

Abstract
Immunomodulatory drugs (IMiDs) are thalidomide analogues, which possess pleiotropic anti-myeloma properties including immune-modulation, anti-angiogenic, anti-inflammatory and anti-proliferative effects. Their development was facilitated by an improved understanding in myeloma (MM) biology and initiated a profound shift in the therapeutic approach towards MM. Despite the diverse effects of IMiDs in vitro, the relative contribution of each effect towards their ultimate anti-MM activity is still unclear. Based on in vitro data, it appears that anti-proliferative effects and downregulation of crucial cytokines are their most important anti-MM attributes. Although the co-stimulatory effects on T and NK cells have been heralded as a unique and important property of IMiDs towards enhancing anti-MM immune activity, these in vitro effects have yet to be firmly corroborated in vivo. Much is yet to be elucidated regarding the complex interplay of immunomodulatory cytokines that occurs in vivo, which ultimately dictates the net effects of IMiDs in MM—the understanding of which is necessary to facilitate optimal manipulation of these drugs in future MM management.
Keywords: myeloma, lenalidomide, IMiDs, immunomodulatory
Introduction
In multiple myeloma (MM), the interplay between malignant plasma cells and their micro-environment is crucial to tumour growth and progression. The notion that survival of malignant cells is dependent on the microenvironment and evasion of the host’s anti-tumour immune response, is fundamental to the understanding of the role of the immunomodulatory drug (IMiD) class. IMiDs are a group of compounds that are analogues of thalidomide, a glutamic acid derivative with anti-angiogenic properties and potent anti-inflammatory effects owing to its anti-tumour necrosis factor (TNF)α activity. Thalidomide analogues were initially synthesized with the aim of optimizing both anti-TNFα and anti-angiogenic properties while reducing toxicities. The two leading IMiD compounds, lenalidomide (CC-5013; IMiD3; Revlimid) and pomalidomide (CC-4047; IMiD1; Acti-mid) were the first to enter clinical trials in MM in 1999,1 and are now the subject of clinical evaluation in other haematological malignancies.
The precise cellular targets and the exact mechanism of actions of IMiDs in MM remains unclear, however preclinical studies have unveiled multiple effects including anti-proliferative, T-cell co-stimulatory, anti-angiogenic and anti-inflammatory effects.2 Coupled with our increased understanding of tumour immunology and the importance of tumour–microenvironment interactions in many malignancies including MM, the discovery of this class of drugs represents a major step forward in the progress in cancer treatment. Here, we provide a review on the development of IMiDs, and explore their mechanisms of action, with a particular focus on immune modulation including their effects on regulatory T cells (Tregs), disruption of plasma cell (PC)–microenvironment interactions, and direct anti-tumour effects.
Development of thalidomide analogues
Thalidomide (α-N-phthalimido-glutarimide), is a synthetic derivative of glutamic acid, which was infamous for causing birth defects when used as an antiemetic in pregnancy in the late 1950s and early 1960s. Despite its withdrawal from most markets at this point, it was serendipitously found to be effective in the treatment of erythema nodosum leprosum, a cutaneous inflammatory complication of leprosy characterized by high levels of serum TNFα.3 Thalidomide’s ability to inhibit TNFα production by activated human monocytes was subsequently shown in 1991.4 Apart from this anti-inflammatory property, thalidomide was also found to have anti-angiogenic5 and immunomodulatory properties including T-cell co-stimulation, and activation of NK cells.6 This resulted in a surge of interest in thalidomide as a potential anti-cancer therapy upon a con-current appreciation of the role of angiogenesis and tumour–microenvironment interaction in tumour growth. As such, a formal medicinal chemistry programme was then initiated by the Celgene Corporation to search for analogues with increased potency but less toxicity compared with thalidomide. The two first-in-class IMiDs were lenalidomide and pomalidomide. Both are derived by adding an amino group to the fourth carbon of the phthaloyl ring of thalidomide. Pomalidomide essentially has a combined chemical structure of thalidomide and lenalidomide (Figure 1).
Figure 1.
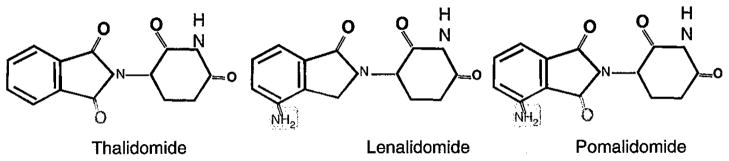
Chemical structure of thalidomide and its analogues, lenalidomide (CC-5013) and pomalidomide (CC-4047). The two first-in-class immunomodulatory drugs were lenalidomide and pomalidomide. Both are derived by adding an amino group to the fourth carbon of the phthaloyl ring of thalidomide. Pomalidomide essentially has a combined chemical structure of thalidomide and lenalidomide.
The initial focus and primary selection of these thalidomide analogues was based on the degree of their inhibition of TNFα production by activated human peripheral blood mononuclear cells (PBMC).7 These thalidomide analogues, called IMiDs (Celgene’s class of immunomodulatory compounds) are not only up to 50000-fold more potent at TNFα inhibition in vitro compared with thalidomide, they are also much more potent than thalidomide in their ability to co-stimulate T cells.8,9 In the context of T-cell co-stimulation, unlike monocytes, IMiDs conversely increase TNFα.6 As such, the immunomodulatory effects of IMiDs may vary in different diseases depending on the type of cell stimulated.
As anti-tumour agents, the emphasis in the development of lenalidomide and pomalidomide focused not only on their immunomodulatory activities, but also on their ability to disrupt tumour–microenvironment interactions and their improved direct tumour anti-proliferative activity compared with thalidomide (Table 1).
Table 1.
Summary of the major mechanisms of action of ImiDs
| Effect | Mechanism | Relative potency+ = potency factor of 10
|
||
|---|---|---|---|---|
| Thalidomide | Lenalidomide | Pomalidomide | ||
| Immune modulation | ||||
| CD4+ and CD8+ T-cell co-stimulation |
|
+ | ++++ | +++++ |
| Tregs suppression |
|
− | + | + |
| Th1 cytokine production |
|
+ | ++++ | +++++ |
| NK and NKT cell activation |
|
+ | ++++ | +++++ |
| Antibody-dependent cellular cytotoxicity (ADCC) |
|
− | ++++ | ++++ |
| Interference with tumour micro–environment interactions | ||||
| Anti-angiogenesis |
|
++++ | +++ | +++ |
| Anti-inflammatory properties |
|
+ | ++++ | +++++ |
| Downregulation of adhesion molecules |
|
|||
| Anti-osteoclastogenic properties |
|
|||
| Direct anti-tumour effects | ||||
| Anti-proliferative activity |
|
+ | +++ | +++ |
IMiDs and immune modulation in multiple myeloma Multiple myeloma induces immune paresis
Survival of myeloma cells, as with other tumours, is partially facilitated by the impaired endogenous immune surveillance against tumour antigens10 (Figure 2). Several mechanisms may contribute to myeloma cell ‘tolerance’, including myeloma-derived cytokines such as transforming growth factor (TGF)-β (which suppresses B cells and T cells via inhibition of IL-2 autocrine pathways11), inadequate antigen presentation,12 resistance to NK cell lysis, and defective T, B and NK cells.13,14 Impairment in both humoral and cellular immunity in MM is associated with impaired B-cell differentiation and antibody responses,14,15 reduced T-cell numbers specifically CD4+ T cells,16,17 abnormal Th1/Th2 CD4+ T-cell ratio,16 impaired cytotoxic T-cell responses,18 dysfunction of NK19 and NKT20 cells, and defective dendritic cell (DC) function. Expression of co-stimulatory molecules on DC for example, are reduced in patients with MM and are completely absent in progressive disease.14 Impairment of T-cell activation by DC is also mediated by MM-induced production of TGFβ,14 IL-10,14 IL-621 and VEGF,22 ultimately leading to poor antigen presentation and suboptimal tumour-specific immune response. In addition, dysregulation of regulatory T cells (Treg) have been reported, although the role of Treg in the pathophysiology of MM remains unclear.17,23
Figure 2.

Multiple Myeloma induces immunoparesis. Plasma cell derived cytokines including transforming growth factor (TGF)-β, interleukin (IL)-10, IL-6 and VEGF mediate suppression of B and T lymphocytes, and impair T-cell co-stimulation by dendritic cells (DC), ultimately leading to poor tumor-specific immune response.
Tregs are a group of immuno-suppressive T cells responsible for the maintenance of peripheral tolerance, and have been implicated in the suppression of tumour immunity.24 They are CD4+ T cells expressing high levels of CD25, and with nuclear expression of transcription factor forkhead box P3 (FOXP3).25 Tregs have been shown to inhibit tumour-specific T-cell functions as well as the cytolytic effects of NK and CD8+ T cells26,27 Their expansion correlates to poor prognosis in some solid tumours.28 However, the situation is less clear in lymphoproliferative disorders including MM.17,29 Unlike solid tumours, decreased Treg have been associated with refractory disease, large cell transformation and poor prognosis in follicular and Hodgkin’s lymphoma.30,31 Conflicting reports are seen for MM patients. Some studies showed lower numbers and function of Foxp3 + Tregs in MM patients compared with healthy volunteers.17,23 This would also appear to correlate with the notion that high IL-6 levels may downregulate Treg activity.32 However, other studies have reported increased CD4 + CD25hi Foxp3 + Treg with inhibitory functions in MM patients.33,34 Possible confounding factors in these studies include the fact that Tregs were activated ex vivo before functional assessments, and uncertainty as to whether results are confounded by the in vitro expansion of Tregs. Another caveat is that Tregs quantification are in percentages rather than absolute numbers, which may be misleading when there is a marked reduction in CD4+ T cells, as may occur in MM.17 Furthermore, it is unclear whether any or all of the MM patients included in these studies were on treatment at the time of Treg assessments. Clearly, although normal Tregs are important in the homoeostasis of immune response following specific immunity, how their modulation effects myeloma outcome remains uncertain. In any case, tumour-specific immune evasion represents one key aspect in PC pathogenesis and proliferation, and an understanding of this may be critical in facilitating the optimal use of IMiDs.
Immune modulation by IMiDs
In vitro studies have shown that IMiDs augment both the adaptive and innate immune system via the co-stimulation of T cells and augmentation of NK and NKT cells. However, the ultimate in vivo effects of IMiDs in MM may be more complex than that presented below, depending on the types of stimulus and cytokine milieu present at the disease site.
IMiDs co-stimulate T cells
T-cell activation requires an antigen-specific T-cell receptor signal in conjunction with co-stimulation provided by professional antigen-presenting cells. Thalidomide and IMiDs are only able to stimulate T cells that have been partially activated by either anti-CD3 or DC; their presence abrogates the requirement of a secondary co-stimulation signal from APCs to allow T cell activation.35,36 In the presence of thalidomide, partially activated CD3 + T cells selected from human PBMC had pronounced proliferation and enhanced production of Th1 type cytokines, IL-2 and IFNγ compared with control.6,35 By contrast, Th2-type cytokines, IL-4 and IL-10 were downregulated.6 Although it was initially shown that thalidomide-induced co-stimulation occurred preferentially in CD8+ T cells, later reports showed that both CD8+ and CD4+ T cells were equally stimulated.35 Compared with thalidomide, lenalidomide is 50–2000 times more potent in inducing T-cell proliferation, and 300 to >1200 times more potent in augmenting T-cell IL-2 and IFNγ production.9,35,37 Pomalidomide appears more potent than lenalidomide with regards to T-cell co-stimulation,9,38 and similarly increases Th1 type cytokines while displaying inhibitory effects on Th2-type cytokines.39 The clinical relevance of the higher in vitro potency of pomalidomide compared with lenalidomide is unclear, given that the former is given at a much lower dose in the clinical setting (maximal-tolerated dose 2 mg p.o. daily), resulting in a plasma drug concentration 10–100 times lower compared with lenalidomide when this is given at a dose of 25 mg p.o. daily.40
Importantly, the T-cell co-stimulatory effects of IMiDs were confirmed in an in vivo setting, whereby IMiDs were shown to enhance tumour-specific Th1-type immune response following tumour cell vaccination. Using an autologous colorectal cancer and an allogeneic melanoma murine model, it was demonstrated that tumour cell vaccination provided partial immunity from a subsequent live challenge with tumour cells, and that this protection was strongly enhanced by the incorporation of pomalidomide into the vaccination protocol.39 The presence of pomalidomide was associated with enhanced tumour-specific generation of IFN-γ and IL-2 from both CD4 and CD8T cells (mainly CD4 +).39 At this point it should be noted that there is a difference between the metabolism of thalidomide between humans and rodents; active hydroxylated thalidomide metabolites are produced in humans, but not in rodents, possibly explaining the greater in vivo efficacy of thalidomide in humans.41 Conversely, metabolism of lenalidomide and pomalidomide, does not appear to contribute to species differences in their pharmacological activity.
In MM, the degree of which IMiDs can improve the inherent immunoparesis is not clear, and dissecting out this issue is compounded by the common use of the immunosuppressant dexamethasone in combination with thalidomide or lenalidomide in the treatment of MM. Certainly, T and B lymphocytes remain significantly depressed in MM patients who responded to lenalidomide–dexamethasone combination,42 and there is emerging evidence that NK cell function declines with treatment.43,44 Whether this can be attributed solely to the concomitant use of dexamethasone, or whether the inherent dysfunctional T, B and NK cells in MM patients have a contributory role, is currently unclear.
The precise targets and mechanism by which IMiDs induce T-cell proliferation and activation is unknown. The mechanism of T-cell co-stimulation by the IMiDs involves enhanced transcriptional activity of activated protein-1 (AP-1), a key driver for IL-2 production.9,38 Pomalidomide was shown to enhance the DNA-binding activity of AP-1 but not nuclear factor (NF)-κB, Octomer-1 (OCT-1), or nuclear factor of activated T cells.45 Treatment of T cells by lenalidomide also results in increased tyrosine phosphorylation of CD28 on T cells,35,36,46 and activation of the PI3K-signalling pathway and the nuclear translocation of the transcription factor nuclear factor of activated T cells -2.35,36,46 Recently, the T-cell signalling adapter protein CD3-epsilon-associated protein has been shown to be required for lenalidomide-induced IL-2 upregulation in both primary human T cells and the Jurkat cell line.47 This increased transcription and production of IL-2 in turn, promotes NK cell proliferation and function, thus also enhancing the activity of the innate immune system.35,36,46
IMiDs alter FOXP3 + regulatory T cells
Interestingly, despite increased IL-2, it has been suggested that IMIDs inhibit IL-2-mediated generation of Tregs. In a recent study, when a PBMC population was maintained in IL-2 over a period of 7 days, the addition of lenalidomide (IC50 10μM) or pomalidomide (IC50 1 μM) in the incubation significantly decreased the proportion/number of Tregs in the population, compared with untreated controls. This was associated with a downregulation of the Tregs transcription factor, FOXP3, by up to 50% among the purified Treg population. Similar observations were seen with regard to Treg suppressor function. In contrast to their derivatives, thalidomide had no effect on Treg.48
Perceivably, IMiDs-mediated Treg suppression may help to promote tumour-specific immunity and improve tumour control. However, this concept does not explain reports of a relative increase in Treg numbers towards normal values in MM patients who are responding to treatment with lenalidomide or thalidomide, where previous values were markedly suppressed.17,34 One explanation is that the effects of lenalidomide in vitro are likely to be considerably less complex than that seen in vivo, in which IL-6 can inhibit Tregs in the setting of progressive disease, and in which the complex cytokine milieu needs to be taken into consideration.32 Furthermore, as Treg are actively sequestered within the tumour microenvironment by chemokines including stromal-derived factor-1α (SDF-1α),49 their peripheral blood quantification during disease states may not be representative of total Treg numbers. A recent report of the ability of lenalidomide to downregulate CXCR4, a receptor for SDF-1α, provides another feasible explanation to the mobilization of Tregs from the tumour micro-environment into the periphery, thus accounting for their increased numbers in the setting of a treatment with IMiDs. Future studies assessing the ‘absolute’ quantity and function of Tregs in both bone marrow/tumour microenvironment and peripheral blood, in an in vivo setting, in patients treated with IMiDs are clearly required. Ideally this should be measured in patients naive to previous immunomodulatory drugs, with observations made before and after subsequent treatment with these agents.
IMiDs enhances NK and NKT cells
The effects of IMiDs on the innate immune system, via enhancement of γδT cells, NK cells, and NKT cell function have been well documented.35,46,50 NKT cells are T lymphocytes which bear NK cell surface markers and recognizes glycolipid antigens (such as α-GalCer) in the context of the major histocompatibility class I-like CD1d molecules.51 Their anti-tumour effects involve direct cytotoxic properties, IFNγ production and activation of NK cells and DC. Normally, DCs loaded with the NKT ligand α-GalCer can activate and expand NKT cells; the addition of lenalidomide not only increases the degree of DC-induced NKT cell expansion, but also NKT cell secretion of IFNγ.52 NKT cell expansion in turn, partially accounts for the activation and proliferation of NK cells associated with IMiDs53 and perhaps also CD4 and CD8T cells.
NK cells have an important role in innate immunity, in killing both tumour and virus-infected cells. Thalidomide, lenalidomide and pomalidomide can increase NK cell proliferation with subsequent enhanced death of MM cell lines and primary patient cancer cells in the presence of IL-2;35 however, only lenalidomide and pomalidomide (but not thalidomide) have been shown to enhance antibody-dependent cellular cytotoxicity (ADCC) and natural cytotoxicity of NK cells in addition to their increase in proliferation.46,54 ADCC is a process whereby immunoglobulins attached to tumour antigens activate Fc-γ receptors on NK cells. This cross-linking triggers tumour cell cytotoxicity via perforin and granzymes released by NK cells, as well as tumour cell apoptosis induced by death ligands: FasL (Fas ligand) and TRAIL (tumour necrosis factor-related apoptosis-inducing ligand) that are expressed on some NK cell populations. The enhanced NK cell ADCC in the presence of lenalidomide and pomalidomide corresponds to increased NK cell FasL and granzyme B (but not perforin) expression.54
Through ADCC augmentation, IMiDs also enhance the cytotoxicity effects of monoclonal antibodies (of lgG1 isotype) including anti-CD40mAb (SGN-40)55 and anti-CD20mAb (rituximab). lenalidomide’s augmentation of NK cell-specific death of rituximab-coated CD20 + cell lines was shown to be associated with increased NK cell expression of IL-8, MCP-1 and GM-CSF and decreased IL-6 expression.54 More recently, however, lenalidomide was shown to downregulate CD20 surface antigens in chronic lymphocytic leukaemia resulting in a net diminished rituximab-mediated ADCC.56 Thus, lenalidomide’s net effects on ADCC are at present not fully understood and may be disease specific. It is interesting to note that the in vitro augmentation of ADCC on NK cells by IMiDs requires both antibody (Ab) binding to Fc-γ receptors on NK cells, as well as the presence of IL-2.46,55 The presence of either IL-2R Ab, or cyclosporin-A have previously been shown to abrogate IMiDs-induced NK cell-mediated cytotoxicity, thus showing that IMIDs-induced proliferation and activation of NK cells is indeed IL-2 dependent.46
Overall, in vitro studies so far support that IMiDs stimulate T cell and NKT cell production of IL-2 and IFNγ, resulting in potentiation of NK-cell proliferation and cytotoxicity (Figure 3). NK cells in turn produce T-cell- and DC-recruiting cytokines including monocyte chemotactic protein (MCP-1) and granulocyte macrophage colony-stimulating factor (GM-CSF) in response to Ab-coated target cells.57 This results in further chemotactic attraction of tumour-specific T cells in the presence of IMiDs.
Figure 3.

Summary of the immunomodulatory effects of immunomodulatory drugs. BMSC: bone marrow stromal cells; APC: antigen-presenting cells; IL: interleukin; TGF: transforming growth factor; TNF: tumor necrosis factor; VEGF: vascular endothelia growth factor; ADCC: antibody-dependent cellular toxicity; MHC: major histocompatibility complex; TCR: T cell receptor; NF-κB: Nuclear factor kappa B; PI3k: phosphoinositide 3-kinase; NFAT: nuclear factor of activated T cell; IFN: interferon; NK: natural killer.
IMiDs and the microenvironment in multiple myeloma Tumour–microenvironment interactions in multiple myeloma
Apart from immune evasion, plasma cell (PC)–bone marrow stromal cells (BMSC) interactions are now recognized as critical to PC survival. The initial homing of malignant PCs to the BM is mediated via the interaction between the chemokine SDF (stromal-derived growth factor)-1α within the BM and the chemokine receptor CXCR4 on PCs.58 SDF-1α upregulates PC adhesion to BMSC via modulation of adhesion molecules including VLA-4 (very late antigen) and LFA-1 (leukocyte function-associated antigen) on PC and VCAM-1 (vascular cell adhesion molecule) and ICAM-1 (intercellular adhesion molecule) on BMSC. PC adhesion to ECM proteins is mediated via syndecan-1 (CD138) and VLA-4 on PC to collagen and fibronectin. These interactions not only entrench PCs securely in the microenvironment, but also results in NF-κb activation and the production of cytokine and adhesion molecules necessary for PC growth.
NF-κB is a transcription factor that has important growth and anti-apoptotic roles in both normal and malignant cells.59 In MM, NF-κB activation induced by PC-BMSC binding results in both the upregulation of intracellular adhesion molecules60 and transcription of a myriad of cytokines including: vascular endothelial growth factors (VEGF), basic fibroblast growth factors (βFGF), TGF-β, TNFα, insulin like growth factor-1, interleukin (IL)-10 and IL-6, which all cooperate to promote PCs proliferation.61 Importantly, IL-6, a critical growth factor for normal B-cell and PC development, is further upregulated by VEGF, TGFβ and TNFα.62,63 This in turn promotes production, from BMSC, of further TNFα and the angiogenic factors VEGF and βFGF in a dose-dependent manner. Overall, PC–BMSC-induced cytokines provide a positive feedback loop for further PC growth and survival.
A number of signalling pathways that are involved with the above process have been elucidated. Binding of the above cytokines to their respective receptors on PCs triggers a number of proliferative/anti-apoptotic signalling pathways including MAPK (Ras/Raf/mitogen-activated protein kinase), JAK (Janus kinase)/STAT (signal transducers and activators of transcription), IKK (IκB kinase)-α/NF-κβ and PI-3K (phosphatidylinositol-3 kinase)/Akt pathways.64–66 These signalling cascades culminate in increased transcription of anti-apoptotic molecules including cyclin D1, Bcl-2 family members, and caspase inhibitors such as FLIP (FLICE inhibitor protein) and clAP-2 (cellular inhibitor of apoptosis protein 2). They also promote the autocrine secretion of VEGF and IL-6 from PC, which in turn further upregulate cytokine production and adhesion molecules expression from BMSCs.67 These bi-directional PC-BMSC interactions ultimately result in a vicious cycle with ongoing cytokine production and PC proliferation (Figure 4).
Figure 4.

Disruption of plasma cell–microenvironment interactions by immunomodulatory drugs. Anti-myeloma activity is mediated via downregulation of adhesion molecules, cytokine modulation, anti-angiogenesis, anti-osteoclastogenesis, as well as having direct anti-proliferative effects on malignant plasma cells.
IMiDs mediate disruption of myeloma cell–microenvironment interactions
Disruption of the MM–microenvironment interactions by IMiDs have a major role in their anti-myeloma activity and is mediated via several mechanisms including anti-angiogenesis, anti-inflammatory effects, modulation of cytokine production and downregulation of adhesion molecules.
IMiDs possess anti-angiogenic properties
The relative importance of anti-angiogenesis in MM treatment is uncertain. Although increased micro-vascular density correlates with MM progression, reduction in micro-vascular density and plasma VEGF levels are not always seen with thalidomide response in relapsed MM.68 All IMiDs have anti-angiogenic activity although it is generally considered that thalidomide has predominant anti-angiogenic activity whereas lenalidomide and pomalidomide have far greater immune enhancing effects.69 It is interesting to note that the anti-angiogenic effects of thalidomide are mediated through its hydroxylated metabolites,70 the formation of which are species specific, and are seen in humans and rabbits but not in rats. This possibly accounts for the lesser in vivo sensitivity to thalidomide-induced teratogenicity in rats compared with rabbits and humans. It appears that anti-angiogenesis occurs via the modulation of chemotactic factors involved in endothelial cell migration including TNFα, VEGF and βFGF from BMSCs rather than a direct inhibition of endothelial cell proliferation.69 Certainly, when MM cell lines were allowed to adhere to BMSC, an increased secretion in VEGF and βFGF (from both MM cells and BMSC) were seen, that was abrogated by the addition of thalidomide or pomalidomide in the in vitro culture system.63 IMiDs-induced anti-angiogenesis appeared to correlate with a reduction in Akt phosphorylation in response to both VEGF and βFGF,71 suggesting the interference of the PI3K/Akt-signalling pathway as a mode of action. Importantly, the ability to inhibit VEGF and βFGF production by IMiDs is likely to have multiple effects beyond that of anti-angiogenesis as these growth factors have multiple other biological effects including upregulation of pro-inflammatory cytokines, including 1L-6 production by BMSC.62,72
IMiDs possess anti-inflammatory properties
The relative contribution of anti-inflammatory properties of IMiDs towards their anti-myeloma activity is uncertain. Many cytokines inhibited by IMiDs have dual pro-inflammatory and pro-myeloma properties. For example, cyclo-oxygenase (COX)-2 is involved in the pathogenesis of various cancers including myeloma.73,74 COX-2 catalyses arachidonic acids into various pro-inflammatory prostaglandins (PG), one of which is PG-E2 which promote tumour angiogenesis and IL-6 production.75 Indeed COX-2 inhibitors may have a potential therapeutic role in MM as they can induce apoptosis in MM cell lines.74,76 Thalidomide, lenalidomide and pomalidomide are all able to inhibit the expression of COX-2, but not COX-1 enzymes, in stimulated PBMCs.77 These drugs shorten the half-life of COX-2 mRNA in a dose-dependent manner, with a net reduction in PGE2 production. IMiDs-mediated reduction in COX-2 expression is dependent on IMiDs-induced increase in IL-10 because addition of anti-IL-10 neutralizing antibody counteracted IMiD-mediated inhibition of COX-2.
Outside the context of anti-myeloma activity, IMiDs have other broad anti-inflammatory properties including: inhibition of macrophage inflammatory protein-α and GM-CSF9 and downregulation of TNFα production from LPS-stimulated monocytes.37 Compared with thalidomide, inhibition of TNFα was 2000-fold more potent with lenalidomide and 20000-fold more potent with pomalidomide78 (Table 1). IMiDs therefore have significant therapeutic potential in inflammatory conditions including Crohn’s and rheumatoid arthritis.79
IMiDs downregulate adhesion molecules
Adhesion molecules that facilitate PC-BMSC interactions, are upregulated by TNFα. Inhibition of TNFα by IMiDs ultimately inhibit a positive feedback loop, which upregulates expression of cell surface adhesion molecules on both BMSC and PC, including LFA-1, ICAM-1, VCAM-1 and VLA-4.60,80 Importantly, the downregulation of PC adherence to BMSCs results in reduction in pro-survival cytokines produced by BMSCs. In this respect, IMiDs can overcome the cellular adhesion-mediated drug resistance by malignant PCs.81
IMiDs possess anti-osteoclastogenic properties
Although the clinical evidence for the protective effects of IMiDs on MM-related bony disease is not strong, there is emerging data regarding their anti-osteoclastogenesis potential in vitro. Indeed the direct inhibition of osteoclasts maturation by IMiDs have been demonstrated in a dose-dependent manner.82 Inhibition of osteoclast-mediated bone resorption was associated with a reduction in the osteoclast expression of Cathepsin K—a protease involved in the bony matrix degradation, and αVβ3-integrin, a marker of OCL differentiation.83 These are probably a result of downregulation of important transcription factors including PU.1, which is important in the formation of osteoclast precursors.82,84 Unlike proteasome inhibitors, IMiDs have not been shown to stimulate osteoblasts.85
Direct anti-tumour effects of IMiDs
Apart from modulation of the microenvironment and immune response, IMiDs also exert direct anti-proliferative effect on PC via inhibition of the cyclin-dependent kinase pathway, activation of Fas-mediated cell death, and downregulation of anti-apoptotic proteins. In one study, the inhibition of MM cell proliferation by various IMiDs occurred at IC50 of 0.1–1.0 μM, which are readily achievable in serum levels.86 Dexamethasone synergises the anti-proliferative effects of IMiDs whereas the addition of exogenous IL-6, a specific inhibitor of dexamethasone-induced apoptosis, mitigated the inhibition of DNA synthesis.86 The IMiD-treated cell lines in this study were demonstrated to be in G1 cell cycle arrest with subsequent apoptosis. In most tumour cell lines examined thus far, lenalidomide induces expression of the cyclin-dependent kinase inhibitors p21, p27 and p15, the early growth response genes Egr-1, 2 and 3, and SPARC (Secreted protein acidic and rich in cysteine).43 The upregulation of p21 by lenalidomide in MM and lymphoma cells results in the inhibition of cyclin-dependent kinase activity and reduced phosphorylation of the retinoblastoma proteins, thereby causing cell cycle arrest in G0/G1 phase. SPARC upregulation was observed in lenalidomide-treated erythroblasts from MDS patients with del5q cytogenetic abnormality,87 as well as in IMiD-treated MM cells. The involvement of various tumour suppressor genes in the mechanism of IMiD-induced cell cycle arrest and apoptosis suggests that the anti-proliferative activity of IMiDs is dependent upon changes in gene expression.
After long-term treatment of MM cells with IMiDs, it was demonstrated that IMiDs treatment downregulated the activity of the transcription factor, NF-κB in myeloma cell lines. This in-turn resulted in reduced expression of anti-apoptotic proteins including clAP2 (cellular inhibitor of apoptosis protein 2)88 and FLIP (FLICE inhibitor protein).89 These anti-apoptotic proteins were previously shown to inhibit caspase-8 activation,90,91 and indeed, both lenalidomide, pomalidomide, and to a lesser extent, thalidomide were able to induce caspase-8 in both MM cell lines and patients’ MM cells (Figure 5). Activation of caspase 3, 8 and 9 was observed in various MM cell lines92 suggesting a degree of heterogeneity in the tumour cell response to IMiDs. The caspase-8-dependence of IMiD-induced cell death was shown by the fact that addition of specific caspase 8 inhibitor, but not caspase-9 inhibitor, attenuated the apoptosis in one cell line (MM.1S). As expected, IMiDs synergises the pro-apoptotic effect in MM cells when combined with other activators of caspase-8, including Fas and TRAIL/Apo2L. Synergism in apoptosis was also demonstrated when IMiDs were used in combination with dexamethasone and proteasome inhibitors, using the latter’s predominantly caspase-9-dependent apoptotic activity (Figure 5), and this synergy has also been demonstrated in the clinical setting.
Figure 5.

Inhibition of caspase-8 by IMiDs. TNF: tumor necrosis factor, TRAIL: TNF-related apoptosis-inducing ligand; FasL: Fas ligand; NF-κB: Nuclear factor kappa B; IL:interleukin; IGF: insulin-like growth factor.
Summary
The introduction of IMiDs has coincided with the beginning of a major shift in the understanding of effective anti-cancer treatments. Anti-cancer therapy is moving towards a three-pronged approach: induction of direct tumour cell apoptosis, interference with the tumour cell–microenvironment interactions, and enhancement of anti-tumour immune response. The multifaceted actions of IMiDs (Table 1) explain their efficacy as single agents or as combination partners in which capitalization on synergistic antitumour mechanisms is possible, not only against MM but other malignancies including MDS, CLL and potentially in a range of solid tumours. As anti-myeloma agents, perhaps the most important mechanism of actions of IMiDs are their anti-proliferative effects and their disruption of MM–BMSC interactions, as many have irrefutably shown their ability to downregulate crucial cytokines and growth factors necessary for myeloma growth. Naturally, the immunomodulatory properties of IMiDs, as shown in vitro could also be important and distinguishes the IMiDs as uniquely positioned to enhance immune-mediated anti-tumour activity. However, these changes have not been firmly correlated with clinical outcome or fully corroborated with in vivo changes in patients receiving these drugs—in which complex cytokine and cellular interactions may well dictate the ultimate immune effects of IMiDs. Although anti-angiogenesis is a potent property of IMiDs, the importance of this attribute in anti-MM activity may be of lesser importance. No clinical studies to date have examined the bone marrow anti-angiogenic effects of lenalidomide or pomalidomide in MM patients, although in del5q MDS patients who respond to lenalidomide therapy, a decrease in bone marrow micro-vascular density has been reported.
Despite our improved understanding of some of the mechanisms of action of IMiDs, many questions remain unanswered. For example, are the diverse effects of IMiDs necessarily always cooperative in their anti-tumour actions in an in vivo setting, and do these agents have divergent effects in different disease setting? The fact that dose response in different disease settings varies, implies that this may be the case. Ultimately, MM remains an incurable disease. Future advances in the use of these agents in MM will aim to optimize the ‘three pronged approach’ by exploring synergies between IMiDs, other novel therapeutic agents and conventional chemotherapies.
Acknowledgments
We acknowledge the insightful comments and critical review of the article by A/Prof S Opat, P Schafer, and Dr H Tran. Dr Hang Quach received funding support from the Vincent Fairfax Scholarship (Royal Australasian College of Physicians), and the Peter MacCallum Foundation (Morris Family Grant).
Footnotes
Conflict of interest
Drs Hang Quach, Paul Neeson and Mark J Smyth declare no potential conflict of interest. Professors H Miles Prince and A Keith Stewart have both participated in advisory boards and received research funding from Celgene Corporation. Dr Simon Harrison has received research funding from Celgene Corporation.
References
- 1.van Rhee F, Dhodapkar M, Shaughnessy JD, Jr, Anaissie E, Siegel D, Hoering A, et al. First thalidomide clinical trial in multiple myeloma: a decade. Blood. 2008;112:1035–1038. doi: 10.1182/blood-2008-02-140954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galustian C, Labarthe MC, Bartlett JB, Dalgleish AG. Thalidomide-derived immunomodulatory drugs as therapeutic agents. Exp Opin Biolog Ther. 2004;4:1963–1970. doi: 10.1517/14712598.4.12.1963. [DOI] [PubMed] [Google Scholar]
- 3.Lyer G. WHO co-ordinated short-term double-blind trial with thalidomide in the treatmetn of acute lepra reactions with male lepromatous patients. Bull World Health Organization. 1971;45:719–732. [PMC free article] [PubMed] [Google Scholar]
- 4.Sampaio EP, Sarno EN, Galilly R, Cohn ZA, Kaplan G. Thalidomide selectively inhibits tumor necrosis factor alpha production by stimulated human monocytes. J Exp Med. 1991;173:699–703. doi: 10.1084/jem.173.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D’Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci USA. 1994;91:4082–4085. doi: 10.1073/pnas.91.9.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haslett PA, Corral LG, Albert M, Kaplan G. Thalidomide costimulates primary human T lymphocytes, preferentially inducing proliferation, cytokine production, and cytotoxic responses in the CD8+ subset. J Exp Med. 1998;187:1885–1892. doi: 10.1084/jem.187.11.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corral LG, Muller GW, Moreira AL, Chen Y, Wu M, Stirling D, et al. Selection of novel analogs of thalidomide with enhanced tumor necrosis factor alpha inhibitory activity. Mol Med (Cambridge, Mass) 1996;2:506–515. [PMC free article] [PubMed] [Google Scholar]
- 8.Muller GW, Corral LG, Shire MG, Wang H, Moreira A, Kaplan G, et al. Structural modifications of thalidomide produce analogs with enhanced tumor necrosis factor inhibitory activity. J Med Chem. 1996;39:3238–3240. doi: 10.1021/jm9603328. [DOI] [PubMed] [Google Scholar]
- 9.Corral LG, Haslett PA, Muller GW, Chen R, Wong LM, Ocampo CJ, et al. Differential cytokine modulation and T cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-alpha. J Immunol. 1999;163:380–386. [PubMed] [Google Scholar]
- 10.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 11.Urashima M, Ogata A, Chauhan D, Hatziyanni M, Vidriales MB, Dedera DA, et al. Transforming growth factor-beta1: differential effects on multiple myeloma versus normal B cells. Blood. 1996;87:1928–1938. [PubMed] [Google Scholar]
- 12.Brimnes MK, Svane IM, Johnsen HE. Impaired functionality and phenotypic profile of dendritic cells from patients with multiple myeloma. Clin Exp Immunol. 2006;144:76–84. doi: 10.1111/j.1365-2249.2006.03037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smyth MJ, Godfrey DI, Trapani JA. A fresh look at tumor immunosurveillance and immunotherapy. Nat Immunol. 2001;2:293–299. doi: 10.1038/86297. [DOI] [PubMed] [Google Scholar]
- 14.Brown RD, Pope B, Murray A, Esdale W, Sze DM, Gibson J, et al. Dendritic cells from patients with myeloma are numerically normal but functionally defective as they fail to up-regulate CD80 (B7-1) expression after huCD40LT stimulation because of inhibition by transforming growth factor-beta1 and interleukin-10. Blood. 2001;98:2992–2998. doi: 10.1182/blood.v98.10.2992. [DOI] [PubMed] [Google Scholar]
- 15.Rawstron AC, Davies FE, Owen RG, English A, Pratt G, Child JA, et al. B-lymphocyte suppression in multiple myeloma is a reversible phenomenon specific to normal B-cell progenitors and plasma cell precursors. Br J Haematol. 1998;100:176–183. doi: 10.1046/j.1365-2141.1998.00525.x. [DOI] [PubMed] [Google Scholar]
- 16.Ogawara H, Handa H, Yamazaki T, Toda T, Yoshida K, Nishimoto N, et al. High Th1/Th2 ratio in patients with multiple myeloma. Leukaemia Res. 2005;29:135–140. doi: 10.1016/j.leukres.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 17.Quach H, Ritchie D, Neeson P, Harrison S, Tai T, Tainton K, et al. Regulatory T cells (Treg) are Depressed in Patients with Relapsed/Refractory Multiple Myeloma (MM) and Increases Towards Normal Range in Responding Patients Treated with Lenalidomide (LEN) [abstract] Blood. 2008;112:1696a. [Google Scholar]
- 18.Maecker B, Anderson KS, von Bergwelt-Baildon MS, Weller E, Vonderheide RH, Richardson PG, et al. Viral antigen-specific CD8+ T-cell responses are impaired in multiple myeloma. Br J Haematol. 2003;121:842–848. doi: 10.1046/j.1365-2141.2003.04375.x. [DOI] [PubMed] [Google Scholar]
- 19.Jarahian M, Watzl C, Issa Y, Altevogt P, Momburg F. Blockade of natural killer cell-mediated lysis by NCAM140 expressed on tumor cells. Int J Cancer. 2007;120:2625–2634. doi: 10.1002/ijc.22579. [DOI] [PubMed] [Google Scholar]
- 20.Dhodapkar MV, Geller MD, Chang DH, Shimizu K, Fujii S, Dhodapkar KM, et al. A reversible defect in natural killer T cell function characterizes the progression of premalignant to malignant multiple myeloma. J Exp Med. 2003;197:1667–1676. doi: 10.1084/jem.20021650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ratta M, Fagnoni F, Curti A, Vescovini R, Sansoni P, Oliviero B, et al. Dendritic cells are. functionally defective in multiple myeloma: the role of interleukin-6. Blood. 2002;100:230–237. doi: 10.1182/blood.v100.1.230. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi A, Kono K, Ichihara F, Sugai H, Fujii H, Matsumoto Y. Vascular endothelial growth factor inhibits maturation of dendritic cells induced by lipopolysaccharide, but not by proinflammatory cytokines. Cancer Immunol Immunother. 2004;53:543–550. doi: 10.1007/s00262-003-0466-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prabhala RH, Neri P, Bae JE, Tassone P, Shammas MA, Allam CK, et al. Dysfunctional T regulatory cells in multiple myeloma. Blood. 2006;107:301–304. doi: 10.1182/blood-2005-08-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Curiel TJ. Tregs and rethinking cancer immunotherapy. J Clin Invest. 2007;117:1167–1174. doi: 10.1172/JCI31202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Curiel TJ. Regulatory T-cell development: is Foxp3 the decider? Nat Med. 2007;13:250–253. doi: 10.1038/nm0307-250. [DOI] [PubMed] [Google Scholar]
- 26.Yang ZZ, Novak AJ, Ziesmer SC, Witzig TE, Ansell SM. Attenuation of CD8(+) T-cell function by CD4(+)CD25(+) regulatory T cells in B-cell non-Hodgkin’s lymphoma. Cancer Res. 2006;66:10145–10152. doi: 10.1158/0008-5472.CAN-06-1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trzonkowski P, Szmit E, Mysliwska J, Dobyszuk A, Mysliwski A. CD4+CD25+ T regulatory cells inhibit cytotoxic activity of T CD8+ and NK lymphocytes in the direct cell-to-cell interaction. Clin Immunol (Orlando, Fla) 2004;112:258–267. doi: 10.1016/j.clim.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 28.Beyer M, Schultze JL. Regulatory T cells in cancer. Blood. 2006;108:804–811. doi: 10.1182/blood-2006-02-002774. [DOI] [PubMed] [Google Scholar]
- 29.Joshua DE, Brown RD, Ho PJ, Gibson J. Regulatory T cells and multiple myeloma. Clin Lymphoma Myeloma. 2008;8:283–286. doi: 10.3816/CLM.2008.n.039. [DOI] [PubMed] [Google Scholar]
- 30.Carreras J, Lopez-Guillermo A, Fox BC, Colomo L, Martinez A, Roncador G, et al. High numbers of tumor-infiltrating FOXP3-positive regulatory T cells are associated with improved overall survival in follicular lymphoma. Blood. 2006;108:2957–2964. doi: 10.1182/blood-2006-04-018218. [DOI] [PubMed] [Google Scholar]
- 31.Alvaro T, Lejeune M, Salvado MT, Bosch R, Garcia JF, Jaen J, et al. Outcome in Hodgkin’s lymphoma can be predicted from the presence of accompanying cytotoxic and regulatory T cells. Clin Cancer Res. 2005;11:1467–1473. doi: 10.1158/1078-0432.CCR-04-1869. [DOI] [PubMed] [Google Scholar]
- 32.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 33.Beyer M, Kochanek M, Giese T, Endl E, Weihrauch MR, Knolle PA, et al. In vivo peripheral expansion of naive CD4+CD25high FoxP3+ regulatory T cells in patients with multiple myeloma. Blood. 2006;107:3940–3949. doi: 10.1182/blood-2005-09-3671. [DOI] [PubMed] [Google Scholar]
- 34.Feyler S, von Lilienfeld-Toal M, Jarmin S, Marles L, Rawstron A, Ashcroft AJ, et al. CD4(+)CD25(+)FoxP3(+) regulatory T cells are increased whilst CD3(+)CD4(−)CD8(−)alphabetaTCR(+) Double Negative T cells are decreased in the peripheral blood of patients with multiple myeloma which correlates with disease burden. Br J Haematol. 2009;144:686–695. doi: 10.1111/j.1365-2141.2008.07530.x. [DOI] [PubMed] [Google Scholar]
- 35.Davies FE, Raje N, Hideshima T, Lentzsch S, Young G, Tai YT, et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood. 2001;98:210–216. doi: 10.1182/blood.v98.1.210. [DOI] [PubMed] [Google Scholar]
- 36.LeBlanc R, Hideshima T, Catley LP, Shringarpure R, Burger R, Mitsiades N, et al. Immunomodulatory drug costimulates T cells via the B7-CD28 pathway. Blood. 2004;103:1787–1790. doi: 10.1182/blood-2003-02-0361. [DOI] [PubMed] [Google Scholar]
- 37.Corral LG, Kaplan G. Immunomodulation by thalidomide and thalidomide analogues. Ann Rheum Dis. 1999;58 (Suppl 1):1107–1113. doi: 10.1136/ard.58.2008.i107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schafer PH, Gandhi AK, Loveland MA, Chen RS, Man HW, Schnetkamp PP, et al. Enhancement of cytokine production and AP-1 transcriptional activity in T cells by thalidomide-related immunomodulatory drugs. J Pharmacol Exp Therap. 2003;305:1222–1232. doi: 10.1124/jpet.102.048496. [DOI] [PubMed] [Google Scholar]
- 39.Dredge K, Marriott JB, Todryk SM, Muller GW, Chen R, Stirling DI, et al. Protective antitumor immunity induced by a costimulatory thalidomide analog in conjunction with whole tumor cell vaccination is mediated by increased Th1-type immunity. J Immunol. 2002;168:4914–4919. doi: 10.4049/jimmunol.168.10.4914. [DOI] [PubMed] [Google Scholar]
- 40.Pomalidomide Investigators’ Brochure. Celgene Corporation; 2008. Study CC-4047-1398/142: A Phase-1, Single-blind, placebo controlled, Ascending Single Oral Dose, Safety, Tolerability, Pharmakokinetic and Pharmacodynamic Study in Healthy Male Subjects; pp. 1–164. version 11. [Google Scholar]
- 41.Lu J, Palmer BD, Kestell P, Browett P, Baguley BC, Muller G, et al. Thalidomide metabolites in mice and patients with multiple myeloma. Clin Cancer Res. 2003;9:1680–1688. [PubMed] [Google Scholar]
- 42.Quach H, Ritchie D, Neeson P, Tai T, Tainton K, Lynch K, et al. Lymphoid subsets and regulatory T cell profiles in patients with relapsed multiple myeloma in a subset of patients enrolled in the REVLITE trial [abstract]. Haematology Society of Australia and New Zealand Annual Scientific Meeting; Perth, Australia. 2008. [Google Scholar]
- 43.Schafer P, Gandhi AK, Zhang L, Kang J, Capone L, Bartlett JB. Opposing Effects of Dexamethasone on Lenalidomide Activity in Multiple Myeloma: Additive/Synergistic Effects on Anti-Proliferative Activity on Myeloma Cells and Antagonistic Effects on Immune Function [abstract] Blood. 2008;112:2761a. [Google Scholar]
- 44.Quach H, Hsu A, Ritchie D, Neeson P, Lynch K, Harrison S, et al. In Vivo Antagonistic Effects of Dexamethasone on Lenalidomide-Induced NK cell Activation [abstract]. 51st Annual Meeting of the American Society of Hematology; New Orleans, LA. 2009. p. Abstract 1639. [Google Scholar]
- 45.Payvandi F, Wu L, Naziruddin SD, Haley M, Parton A, Schafer PH, et al. Immunomodulatory drugs (IMiDs) increase the production of IL-2 from stimulated T cells by increasing PKC-theta activation and enhancing the DNA-binding activity of AP-1 but not NF-kappaB, OCT-1, or NF-AT. J Interferon Cytokine Res. 2005;25:604–616. doi: 10.1089/jir.2005.25.604. [DOI] [PubMed] [Google Scholar]
- 46.Hayashi T, Hideshima T, Akiyama M, Podar K, Yasui H, Raje N, et al. Molecular mechanisms whereby immunomodulatory drugs activate natural killer cells: clinical application. Br J Haematol. 2005;128:192–203. doi: 10.1111/j.1365-2141.2004.05286.x. [DOI] [PubMed] [Google Scholar]
- 47.Gandhi AK, Rogovitz A, Lopez-Girona A, Mendy D, Peter HS. Stimulation of T cells by Lenalidomide Involves Putative Lenalidomide Binding Protein Cd3-Epsilon-Associated Protein and GDP-Mannose Pyrophosphorylase a [abstract] Blood. 2008;112:2606a. [Google Scholar]
- 48.Galustian C, Meyer B, Labarthe MC, Dredge K, Klaschka D, Henry J, et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol Immunother. 2009;58:1033–1045. doi: 10.1007/s00262-008-0620-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alsayed Y, Ngo H, Runnels J, Leleu X, Singha UK, Pitsillides CM, et al. Mechanisms of regulation of CXCR4/SDF-1 (CXCL12)-dependent migration and homing in multiple myeloma. Blood. 2007;109:2708–2717. doi: 10.1182/blood-2006-07-035857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Galustian C, Klaschka D, Labarthe MC, Bartlett JB, Dalgleish AG. The immunomodulatory drug (IMID(R)) CC-4047 enhances the proliferation and anti-tumour fuction of gamma delta T cells. J Immunother. 2004;27:S50. [Google Scholar]
- 51.Kronenberg M. Toward an understanding of NKT cell biology: progress and paradoxes. Ann Rev Immunol. 2005;23:877–900. doi: 10.1146/annurev.immunol.23.021704.115742. [DOI] [PubMed] [Google Scholar]
- 52.Fujii S, Shimizu K, Steinman RM, Dhodapkar MV. Detection and activation of human Valpha24+ natural killer T cells using alpha-galactosyl ceramide-pulsed dendritic cells. J Immunolog Methods. 2003;272:147–159. doi: 10.1016/s0022-1759(02)00497-0. [DOI] [PubMed] [Google Scholar]
- 53.Carnaud C, Lee D, Donnars O, Park SH, Beavis A, Koezuka Y, et al. Cutting edge: Cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J Immunol. 1999;163:4647–4650. [PubMed] [Google Scholar]
- 54.Wu L, Adams M, Carter T, Chen R, Muller G, Stirling D, et al. lenalidomide enhances natural killer cell and monocyte-mediated antibody-dependent cellular cytotoxicity of rituximab-treated CD20+ tumor cells. Clin Cancer Res. 2008;14:4650–4657. doi: 10.1158/1078-0432.CCR-07-4405. [DOI] [PubMed] [Google Scholar]
- 55.Tai YT, Li XF, Catley L, Coffey R, Breitkreutz I, Bae J, et al. Immunomodulatory drug lenalidomide (CC-5013, IMiD3) augments anti-CD40 SGN-40-induced cytotoxicity in human multiple myeloma: clinical implications. Cancer research. 2005;65:11712–11720. doi: 10.1158/0008-5472.CAN-05-1657. [DOI] [PubMed] [Google Scholar]
- 56.Lapalombella R, Yu B, Triantafillou G, Liu Q, Butchar JP, Lozanski G, et al. Lenalidomide down-regulates the CD20 antigen and antagonizes direct and antibody-dependent cellular cytotoxicity of rituximab on primary chronic lymphocytic leukemia cells. Blood. 2008;112:5180–5189. doi: 10.1182/blood-2008-01-133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roda JM, Parihar R, Magro C, Nuovo GJ, Tridandapani S, Carson WE., III Natural killer cells produce T cell-recruiting chemokines in response to antibody-coated tumor cells. Cancer Res. 2006;66:517–526. doi: 10.1158/0008-5472.CAN-05-2429. [DOI] [PubMed] [Google Scholar]
- 58.Hideshima T, Chauhan D, Hayashi T, Podar K, Akiyama M, Gupta D, et al. The biological sequelae of stromal cell-derived factor-1 alpha in multiple myeloma. Mol Cancer Therap. 2002;1:539–544. [PubMed] [Google Scholar]
- 59.Li ZW, Chen H, Campbell RA, Bonavida B, Berenson JR. NF-kappaB in the pathogenesis and treatment of multiple myeloma. Curr Opin Hematol. 2008;15:391–399. doi: 10.1097/MOH.0b013e328302c7f4. [DOI] [PubMed] [Google Scholar]
- 60.Hideshima T, Chauhan D, Schlossman R, Richardson P, Anderson KC. The role of tumor necrosis factor alpha in the pathophysiology of human multiple myeloma: therapeutic applications. Oncogene. 2001;20:4519–4527. doi: 10.1038/sj.onc.1204623. [DOI] [PubMed] [Google Scholar]
- 61.Chauhan D, Uchiyama H, Akbarali Y, Urashima M, Yamamoto K, Libermann TA, et al. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-kappa B. Blood. 1996;87:1104–1112. [PubMed] [Google Scholar]
- 62.Dankbar B, Padro T, Leo R, Feldmann B, Kropff M, Mesters RM, et al. Vascular endothelial growth factor and interleukin-6 in paracrine tumor-stromal cell interactions in multiple myeloma. Blood. 2000;95:2630–2636. [PubMed] [Google Scholar]
- 63.Gupta D, Treon SP, Shima Y, Hideshima T, Podar K, Tai YT, et al. Adherence of multiple myeloma cells to bone marrow stromal cells upregulates vascular endothelial growth factor secretion: therapeutic applications. Leukemia. 2001;15:1950–1961. doi: 10.1038/sj.leu.2402295. [DOI] [PubMed] [Google Scholar]
- 64.Ogata A, Chauhan D, Teoh G, Treon SP, Urashima M, Schlossman RL, et al. IL-6 triggers cell growth via the Ras-dependent mitogen-activated protein kinase cascade. J Immunol. 1997;159:2212–2221. [PubMed] [Google Scholar]
- 65.Chauhan D, Kharbanda S, Ogata A, Urashima M, Teoh G, Robertson M, et al. Interleukin-6 inhibits Fas-induced apoptosis and stress-activated protein kinase activation in multiple myeloma cells. Blood. 1997;89:227–234. [PubMed] [Google Scholar]
- 66.Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, et al. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem. 2002;277:16639–16647. doi: 10.1074/jbc.M200360200. [DOI] [PubMed] [Google Scholar]
- 67.Uchiyama H, Barut BA, Chauhan D, Cannistra SA, Anderson KC. Characterization of adhesion molecules on human myeloma cell lines. Blood. 1992;80:2306–2314. [PubMed] [Google Scholar]
- 68.Mileshkin L, Honemann D, Gambell P, Trivett M, Hayakawa Y, Smyth M, et al. Patients with multiple myeloma treated with thalidomide: evaluation of clinical parameters, cytokines, angiogenic markers, mast cells and marrow CD57+ cytotoxic T cells as predictors of outcome. Haematologica. 2007;92:1075–1082. doi: 10.3324/haematol.11208. [DOI] [PubMed] [Google Scholar]
- 69.Dredge K, Marriott JB, Macdonald CD, Man HW, Chen R, Muller GW, et al. Novel thalidomide analogues display anti-angiogenic activity independently of immunomodulatory effects. Br J Cancer. 2002;87:1166–1172. doi: 10.1038/sj.bjc.6600607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Price DK, Ando Y, Kruger EA, Weiss M, Figg WD. 5′-OH-thalidomide, a metabolite of thalidomide, inhibits angiogenesis. Ther Drug Monit. 2002;24:104–110. doi: 10.1097/00007691-200202000-00017. [DOI] [PubMed] [Google Scholar]
- 71.Dredge K, Horsfall R, Robinson SP, Zhang LH, Lu L, Tang Y, et al. Orally administered lenalidomide (CC-5013) is anti-angiogenic in vivo and inhibits endothelial cell migration and Akt phosphorylation in vitro. Microvascular Res. 2005;69:56–63. doi: 10.1016/j.mvr.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 72.Costes V, Portier M, Lu ZY, Rossi JF, Bataille R, Klein B. Interleukin-1 in multiple myeloma: producer cells and their role in the control of IL-6 production. BrJ Haematol. 1998;103:1152–1160. doi: 10.1046/j.1365-2141.1998.01101.x. [DOI] [PubMed] [Google Scholar]
- 73.Masferrer JL, Leahy KM, Koki AT, Zweifel BS, Settle SL, Woerner BM, et al. Antiangiogenic and antitumor activities of cyclooxygenase-2 inhibitors. Cancer Res. 2000;60:1306–1311. [PubMed] [Google Scholar]
- 74.Prince HM, Mileshkin L, Roberts A, Ganju V, Underhill C, Catalano J, et al. A multicenter phase II trial of thalidomide and celecoxib for patients with relapsed and refractory multiple myeloma. Clin Cancer Res. 2005;11:5504–5514. doi: 10.1158/1078-0432.CCR-05-0213. [DOI] [PubMed] [Google Scholar]
- 75.Hinson RM, Williams JA, Shacter E. Elevated interleukin 6 is induced by prostaglandin E2 in a murine model of inflammation: possible role of cyclooxygenase-2. Proc Natl Acad Sci USA. 1996;93:4885–4890. doi: 10.1073/pnas.93.10.4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang M, Abe Y, Matsushima T, Nishimura J, Nawata H, Muta K. Selective cyclooxygenase 2 inhibitor NS-398 induces apoptosis in myeloma cells via a Bcl-2 independent pathway. Leuk lymphoma. 2005;46:425–433. doi: 10.1080/10428190400015691. [DOI] [PubMed] [Google Scholar]
- 77.Payvandi F, Wu L, Haley M, Schafer PH, Zhang LH, Chen RS, et al. Immunomodulatory drugs inhibit expression of cyclooxygenase-2 from TNF-alpha, IL-1beta, and LPS-stimulated human PBMC in a partially IL-10-dependent manner. Cell Immunol. 2004;230:81–88. doi: 10.1016/j.cellimm.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 78.Muller GW, Chen R, Huang SY, Corral LG, Wong LM, Patterson RT, et al. Amino-substituted thalidomide analogs: potent inhibitors of TNF-alpha production. Bioorg Med Chem Lett. 1999;9:1625–1630. doi: 10.1016/s0960-894x(99)00250-4. [DOI] [PubMed] [Google Scholar]
- 79.Akobeng AK, Stokkers PC. Thalidomide and thalidomide analogues for maintenance of remission in Crohn’s disease. Cochrane Database Syst Rev. 2009;(2):CD007351. doi: 10.1002/14651858.CD007351.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Geitz H, Handt S, Zwingenberger K. Thalidomide selectively modulates the density of cell surface molecules involved in the adhesion cascade. Immunopharmacology. 1996;31:213–221. doi: 10.1016/0162-3109(95)00050-x. [DOI] [PubMed] [Google Scholar]
- 81.Hideshima T, Bergsagel PL, Kuehl WM, Anderson KC. Advances in biology of multiple myeloma: clinical applications. Blood. 2004;104:607–618.0. doi: 10.1182/blood-2004-01-0037. [DOI] [PubMed] [Google Scholar]
- 82.Breitkreutz I, Raab MS, Vallet S, Hideshima T, Raje N, Mitsiades C, et al. Lenalidomide inhibits osteoclastogenesis, survival factors and bone-remodeling markers in multiple myeloma. Leukemia. 2008;22:1925–1932. doi: 10.1038/leu.2008.174. [DOI] [PubMed] [Google Scholar]
- 83.Rieman DJ, McClung HA, Dodds RA, Hwang SM, Holmes MW, James IE, et al. Biosynthesis and processing of cathepsin K in cultured human osteoclasts. Bone. 2001;28:282–289. doi: 10.1016/s8756-3282(00)00445-2. [DOI] [PubMed] [Google Scholar]
- 84.Anderson G, Gries M, Kurihara N, Honjo T, Anderson J, Donnenberg V, et al. Thalidomide derivative CC-4047 inhibits osteoclast formation by down-regulation of PU. 1. Blood. 2006;107:3098–3105. doi: 10.1182/blood-2005-08-3450. [DOI] [PubMed] [Google Scholar]
- 85.Heider U, Kaiser M, Muller C, Jakob C, Zavrski I, Schulz CO, et al. Bortezomib increases osteoblast activity in myeloma patients irrespective of response to treatment. Euro J Haematol. 2006;77:233–238. doi: 10.1111/j.1600-0609.2006.00692.x. [DOI] [PubMed] [Google Scholar]
- 86.Hideshima T, Chauhan D, Shima Y, Raje N, Davies FE, Tai YT, et al. Thalidomide and its analogs overcome drug resistance of human multiple myeloma cells to conventional therapy. Blood. 2000;96:2943–2950. [PubMed] [Google Scholar]
- 87.Pellagatti A, Jadersten M, Forsblom AM, Cattan H, Christensson B, Emanuelsson EK, et al. Lenalidomide inhibits the malignant clone and up-regulates the SPARC gene mapping to the commonly deleted region in 5q- syndrome patients. Proc Natl Acad Sci USA. 2007;104:11406–11411. doi: 10.1073/pnas.0610477104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis C-IAP2 is under NF-kappaB control. Proc Natl Acad Sci USA. 1997;94:10057–10062. doi: 10.1073/pnas.94.19.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kreuz S, Siegmund D, Scheurich P, Wajant H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol Cell Biol. 2001;21:3964–3973. doi: 10.1128/MCB.21.12.3964-3973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mitsiades N, Mitsiades CS, Poulaki V, Anderson KC, Treon SP. Intracellular regulation of tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human multiple myeloma cells. Blood. 2002;99:2162–2171. doi: 10.1182/blood.v99.6.2162. [DOI] [PubMed] [Google Scholar]
- 91.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science (New York, NY) 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 92.Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Richardson PG, Hideshima T, et al. Apoptotic signaling induced by immunomodulatory thalidomide analogs in human multiple myeloma cells: therapeutic implications. Blood. 2002;99:4525–4530. doi: 10.1182/blood.v99.12.4525. [DOI] [PubMed] [Google Scholar]