3-positive neurons within each ROI. The relative number of positive cells is compared to the total number of nuclei found from the SYTOX spots analysis; for example, the ratio of NeuN+ cells to total cells was used to calculate neuron fraction.
FIG. 8.

Neuron Fraction in CA3 at PID2. (A) Schematic representation of the regions of interest (ROIs: CA1, CA3, DG) drawn for export to Imaris. (B) Representative image of colored spheres resulting from spots analysis of a CA3 ROI. Caspase+ cells are shown in this schematic as blue spheres, NeuN+as red spheres; cells positive for both markers are shown as purple spheres, for computerized counting of all spheres present in the ROI. (C) Representative image of Ipsilateral CA3 at higher magnification (60X). NeuN positive cells are shown in blue, while SYTOX positive are shown in red. CA3 neuron fraction increased in EPOCCI rats, relative to VEHCC. CA3 neuron fraction in EPOCCI rats did not differ from SHAMVEH. Scale bar: 60 μm. Color image is available online at www.liebertpub.com/neu
Statistics
All data are expressed as mean±SE and/or as percentage of control. Data were analyzed using one-way ANOVA, treating each of the three groups as an independent group. For hematocrit and cognitive testing, the three groups were EPOCCI, VEHCCI, and SHAMVEH. For histology and tissue analyses, since samples were obtained from the hemisphere ipsilateral and contralateral to injury from each rat, comparisons between the three groups were limited to samples from the same hemisphere. Thus, comparisons were made between ipsilateral hemispheres from EPOCCI, VEHCCI, and SHAMVEH or between contralateral hemispheres from EPOCCI, VEHCCI, and SHAMVEH. Post Hoc testing was done using Fisher's Protected Least Significant Difference and p<0.05 was defined as the cutoff for statistical significance, using Statview® software.
Results
Overall survival was not different between the three study groups, ranging from 97%–98.5%. Similarly, baseline weight and weight gain after injury were not different between groups. As shown in Figure 1, Rh EPO increased rat pup hematocrit after CCI, relative to vehicle, only at PID7 (40±1.2% and 35±0.6%, respectively) without attaining values typically reported for polycythemia in rats (45%–65%).44,45
Fig. 1.

Hematocrit levels in EPOCCI, VEHCCI, and SHAMVEH rat pups over time after CCI or sham surgery. The graph depicts hematocrit levels in EPOCCI (gray), VEHCCI (black), and SHAMVEH (dotted white) rat pups immediately after CCI or SHAM surgery (post injury day (PID) 0) and at PID 3, 7, and 14. EPO administration increased hematocrit only at PID7 without producing polycythemia. N=4–6/group, *p<0.05 relative to VEHCCI.
Exogenous and total EPO levels
ELISA demonstrated that Rh EPO is present in rat pup brain. Exogenous EPO (Rh EPO, Procrit®) levels were measured in each hemi-forebrain per rat (including the hippocampus) because hippocampus alone did not yield sufficient tissue to detect Rh EPO using ELISA.
Rh EPO levels in forebrain were detectable in EPOCCI, but not VEHCCI, rats. Rh EPO levels in EPOCCI rats were not significantly different between hemispheres ipsilateral and contralateral to injury (3.7±0.7 mU/mg vs. 2.4±0.7 mU/mg protein, p=0.1). Uncorrected for protein content, Rh EPO levels were 49±9.2 mU/mL and 27±3.8 mU/mL in EPOCCI ipsilateral and contralateral to injury, respectively.
Immunoblot results suggest that Rh EPO levels in rat brain were very low relative to endogenous rat EPO. Concentration of total EPO (endogenous and exogenous) in ipsilateral rat hippocampus was assayed using the anti-EPO antibody, sc-7956. This antibody detected endogenous rat EPO (36kDa) in the hippocampus (Fig. 2A) as well as Rh EPO (40kDa) in diluted Procrit ® samples (Fig. 2B). As shown in Figure 2A, the 40 kDa band representing Rh EPO was absent in all rat hippocampi (EPOCCI and VEHCCI). Data shown in Figure 2B suggest that this absent band does not mean Rh EPO is truly missing but, rather, present in low concentrations relative to rat EPO in the brain. As shown in Figure 2B, Procrit ® dilutions of 20–50 mU/mL (corresponding to the EPOCCI ELISA results) are detectable by immunoblot, but the 20 mU/ml band is faint. Correction for protein loading suggests that Rh EPO levels in the hippocampal samples used for immunoblot were actually in the 4–10 mU/ml range (arrow in Fig. 2B) which would likely be undetectable by immunoblot using standard protein loading. As shown in Figure 2A, levels of endogenous EPO (36 kDa) in the injured hippocampus were not significantly different between EPOCCI and VEHCCI rats (0.046±0.007 vs. 0.039±0.005, p=0.44).
FIG. 2.
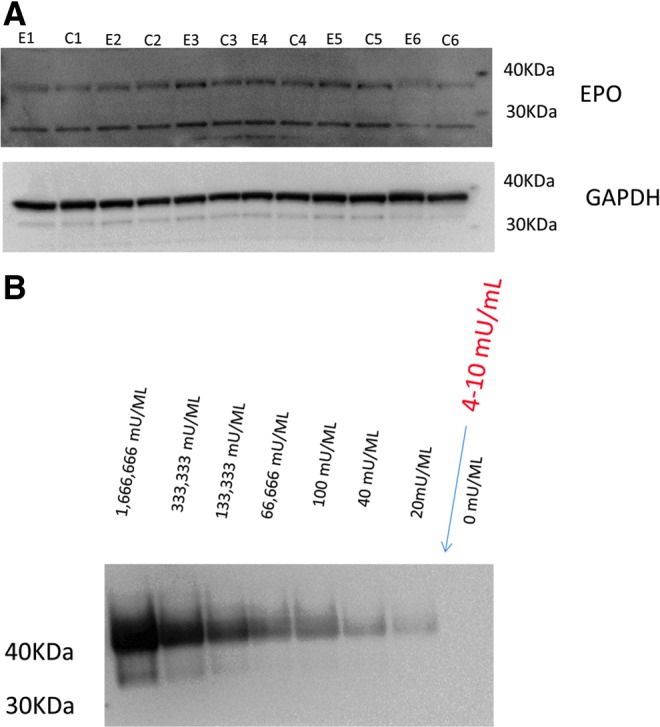
Rat hippocampus immunoblot only detected endogenous rat EPO. Immunoblot of serial Procrit® dilutions suggests that Rh EPO in rat hippocampus is very low relative to endogenous rat EPO. (A) Immunoblot of ipsilateral hippocampus from EPOCCI (E1-6) and VEHCCI (C1-6) shows two bands. EPO levels in EPOCCI did not differ from VEHCCI in either the 36 kDA or the 26 kDA band. (B) Immunoblot of serial Procrit ® dilutions revealed a band for Rh EPO at 40 kDA. As shown, bands are detectable even at dilutions as low as 20 mU/mL. Based on ELISA and BCA results, the expected concentration of Rh EPO in the hippocampal samples used for immunoblots should be in the 4–10 mU/mL range and therefore likely not detectable by this method. Therefore, it is not surprising that we did not find a 40 kDa band in addition to the 36 kDa one shown in A. Color image is available online at www.liebertpub.com/neu
mRNA results
As noted in the methods, statistical analyses were limited to three groups (EPOCCI, VEHCCI, SHAMVEH) using either left-sided (ipsilateral) or right-sided (contralateral) hippocampi. For ease of presentation, results are presented as a percent of the corresponding SHAM hippocampus. GAPDH mRNA expression did not vary between groups at any time.
EPO mRNA was lower at PID1 in EPOCCIipsi relative to VEHCCIipsi (116±14 vs. 307±111% SHAMipsi, p=0.08) and in EPOCCIcontra relative to VEHCCIcontra (64.5±18 vs. 131±36% SHAMipsi, p=0.07) though the decrease did not reach statistical significance (Fig. 3). At PID3, EPO mRNA increased in EPOCCIipsi relative to VEHCCIipsi (163±16 vs. 40±9% SHAMipsi, p<0.0001) at PID3. Neonatal and adult rat pharmacokinetic studies have shown that brain Rh EPO levels are higher in the side ipsilateral to injury, relative to contralateral. These same studies have also shown that brain Rh EPO levels remain elevated in the brain less than 24 h after systemic administration.37,38 Thus, it is possible that a rebound increase in endogenous EPO production occurred 30 h after the last dose of Procrit®, at PID3, limited to the ipsilateral hippocampus. At PID7, EPO mRNA decreased in EPOCCI hippocampi bilaterally (63±9 vs. 173±36% SHAMipsi, p=0.01 and 46±4 vs. 278±79% SHAMcon, p=0.0009), suggesting that EPO mRNA was downregulated by Procrit® administration.
FIG. 3.

Hippocampal EPO mRNA Levels in EPOCCI and VEHCCI rats obtained at post injury days (PID) 1, 2, 3, 7, and 14, presented as a percentage of corresponding Sham mRNA. Contralateral and Ipsilateral hemispheres are abbreviated as contra and ipsi, respectively. EPOCCIcontra and ipsi are shown as light gray and white bars, respectively. VEHCCI contra and ipsi are shown as dark gray and black bars, respectively. Results are presented as percent Sham mRNA±SEM, n=6–8/group. *p<0.05 relative to VEHCCI; &p<0.05 relative to SHAM. EPO mRNA decreased at PID1 in ipsilateral and contralateral EPOCCI relative to VEHCCI, but did not reach statistical significance (p=0.08 and 0.07, respectively). EPO mRNA decreased relative to VEHCCI at PID 7, suggesting that EPO mRNA was downregulated by Procrit® administration. At PID3, EPO mRNA increased in EPOCCIipsi relative to VEHCCIipsi, suggesting a rebound increase in endogenous EPO production at a time during which Procrit® should no longer be present in the brain.
Bad, Bax and Bcl2 mRNA results are shown in Figures 4–6. At PID1, pro-apoptotic Bad mRNA decreased in EPOCCI hippocampi bilaterally (88±2 vs. 113±5% SHAMcontra, p=0.03 and 96±5 vs. 111±4% SHAMipsi, p=0.01) suggesting Procrit® had an anti-apoptotic effect in bilateral hippocampi at PID1 (Fig. 4).
FIG. 4.

Hippocampal Bad mRNA Levels in EPOCCI and VEHCCI rats obtained at post injury days (PID) 1, 2, 3, 7, and 14, presented as a percentage of corresponding Sham mRNA. Contralateral and Ipsilateral hemispheres are abbreviated as contra and ipsi, respectively. EPOCCIcontra and ipsi are shown as light gray and white bars, respectively. VEHCCI contra and ipsi are shown as dark gray and black bars, respectively. Results are presented as percent Sham mRNA±SEM, n=6–8/group. *p<0.05 relative to VEHCCI; &p<0.05 relative to SHAM. Bad mRNA decreased relative to VEHCCI at PID1 and 7 in bilateral hippocampi, suggesting anti-apoptotic effects after Procrit® administration. At PID3, Bad mRNA increased in EPOCCIcontra relative to VECHCCIcontra, suggesting a rebound increase in Bad mRNA at a time during which Procrit® should no longer be present in the brain.
FIG. 5.

Hippocampal Bax mRNA Levels in EPOCCI and VEHCCI rats obtained at post injury days (PID) 1, 2, 3, 7, and 14, presented as a percentage of corresponding Sham mRNA. Contralateral and Ipsilateral hemispheres are abbreviated as contra and ipsi, respectively. EPOCCIcontra and ipsi are shown as light gray and white bars, respectively. VEHCCI contra and ipsi are shown as dark gray and black bars, respectively. Results are presented as percent Sham mRNA±SEM, n=6–8/group. *p<0.05 relative to VEHCCI; &p<0.05 relative to SHAM. Bax mRNA decreased relative to VEHCCI at PID 2 (in contralateral hippocampi) and 7 (ipsilateral hippocampi), but increased relative to VEHCCI in contralateral hippocampi at PID3 and 7.
FIG. 6.

Hippocampal Bcl2 mRNA Levels in EPOCCI and VEHCCI rats obtained at post injury days (PID) 1, 2, 3, 7, and 14, presented as a percentage of corresponding Sham mRNA. Contralateral and Ipsilateral hemispheres are abbreviated as contra and ipsi, respectively. EPOCCIcontra and ipsi are shown as light gray and white bars, respectively. VEHCCI contra and ipsi are shown as dark gray and black bars, respectively. Results are presented as percent Sham mRNA±SEM, n=6–8/group. *p<0.05 relative to VEHCCI; &p<0.05 relative to SHAM. Bcl2 mRNA increased relative to VEHCCI at PID2 (in contralateral hippocampi) but decreased relative to VEHCCI in both hippocampi at PID3.
The most dramatic changes in apoptotic factor mRNA levels occurred at PID2, in Bax and Bcl2 mRNA. EPOCCI Bax mRNA decreased to about half of VEHCCI in contralateral hippocampi (100±12.5 vs. 200±12% SHAMcontra, p=0.003), as shown in Figure 5. EPOCCI Bcl2 mRNA increased by about half of VEHCCI in contralateral hippocampi (193±13 vs. 127±27% SHAMcontra, p=0.02), as shown in Figure 6. PID2 Bad mRNA did not differ between groups. Presented as ratios, the Bax/Bcl2 and Bad/Bcl2 ratios decreased at PID2 in contralateral hippocampi (0.3±0.02 vs. 0.9±0.1, p<0.0001) and (3.5±0.5 vs. 6.9±0.2, p=0.0003) suggesting Rh EPO had anti-apoptotic effects in contralateral hippocampi at PID2.
However, at PID 3, apoptotic factor mRNA levels suggested a pro-apoptotic effect in EPOCCI hippocampi. In contralateral hippocampi, Bad mRNA increased at PID3 (108±2 vs. 88±6% SHAMcontra, p=0.005) and Bax mRNA increased at PID3 (128±3 vs. 99±9% SHAMcontra, p<0.0001) relative to VEHCCI (Figs. 4 and 5). In both hippocampi, Bcl2 mRNA decreased (95±4 vs. 124±7% SHAMipsi, p=0.001 and 87±2 vs. 106±1% SHAMcontra, p<0.001) relative to VEHCCI (Fig. 6). Presented as ratios, in both hippocampi the Bax/Bcl2 ratio (4.3±0.2 vs. 2.6±0.2, p<0.0001 contralateral, and 4.5±0.5 vs. 3.4±0.2, p=0.02 ipsilateral) and Bad/Bcl2 ratio (17.7±0.8 vs. 11.4±1.2, p=0.0004 contralateral, and 20±2 vs. 15±0.9, p=0.03 ipsilateral) increased relative to VEHCCI, suggesting a pro-apoptotic milieu in EPOCCI rat hippocampi at PID3.
At PID7, pro-apoptotic Bad mRNA decreased by about half at PID7 in both EPOCCI hippocampi (46±2 vs. 86±5% SHAMcontra and 36±2 vs. 98±4% SHAMipsi, p<0.0001 for both) (Fig. 4). Accordingly, the Bad/Bcl2 ratio decreased in EPOCCI relative to VEHCCI in both hippocampi (6.1±0.5 vs. 11.7±0.8 in contralateral and 4.6±0.4 vs. 13.5±0.8 in ipsilateral, p<0.0001 for both) at PID7. Pro-apoptotic Bax mRNA changed slightly at PID7, in different directions: increased in contralateral (79±4 vs. 68±2%, p=0.03) and decreased in ipsilateral hippocampi (71±4 vs. 84±3%, p=0.03) (Fig. 5). Apoptotic factor mRNA levels were not significantly different between CCI groups at PID14. Bcl-xL mRNA levels were not significantly different between CCI groups at any time point.
Caspase activity
Results are only comparable between groups for each time point, as described in the methods, because each plate contained only samples from the same time point. Statistical analyses were done between hippocampi from the same side relative to injury, as described in the methods. For simplicity, graphical results are presented as % SHAM values (Fig. 7).
FIG. 7.

Caspase activity in hippocampi ipsilateral to injury at PID1, 2, and 3 after CCI. EPOCCI is shown in gray bars and VEHCCI in black bars. Data are presented as relative luminescence units normalized to SHAMVEH±SEM, n=6–7/group. *p<0.05 relative to EPOCCI; &p<0.05 relative to SHAMVEH. EPO blunted the increase in caspase activity on PID1, but not at PID2. By PID3, caspase activity in both CCI groups was not different from SHAMVEH.
Caspase activity was measured in PID1-3 hippocampi because the most marked mRNA changes occurred at PID2. Additional time points beyond PID3 were not measured because, as shown below, caspase activity in both CCI groups decreased to SHAM levels by PID3.
Rh EPO administration decreased caspase activity in ipsilateral hippocampi at PID1 after CCI relative to VEHCCI (22,980±3000 vs. 35,350±3800 luminescence units in EPOCCI and VEHCCI, respectively, p=0.009) and abrogated the effects of CCI relative to SHAM (22,980±3000 and 19,570±1100 luminescence units in EPOCCI and SHAMVEH, respectively, p=0.44). At PID2, Rh EPO no longer blunted caspase activity as it increased in both EPOCCI and VEHCCI ipsilateral hippocampi relative to SHAMVEH (96,980±17,000 and 90,000±6000 vs. 51,300±5000 luminescence units, respectively, p=0.01 and p=0.03, respectively, relative to the corresponding SHAMVEH hippocampus). At PID3, caspase activity in both groups had normalized relative to SHAM. Caspase activity did not differ between groups at any time in contralateral hippocampi.
NOR results
Higher novel exploration time represents better memory, while scores of 50% are no better than those achieved by chance alone. Rh EPO improved recognition memory in rat pups after CCI. EPOCCI rats scored higher than VEHCCI rats (66.4±5 vs. 44.2±5%, p=0.01). In contrast, VEHCCI rats explored the novel object less than 50% of the time and less than SHAMVEH (44±5 vs. 62±5%, p=0.03) at PID14.
Histology
Histological analysis was performed at PID2 to determine if Rh EPO treatment was associated with increased hippocampal neuron fraction following decreased hippocampal caspase activity in EPOCCI rats at PID1. Further, this time point was chosen to detect any hippocampal subregion differences in caspase activation between CCI groups, as these would not necessarily be detected by measuring total hippocampal caspase activity. There were no differences in total cell counts (as determined by nuclear staining) or volumes between groups in any hippocampal subregion.
At PID2, neuron fraction increased in EPOCCI relative to VEHCCI rats (0.94±0.02 vs. 0.82±0.04, p=0.02) resulting in levels which did not differ from SHAM (0.94±0.02 vs. 0.95±0.01) in the ipsilateral CA3 hippocampal subregion. Ipsilateral CA3 volumes for EPOCCI, VEHCCI, and SHAMVEH did not differ (315,000±22,000, 351,000±57,000, and 342,000±8,542 μ3). Representative images of ipsilateral CA3 are shown in Figure 8C. While it did not reach statistical significance, neuron fraction also appeared to increase in the ipsilateral CA1 subregion relative to VEHCCI (0.97±0.04 vs. 0.9±0.02), p=0.1. Neuron fraction did not differ for any contralateral region or for DG.
Rh EPO did not blunt the increase in caspase 3-positive cells in CCI groups relative to SHAM at PID2. CCI increased the fraction of caspase 3-positive cells relative to SHAM in ipsilateral CA1 (0.36±0.09 and 0.34±0.05 vs. 0.071±0.04, p=0.02 for each CCI group relative to SHAM) and in ipsilateral DG (0.19±0.04 and 0.18±0.03 vs, 0.05±0.02, p<0.05 for each CCI group relative to SHAM). CCI increased caspase 3-positive cell fraction relative to SHAM in contralateral CA3 (0.22±0.03 and 0.22±0.05 vs. 0.06±0.03, p<0.05 for each CCI group relative to SHAM), but CCI had no effect on caspase 3-positive cell fraction in the ipsilateral CA3. Representative images of contralateral CA3 are shown in Figure 9.
FIG. 9.
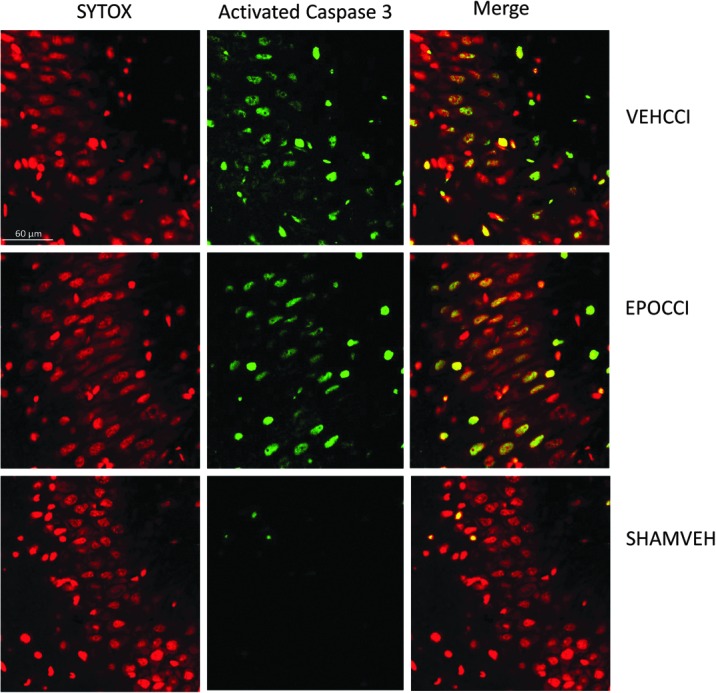
Caspase+cell fraction in CA3 at PID2. Representative image of contralateral CA3 at higher magnification (60X). Caspase positive cells are shown in green, while SYTOX positive are shown in red. At PID2, CA3 caspase+cell fraction increased in EPOCCI and VEHCCI rats relative to SHAM in contralateral CA3, not blunted by Rh EPO. Scale bar: 60 μm. Color image is available online at www.liebertpub.com/neu
Caspase3-positive cell fraction did not differ between EPOCCI and VEHCCI in any subregion. In contralateral DG, caspase 3-positive cell fraction increased in EPOCCI relative to SHAMVEH (0.19±0.05 vs. 0.06±0.04, p=0.04) but was not different from VEHCCI (0.19±0.05 vs. 0.14±0.02, p=0.4). In contralateral CA1, a reverse trend was seen in which caspase 3-positive cell fraction increased (without reaching statistical significance) in VEHCCI relative to SHAM (0.28±0.04 vs. 0.14±0.08, p=0.07). Again, EPOCCI was not different from VEHCCI (0.2±0.03 vs. 0.28±0.04, p=0.3)
While CCI did not increase the fraction of caspase-3+/NeuN+ cells in any hippocampal subregion in a statistically significant fashion, both CCI groups showed trends towards increased caspase-3+/NeuN+fraction (i.e., in ipsilateral CA1, CA3, and DG, p values were 0.07/0.08, 0.1/0.2, and 0.1/0.2, respectively, vs. SHAMVEH).
Discussion
We found that Rh EPO improved cognitive outcome, decreased caspase activity, and increased hippocampal neuronal fraction in rat pups after TBI. To our knowledge, this is the first reported study of Rh EPO administration in a developmental model of TBI.
Rh EPO was well tolerated in the 17-day-old rat pup after CCI. It did not adversely affect weight gain or survival nor did it produce polycythemia. The latter is particularly important since large doses (5000 u/kg) of Rh EPO were needed to attain measurable levels in the brain. Albeit relatively low levels of exogenous relative to endogenous EPO in the brain, the timing of Rh EPO administration coincided with a decrease in hippocampal EPO mRNA levels, suggesting that exogenous EPO affected hippocampal gene expression.
Recognition memory is an important function of the hippocampus and perirhinal cortex.40,46,47 TBI impairs recognition memory in adult48 and pediatric patients,49 as well as in immature rodents.50 Rh EPO normalized recognition memory, measured by performance on the NOR task, after CCI. The effects of Rh EPO on NOR performance persisted to day 60 after CCI in our model (unpublished data). Our results are consistent with studies in adult rats showing that Rh EPO improved cognitive function.12,14
Rh EPO decreased Bad mRNA in bilateral hippocampi at PID1, the hippocampal Bax/Bcl2 and Bad/Bcl2 ratios at PID2 (contralaterally) and the hippocampal Bad/Bcl2 ratio at PID7 (bilaterally) after CCI. These results are in keeping with adult rat studies showing that Rh EPO decreased the brain Bax/Bcl2 ratio for the first 5 days after TBI.24,51 In a given tissue, the ratio of pro-apoptotic Bax or Bad, and anti-apoptotic Bcl2, is important in determining the direction of apoptotic forces.52–54 The intrinsic pathway, thought to be the predominant form of apoptotic death in neuronal cells, is regulated by the Bcl2 family and culminates in activation of the executioner caspase 3.35 Most of the mRNA data supported anti-apoptotic effects on the days of Rh EPO administration, followed by a rebound pro-apoptotic effect on PID3. A discrepancy in this pattern is found in the opposing results at PID7 for Bax mRNA. While Bax mRNA increased in contralateral hippocampi, Bax mRNA decreased in ipsilateral hippocampi at PID7. These results are of uncertain significance since, in contrast to the rest of the mRNA data, the changes are small and not mirrored by the Bad or Bcl2 results.
Hippocampal caspase activity increased for the first 2 days after CCI, normalizing to SHAM levels on PID3. Similarly, Rh EPO decreased caspase 3 activation in the adult rat brain after TBI.12 Rh EPO decreased hippocampal caspase activity at PID1, but not at PID2. In light of pharmacokinetic studies in neonatal and adult rats, it is most likely that Rh EPO failed to blunt caspase activity at PID2 because brain Rh EPO levels were lower than on PID1. Based on mRNA data, however, we had expected to find decreased caspase activity at PID2 as well as at PID1. There are several potential reasons for this apparent discordance. For one, it is possible that decreased caspase activity at PID1 resulted from decreased Bad at PID1 alone or in combination with changes in other, unmeasured, pro-apoptotic factors at that time. We did not measure all Bcl-2 family members (i.e., Bid, Bak, CED-9) but instead selected those altered by EPO and/or TBI in previous studies. The unchanged caspase activity at PID2 may have resulted from caspase 3-activating mechanisms overwhelming Rh EPO effects on the Bcl2 factors measured. For example, activation of caspase-8 via the extrinsic (Fas) pathway can directly activate caspase 3.55 To our knowledge, effects of EPO on caspase 8 have not been studied.
While Rh EPO did not affect hippocampal caspase activity at PID2, Rh EPO was associated with increased hippocampal subregion neuronal fraction at this time. We speculate that decreased caspase-dependent apoptosis on PID1 increased hippocampal neuronal fraction at PID2. Alternatively, Rh EPO could have increased neuronal fraction at PID2 via activation of non-apoptotic pathways. EPO is a pleiotropic cytokine that decreases neuronal injury from inflammation56 and excitotoxicity.57 In addition, EPO promotes neural stem cell proliferation and differentiation.20,28
Our data suggest that the amount of exogenous EPO in the rat pup hippocampus after CCI is low relative to endogenous EPO. To our knowledge, ours is the first attempt to directly compare endogenous versus exogenous EPO in the brain. Our results are not unexpected, given other reports that only a small amount of administered EPO actually enters the brain.20,38 However, since administered EPO appears to target neurons preferentially,36 it appears that Rh EPO can exert neuronal protection even when present in small amounts.
Rh EPO has the potential to be a useful therapy in pediatric TBI. For one, it is commonly used in clinical settings. Rh EPO is used to increase hematopoiesis in neonates and in children, albeit at lower doses than what would be used for neuroprotection. However, high dose-Rh EPO in neonates produced serum concentrations shown to be neuroprotective in experimental animals and was well tolerated.58,59 Second, Rh EPO has pre-clinical efficacy in TBI. In our model of pediatric TBI, Rh EPO improved outcome without overt adverse effects. In adult experimental traumatic brain injury, Rh EPO improved functional and/or histologic outcomes.12–14,60,61 Finally, Rh EPO improved neurologic outcome in multiple animal studies of neonatal hypoxic-ischemic brain injury.58,62,63
Our study has a number of limitations. Our findings are limited to 17-day-old male rat pups. While no animal model can replicate the human condition, the results of our study provide useful information for further work in EPO and experimental pediatric TBI. We chose to focus on 17-day-old rat pups to model the infant/toddler age group, a group at high risk for poor cognitive outcomes after TBI.64,65 We did not address various other pathways for EPO neuroprotection. We focused on apoptosis, as this is one of the important ways in which the response of the developing brain to trauma differs from that of the adult. In addition, our results are limited to a moderate injury. We chose to use a 1.5 mm depression, rather than 2 mm, to enable histologic analysis by avoiding the near-total loss of hippocampal tissue we had observed with more severe injury. Finally, our histologic analysis at PID2 did not include glial cell population fraction. We acknowledge that Rh EPO could be associated with both decreased gliosis and increased neuronal fraction in the ipsilateral CA3 hippocampi at PID2, since we found no differences in the denominator used (hippocampal subregion total cell counts) or in hippocampal subregion volumes. We plan to assess glial fraction at later time points after CCI because EPO has variable effects on microglial and astrocytic populations at early time points after injury. EPO clearly decreases gliosis weeks after experimental injuries, including TBI.66–69 Within hours to days after injury, however EPO decreased reactive astrocytes70 and microglia71 in some models but increased astrocyte and microglial survival in others.72–74,75
In summary, Rh EPO administration improved cognitive outcome and hippocampal neuronal fraction after CCI in rat pups, associated with decreased caspase activation in the hippocampus. We speculate that Rh EPO improved cognitive outcome in rat pups after CCI as a result of improved neuronal survival via inhibition of caspase-dependent apoptosis early after injury. Our data suggest that Rh EPO, even at seemingly low levels in the brain, exerts significant neuroprotection in the rat pup after CCI. We are currently conducting additional studies to determine more long-term effects of Rh EPO after CCI. EPO can be modified (asialoEPO, neuroEPO, and carbamylated EPO) to achieve greater brain penetration.76 Thus, modified EPO may show larger treatment effects after developmental TBI. Our findings suggest that further study to assess the effects of modified EPO on cognitive function after developmental TBI is warranted.
Acknowledgments
Statement of Financial Support: funding for this study was provided by the CHRCDA (NIH K12HD001410) and from the Divisions of Neonatology and Pediatric Critical Care Medicine, Department of Pediatrics, University of Utah, Salt Lake City, Utah. Input on histology from Scott Rogers, PhD1 is greatly appreciated.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Langlois JA, Rutland-Brown W, and Thomas KE. (2005). The incidence of traumatic brain injury among children in the United States: Differences by race. J Head Trauma Rehabil 20:229–238 [DOI] [PubMed] [Google Scholar]
- 2.Langlois JA, Rutland-Brown W, and Wald MM. (2006). The epidemiology and impact of traumatic brain injury: A brief overview. J Head Trauma Rehabil 21:375–378 [DOI] [PubMed] [Google Scholar]
- 3.Yeates KO, Taylor HG, Wade SL, Drotar D, Stancin T, and Minich N. (2002). A prospective study of short- and long-term neuropsychological outcomes after traumatic brain injury in children. Neuropsychology 16:514–523 [DOI] [PubMed] [Google Scholar]
- 4.Yeates KO, Armstrong K, Janusz J, Taylor HG, Wade S, Stancin T, and Drotar D. (2005). Long-term attention problems in children with traumatic brain injury. J Am Acad Child Adolesc Psychiatry 44:574–584 [DOI] [PubMed] [Google Scholar]
- 5.Catroppa C, and Anderson V. (2007). Recovery in memory function, and its relationship to academic success, at 24 months following pediatric TBI. Child Neuropsychology 13:240–261 [DOI] [PubMed] [Google Scholar]
- 6.Anderson V, Godfrey C, Rosenfeld JV, and Catroppa C. (2012). 10 years outcome from childhood traumatic brain injury. Int J Dev Neurosci 30:217–224 [DOI] [PubMed] [Google Scholar]
- 7.Anderson VA, Catroppa C, Haritou F, Morse S, and Rosenfeld JV. (2005). Identifying factors contributing to child and family outcome 30 months after traumatic brain injury in children. J Neurol Neurosurg Psychiatry 76:401–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slomine BS, McCarthy ML, Ding R, et al. (2006). Health care utilization and needs after pediatric traumatic brain injury. Pediatrics 117:e663–674 [DOI] [PubMed] [Google Scholar]
- 9.Aitken ME, McCarthy ML, Slomine BS, et al. (2009). Family burden after traumatic brain injury in children. Pediatrics 123:199–206 [DOI] [PubMed] [Google Scholar]
- 10.Mammis A, McIntosh TK, and Maniker AH. (2009). Erythropoietin as a neuroprotective agent in traumatic brain injury Review. Surg Neurol 71:527–531 [DOI] [PubMed] [Google Scholar]
- 11.Dame C, Juul SE, and Christensen RD. (2001). The biology of erythropoietin in the central nervous system and its neurotrophic and neuroprotective potential. Biol Neonate 79:228–235 [DOI] [PubMed] [Google Scholar]
- 12.Yatsiv I, Grigoriadis N, Simeonidou C, et al. (2005). Erythropoietin is neuroprotective, improves functional recovery, and reduces neuronal apoptosis and inflammation in a rodent model of experimental closed head injury. FASEB J 19:1701–1703 [DOI] [PubMed] [Google Scholar]
- 13.Grasso G, Sfacteria A, Meli F, Fodale V, Buemi M, and Iacopino DG. (2007). Neuroprotection by erythropoietin administration after experimental traumatic brain injury. Brain Res 1182:99–105 [DOI] [PubMed] [Google Scholar]
- 14.Mahmood A, Lu D, Qu C, et al. (2007). Treatment of traumatic brain injury in rats with erythropoietin and carbamylated erythropoietin. J Neurosurg 107:392–397 [DOI] [PubMed] [Google Scholar]
- 15.Zhu L, Wang HD, Yu XG, et al. (2009). Erythropoietin prevents zinc accumulation and neuronal death after traumatic brain injury in rat hippocampus: In vitro and in vivo studies. Brain Res 1289:96–105 [DOI] [PubMed] [Google Scholar]
- 16.Polster BM, Robertson CL, Bucci CJ, Suzuki M, and Fiskum G. (2003). Postnatal brain development and neural cell differentiation modulate mitochondrial Bax and BH3 peptide-induced cytochrome c release. Cell Death Differ 10:365–370 [DOI] [PubMed] [Google Scholar]
- 17.Rice D, and Barone S., Jr. (2000). Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environ Health Perspect 108:511–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Digicaylioglu M, Bichet S, Marti HH, Wenger RH, Rivas LA, Bauer C, and Gassmann M. (1995). Localization of specific erythropoietin binding sites in defined areas of the mouse brain. Proc Natl Acad Sci USA 92:3717–3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Juul SE, Yachnis AT, Rojiani AM, and Christensen RD. (1999). Immunohistochemical localization of erythropoietin and its receptor in the developing human brain. Pediatr Dev Pathol 2:148–158 [DOI] [PubMed] [Google Scholar]
- 20.Noguchi CT, Asavaritikrai P, Teng R, and Jia Y. (2007). Role of erythropoietin in the brain. Crit Rev Oncol Hematol 64:159–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakanaka M, Wen TC, Matsuda S, Masuda S, Morishita E, Nagao M, and Sasaki R. (1998). In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Natl Acad Sci USA 95:4635–4640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shein NA, Grigoriadis N, Alexandrovich AG, et al. (2008). Differential neuroprotective properties of endogenous and exogenous erythropoietin in a mouse model of traumatic brain injury. J Neurotrauma 25:112–123 [DOI] [PubMed] [Google Scholar]
- 23.Liao ZB, Zhi XG, Shi QH, and He ZH. (2008). Recombinant human erythropoietin administration protects cortical neurons from traumatic brain injury in rats. Eur J Neurol 15:140–149 [DOI] [PubMed] [Google Scholar]
- 24.Liao ZB, Jiang GY, Tang ZH, et al. (2009). Erythropoietin can promote survival of cerebral cells by downregulating Bax gene after traumatic brain injury in rats. Neurol India 57:722–728 [DOI] [PubMed] [Google Scholar]
- 25.Xiong Y, Mahmood A, Qu C, et al. (2010). Erythropoietin improves histological and functional outcomes following traumatic brain injury in mice in the absence of the neural erythropoietin receptor. J Neurotrauma 27:205–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maiese K, Li F, and Chong ZZ. (2004). Erythropoietin in the brain: Can the promise to protect be fulfilled? Trends Pharmacol Sci 25:577–583 [DOI] [PubMed] [Google Scholar]
- 27.Chong ZZ, Kang JQ, and Maiese K. (2003). Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways. Br J Pharmacol 138:1107–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pavlica S, Milosevic J, Keller M, et al. (2012). Erythropoietin enhances cell proliferation and survival of human fetal neuronal progenitors in normoxia. Brain Res 1452:18–28 [DOI] [PubMed] [Google Scholar]
- 29.Xenocostas A, Cheung WK, Farrell F, et al. (2005). The pharmacokinetics of erythropoietin in the cerebrospinal fluid after intravenous administration of recombinant human erythropoietin. Eur J Clin Pharmacol 61:189–195 [DOI] [PubMed] [Google Scholar]
- 30.Juul SE, McPherson RJ, Farrell FX, Jolliffe L, Ness DJ, and Gleason CA. (2004). Erytropoietin concentrations in cerebrospinal fluid of nonhuman primates and fetal sheep following high-dose recombinant erythropoietin. Biol Neonate 85:138–144 [DOI] [PubMed] [Google Scholar]
- 31.Schober ME, Block B, Beachy JC, Statler KD, Giza CC, and Lane RH. (2010). Early and sustained increase in the expression of hippocampal IGF-1, but not EPO, in a developmental rodent model of traumatic brain injury. J Neurotrauma 27:2011–2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Antunes M, and Biala G. (2012). The novel object recognition memory: Neurobiology, test procedure, and its modifications. Cognit Process 13:93–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bittigau P, Sifringer M, Pohl D, et al. (1999). Apoptotic neurodegeneration following trauma is markedly enhanced in the immature brain. Ann Neurol 45:724–735 [DOI] [PubMed] [Google Scholar]
- 34.Pohl D, Bittigau P, Ishimaru MJ, et al. (1999). N-Methyl-D-aspartate antagonists and apoptotic cell death triggered by head trauma in developing rat brain. Proc Natl Acad Sci USA 96:2508–2513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.D'Amelio M, Cavallucci V, and Cecconi F. (2010). Neuronal caspase-3 signaling: Not only cell death. Cell Death Differ 17:1104–1114 [DOI] [PubMed] [Google Scholar]
- 36.Brines ML, Ghezzi P, Keenan S, et al. (2000). Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci USA 97:10526–10531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Statler PA, McPherson RJ, Bauer LA, Kellert BA, and Juul SE. (2007). Pharmacokinetics of high-dose recombinant erythropoietin in plasma and brain of neonatal rats. Pediatr Res 61:671–675 [DOI] [PubMed] [Google Scholar]
- 38.Lieutaud T, Andrews PJ, Rhodes JK, and Williamson R. (2008). Characterization of the pharmacokinetics of human recombinant erythropoietin in blood and brain when administered immediately after lateral fluid percussion brain injury and its pharmacodynamic effects on IL-1beta and MIP-2 in rats. J Neurotrauma 25:1179–1185 [DOI] [PubMed] [Google Scholar]
- 39.Gonzalez FF, Abel R, Almli CR, Mu D, Wendland M, and Ferriero DM. (2009). Erythropoietin sustains cognitive function and brain volume after neonatal stroke. Develop Neurosci 31:403–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reger ML, Hovda DA, and Giza CC. (2009). Ontogeny of rat recognition memory measured by the novel object recognition task. Develop Psychobiol 51:672–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paxinos G, and Charles W. (2005). The Rat Brain in Stereotaxic Coordinates [Google Scholar]
- 42.Marvizon JC, Perez OA, Song B, Chen W, Bunnett NW. Grady EF, and Todd AJ. (2007). Calcitonin receptor-like receptor and receptor activity modifying protein 1 in the rat dorsal horn: Localization in glutamatergic presynaptic terminals containing opioids and adrenergic alpha2C receptors. Neuroscience 148:250–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pierce DR, Hayar A, Williams DK, and Light KE. (2011). Olivary climbing fiber alterations in PN40 rat cerebellum following postnatal ethanol exposure. Brain Res 1378:54–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swigart RH. (1965). Polycythemia and right ventricular hypertrophy. Circ Res 17:30–38 [DOI] [PubMed] [Google Scholar]
- 45.Rakusan K, Cicutti N, and Kolar F. (2001). Cardiac function, microvascular structure, and capillary hematocrit in hearts of polycythemic rats. Am J Physiol Heart Circ Physiol 281:H2425–2431 [DOI] [PubMed] [Google Scholar]
- 46.Broadbent NJ, Gaskin S, Squire LR, and Clark RE. (2010). Object recognition memory and the rodent hippocampus. Learn Mem 17:5–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scott HL, Tamagnini F, Narduzzo KE, et al. (2012). MicroRNA-132 regulates recognition memory and synaptic plasticity in the perirhinal cortex. Eur J Neurosci 36:2941–2948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McBride J, Zhao X, Nichols T, et al. (2013). Scalp EEG-based discrimination of cognitive deficits after traumatic brain injury using event-related Tsallis entropy analysis. IEEE Trans Biomed Eng 60:90–96 [DOI] [PubMed] [Google Scholar]
- 49.Mandalis A, Kinsella G, Ong B, and Anderson V. (2007). Working memory and new learning following pediatric traumatic brain injury. Dev Neuropsychol 32:683–701 [DOI] [PubMed] [Google Scholar]
- 50.Scafidi S, Racz J, Hazelton J, McKenna MC, and Fiskum G. (2010). Neuroprotection by acetyl-L-carnitine after traumatic injury to the immature rat brain. Develop Neurosci 32:480–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li YX, Ding SJ, Xiao L, Guo W, and Zhan Q. (2008). Desferoxamine preconditioning protects against cerebral ischemia in rats by inducing expressions of hypoxia inducible factor 1 alpha and erythropoietin. Neurosci Bull 24:89–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Agarwal MK, Agarwal ML, Athar M, and Gupta S. (2004). Tocotrienol-rich fraction of palm oil activates p53, modulates Bax/Bcl2 ratio and induces apoptosis independent of cell cycle association. Cell Cycle 3:205–211 [DOI] [PubMed] [Google Scholar]
- 53.Ammar HI, Saba S, Ammar RI, Elsayed LA, Ghaly WB, and Dhingra S. (2011). Erythropoietin protects against doxorubicin-induced heart failure. Am J Physiol Heart Circ Physiol 301:H2413–2421 [DOI] [PubMed] [Google Scholar]
- 54.Lowthert L, Leffert J, Lin A, et al. (2012). Increased ratio of anti-apoptotic to pro-apoptotic Bcl2 gene-family members in lithium-responders one month after treatment initiation. Biol Mood Anxiety Dis 2:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Srinivasan A, Li F, Wong A, et al. (1998). Bcl-xL functions downstream of caspase-8 to inhibit Fas- and tumor necrosis factor receptor 1-induced apoptosis of MCF7 breast carcinoma cells. J Biol Chem 273:4523–4529 [DOI] [PubMed] [Google Scholar]
- 56.Chen G, Shi JX, Hang CH, Xie W, Liu J, and Liu X. (2007). Inhibitory effect on cerebral inflammatory agents that accompany traumatic brain injury in a rat model: A potential neuroprotective mechanism of recombinant human erythropoietin (rhEPO). Neurosci Lett 425:177–182 [DOI] [PubMed] [Google Scholar]
- 57.Dzietko M, Felderhoff-Mueser U, Sifringer M, et al. (2004). Erythropoietin protects the developing brain against N-methyl-D-aspartate receptor antagonist neurotoxicity. Neurobiol Dis 15:177–187 [DOI] [PubMed] [Google Scholar]
- 58.Juul SE, McPherson RJ, Bauer LA, Ledbetter KJ, Gleason CA, and Mayock DE. (2008). A phase I/II trial of high-dose erythropoietin in extremely low birth weight infants: pharmacokinetics and safety. Pediatrics 122:383–391 [DOI] [PubMed] [Google Scholar]
- 59.Wu YW, Bauer LA, Ballard RA, et al. (2012). Erythropoietin for neuroprotection in neonatal encephalopathy: Safety and pharmacokinetics. Pediatrics 130:683–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Verdonck O, Lahrech H, Francony G, et al. (2007). Erythropoietin protects from post-traumatic edema in the rat brain. J Cereb Blood Flow Metab 27:1369–1376 [DOI] [PubMed] [Google Scholar]
- 61.Xiong Y, Lu D, Qu C, Goussev A, Schallert T, Mahmood A, and Chopp M. (2008). Effects of erythropoietin on reducing brain damage and improving functional outcome after traumatic brain injury in mice. J Neurosurg 109:510–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sola A, Wen TC, Hamrick SE, and Ferriero DM. (2005). Potential for protection and repair following injury to the developing brain: A role for erythropoietin? Pediatr Res 57:110R–117R [DOI] [PubMed] [Google Scholar]
- 63.Kim SS, Lee KH, Sung DK, et al. (2008). Erythropoietin attenuates brain injury, subventricular zone expansion, and sensorimotor deficits in hypoxic-ischemic neonatal rats. J Korean Med Sci 23:484–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bittigau P, Sifringer M, Felderhoff-Mueser U, and Ikonomidou C. (2004). Apoptotic neurodegeneration in the context of traumatic injury to the developing brain. Exp Toxicol Pathol 56:83–89 [DOI] [PubMed] [Google Scholar]
- 65.Anderson V, Catroppa C, Morse S, Haritou F, and Rosenfeld J. (2005). Functional plasticity or vulnerability after early brain injury? Pediatrics 116:1374–1382 [DOI] [PubMed] [Google Scholar]
- 66.Hagemeyer N, Boretius S, Ott C, et al. (2012). Erythropoietin attenuates neurological and histological consequences of toxic demyelination in mice. Mol Med 18:628–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gonzalez FF, McQuillen P, Mu D, Chang Y, Wendland M, Vexler Z, and Ferriero DM. (2007). Erythropoietin enhances long-term neuroprotection and neurogenesis in neonatal stroke. Developmental Neuroscience 29:321–330 [DOI] [PubMed] [Google Scholar]
- 68.Zhang Y, Chopp M, Mahmood A, Meng Y, Qu C, and Xiong Y. (2012). Impact of inhibition of erythropoietin treatment-mediated neurogenesis in the dentate gyrus of the hippocampus on restoration of spatial learning after traumatic brain injury. Exp Neurol 235:336–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Y, Zhang ZG, Rhodes K, et al. (2007). Post-ischemic treatment with erythropoietin or carbamylated erythropoietin reduces infarction and improves neurological outcome in a rat model of focal cerebral ischemia. Br J Pharmacol 151:1377–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li Y, Ogle ME, Wallace GC, Lu ZY, Yu SP, and Wei L. (2008). Erythropoietin attenuates intracerebral hemorrhage by diminishing matrix metalloproteinases and maintaining blood-brain barrier integrity in mice. Acta Neurochir Suppl 105:105–112 [DOI] [PubMed] [Google Scholar]
- 71.Sargin D, Hassouna I, Sperling S, Siren AL, and Ehrenreich H. (2009). Uncoupling of neurodegeneration and gliosis in a murine model of juvenile cortical lesion. Glia 57:693–702 [DOI] [PubMed] [Google Scholar]
- 72.Tang Z, Sun X, Huo G, Xie Y, Shi Q, Chen S, Wang X, and Liao Z. (2013). Protective effects of erythropoietin on astrocytic swelling after oxygen-glucose deprivation and reoxygenation: Mediation through AQP4 expression and MAPK pathway. Neuropharmacology 67:8–15 [DOI] [PubMed] [Google Scholar]
- 73.Yamada M, Burke C, Colditz P, Johnson DW, and Gobe GC. (2011). Erythropoietin protects against apoptosis and increases expression of non-neuronal cell markers in the hypoxia-injured developing brain. Journal of Pathology 224:101–109 [DOI] [PubMed] [Google Scholar]
- 74.Liu J, Narasimhan P, Song YS, Nishi T, Yu F, Lee YS, and Chan PH. (2006). Epo protects SOD2-deficient mouse astrocytes from damage by oxidative stress. Glia 53:360–365 [DOI] [PubMed] [Google Scholar]
- 75.Vairano M, Dello Russo C, Pozzoli G, et al. (2002). Erythropoietin exerts anti-apoptotic effects on rat microglial cells in vitro. Eur J Neurosci 16:584–592 [DOI] [PubMed] [Google Scholar]
- 76.Ponce LL, Navarro JC, Ahmed O, and Robertson CS. (2012). Erythropoietin neuroprotection with traumatic brain injury. Pathophysiology 20:31–38 [DOI] [PMC free article] [PubMed] [Google Scholar]