

Abstract
We investigated the phosphorylation signatures of two Rab-GTPase activating proteins TBC1D1 and TBC1D4 in human skeletal muscle in response to physical exercise and physiological insulin levels induced by a carbohydrate rich meal using a paired experimental design. Eight healthy male volunteers exercised in the fasted or fed state and muscle biopsies were taken before and immediately after exercise. We identified TBC1D1/4 phospho-sites that (1) did not respond to exercise or postprandial increase in insulin (TBC1D4: S666), (2) responded to insulin only (TBC1D4: S318), (3) responded to exercise only (TBC1D1: S237, S660, S700; TBC1D4: S588, S751), and (4) responded to both insulin and exercise (TBC1D1: T596; TBC1D4: S341, T642, S704). In the insulin-stimulated leg, Akt phosphorylation of both T308 and S473 correlated significantly with multiple sites on both TBC1D1 (T596) and TBC1D4 (S318, S341, S704). Interestingly, in the exercised leg in the fasted state TBC1D1 phosphorylation (S237, T596) correlated significantly with the activity of the α2/β2/γ3 AMPK trimer, whereas TBC1D4 phosphorylation (S341, S704) correlated with the activity of the α2/β2/γ1 AMPK trimer. Our data show differential phosphorylation of TBC1D1 and TBC1D4 in response to physiological stimuli in human skeletal muscle and support the idea that Akt and AMPK are upstream kinases. TBC1D1 phosphorylation signatures were comparable between in vitro contracted mouse skeletal muscle and exercised human muscle, and we show that AMPK regulated phosphorylation of these sites in mouse muscle. Contraction and exercise elicited a different phosphorylation pattern of TBC1D4 in mouse compared with human muscle, and although different circumstances in our experimental setup may contribute to this difference, the observation exemplifies that transferring findings between species is problematic.
Key points
Phosphorylation signature patterns on TBC1D1 and TBC1D4 proteins in the insulin–glucose pathway were investigated in human skeletal muscle in response to physiological insulin and exercise.
In response to postprandial increase in insulin, Akt phosphorylation of T308 and S473 correlated significantly with sites on TBC1D1 (T596) and TBC1D4 (S318, S341, S704).
Exercise induced phosphorylation of TBC1D1 (S237, T596) that correlated significantly with activity of the α2/β2/γ3 AMPK trimer, whereas TBC1D4 phosphorylation (S341, S704) with exercise correlated with activity of the α2/β2/γ1 AMPK trimer.
TBC1D1 phosphorylation signatures with exercise/muscle contraction were comparable between human and mouse skeletal muscle, and AMPK regulated phosphorylation of these sites in mouse muscle, whereas contraction and exercise elicited different TBC1D4 phosphorylation patterns in mouse compared with human muscle.
Our results show differential phosphorylation of TBC1D1 and TBC1D4 in response to physiological stimuli in human skeletal muscle and indicate that Akt and AMPK may be upstream kinases.
Introduction
Skeletal muscles constitute a major sink for whole body glucose disposal as evidenced by glucose uptake measurements during hyperinsulinaemic euglycaemic clamp conditions in non-insulin resistant individuals (DeFronzo et al. 1981; Richter et al. 1989). GLUT4 is the main glucose transporter in skeletal muscle and adipose tissue located within the endosomal and the trans-Golgi network compartments and to small tubule–vesicular structures (Slot et al. 1991; Ploug et al. 1998). In response to certain stimuli (e.g. muscle contraction, exercise, insulin, or hypoxia; Klip et al. 1987; Douen et al. 1989; Cartee et al. 1991; Kristiansen et al. 1996) these transporters are mobilized to the sarcolemma and/or T-tubules to facilitate glucose uptake. Skeletal muscle thus represents an important tissue for studying the intracellular signalling pathways leading to incorporation of GLUT4 molecules into the cell surface membrane.
The signalling pathway in skeletal muscle leading to glucose uptake in response to contraction is not completely understood. It is thought to involve increases in intracellular Ca2+ and activation of kinases downstream of LKB1 (i.e. 5′-AMP activated protein kinase (AMPK) and sucrose nonfermenting AMPK-related kinase (SNARK); Friedrichsen et al. 2012; Jensen & Richter, 2012). The signalling pathway whereby insulin stimulates glucose uptake has been resolved in greater detail, and is known to involve the insulin receptor – insulin receptor substrate – phosphatidylinositol 3 kinase (IR-IRS-PI3K)–Akt2 axis (Huang & Czech, 2007; Ishikura et al. 2008). Akt2 activation is necessary for insulin-stimulated GLUT4 translocation to the plasma membrane in adipocytes (Hill et al. 1999) and for insulin-stimulated glucose uptake in skeletal muscle (Cho et al. 2001; Bouzakri et al. 2006). In the search for novel Akt targets involved in these processes, an Akt substrate of 160 kDa (AS160), known as TBC1D4 (Nagase et al. 1998), was identified as a Rab-GTPase activating protein (Rab-GAP) (Sano et al. 2003). It is now evident that TBC1D4 has multiple upstream kinases also including AMPK (Kramer et al. 2006a; Treebak et al. 2010) and potentially protein kinase Cζ (Ng et al. 2010).
The current thinking is that Rab-GAPs in the signalling pathways to stimulate glucose uptake repress Rab-GTPase function in the basal state by inhibiting the exchange of GDP to GTP leading to intracellular retention of GLUT4 vesicles (Kane et al. 2002; Sakamoto & Holman, 2008). In response to stimulation Rab-GAPs are phosphorylated, for instance by Akt or AMPK, allowing for the active exchange of GDP to GTP in the Rab-GTPase and thus stimulating GLUT4 mobilization to the plasma membrane (Hoffman & Elmendorf, 2011). In addition to TBC1D4, the TBC1D4 paralogue TBC1D1 has been identified as a functional Rab-GAP. Similarly to TBC1D4, TBC1D1 has inhibitory effects on GLUT4 translocation in 3T3-L1 adipocytes (Roach et al. 2007) and skeletal muscle (An et al. 2010). TBC1D4 and TBC1D1 are involved in regulation of both insulin- and contraction-induced glucose uptake in intact mouse skeletal muscle (Kramer et al. 2006b; An et al. 2010), and TBC1D4 has emerged as a nexus for insulin- and contraction-responsive signals, potentially mediating enhanced insulin action in human and rat skeletal muscle after exercise (Arias et al. 2007; Funai et al. 2009; Treebak et al. 2009b; Pehmøller et al. 2012).
The phosphorylation status of TBC1D1 and TBC1D4 in response to insulin stimulation in human skeletal muscle has been studied only during hyperinsulinaemic euglyacemic clamp conditions and most studies have used the phospho-Akt substrate (PAS) antibody (Karlsson et al. 2005a,b2005b; O'Gorman et al. 2006; Frøsig et al. 2007; Howlett et al. 2007, 2008; Højlund et al. 2008; Hoeg et al. 2011), whereas other studies (Treebak et al. 2009b; Vind et al. 2011; Pehmøller et al. 2012; Vendelbo et al. 2012; Consitt et al. 2013) have used site- and phospho-specific antibodies. No study has investigated whether physiological insulin levels (i.e. insulin levels obtained after a meal) would induce changes in phosphorylation status of the two proteins similarly to those observed during sustained elevated insulin levels as applied during the hyperinsulinaemic clamp condition. Moreover, acute exercise in humans is known to induce changes in phosphorylation status of both TBC1D1 and TBC1D4 (Frøsig et al. 2010; Jessen et al. 2011), but the phosphorylation response to physiological insulin stimulation combined with acute exercise remains to be established. Here we employed the one-legged knee extensor exercise model (Andersen et al. 1985) to investigate skeletal muscle site-specific phosphorylation signature patterns of TBC1D1 and TBC1D4 in response to exercise in the fasted or fed condition of healthy human subjects. This design allows for a paired evaluation of the phosphorylation signatures in skeletal muscle at rest and in response to acute exercise alone, physiological insulin stimulation alone, and acute exercise combined with physiological insulin stimulation. We hypothesized the presence of stimuli-dependent phosphorylation signatures on TBC1D1 and TBC1D4. Identification of these signatures in response to physiological stimuli will increase the understanding of how TBC1D1 and TBC1D4 are regulated and how these proteins may impact on metabolism in human skeletal muscle.
Methods
Ethical approval
The human experiments in this study were conducted in accordance with the Helsinki II Declaration and approved by the ethics committee of Capital Region of Denmark (no. KF1277313). Protocols for animal studies were approved by the Danish Animal Experimental Inspectorate and complied with the European Convention for the protection of Vertebrate Animals used for Experiments and Other Scientific Purposes (Council of Europe 123, Strasbourg, France, 1985), UK regulations, and the policies of The Journal of Physiology.
Subjects and initial tests
Eight healthy moderately trained male subjects (age 27 ± 2 years, weight 83.5 ± 2.6 kg, body mass index 24.8 ± 0.6 kg m−2) volunteered for this study and gave their written informed consent prior to participation. Subjects initially came to the laboratory on two separate days to complete an incremental cycle ergometer test to determine peak oxygen uptake (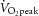 52 ± 2 ml O2 min−1 kg−1), and an incremental one-legged knee extensor exercise test to determine the maximal work load for the knee extensors (51 ± 2 W). Half of the subjects exercised with their dominant leg and the other half with their non-dominant leg.
52 ± 2 ml O2 min−1 kg−1), and an incremental one-legged knee extensor exercise test to determine the maximal work load for the knee extensors (51 ± 2 W). Half of the subjects exercised with their dominant leg and the other half with their non-dominant leg.
Experimental days
Two–three weeks after the initial baseline testing, the subjects came by car or public transportation to the laboratory on two separate occasions. Subjects were instructed to refrain from physical activity 2 days prior to the experiment. On both occasions, subjects came in at 07.00 h after an overnight fast. Upon arrival at the laboratory subjects rested for 30–40 min in the supine position, after which one muscle biopsy was obtained from the vastus lateralis muscle of each leg. Subjects rested for 20 min and then performed a single, 60 min bout of one-legged knee extensor exercise at 80% of peak work load (41 ± 2 W) with two 5 min intervals at 100% of peak work load (51 ± 2 W) after 15 and 35 min. The other leg served as a rested control. On one occasion subjects performed the exercise bout after an overnight fast and on another occasion subjects were given a carbohydrate rich (∼80 E%) meal consisting of cornflakes with skimmed milk corresponding to 5% (∼600–800 kJ) of their daily energy intake. This meal was given after the resting biopsy had been obtained and approximately 15 min before the exercise bout commenced. One participant only took part in the exercise trial in the fed state. Thus n = 7 completed the fasted trial and n = 8 completed the fed trial. Immediately after termination of the exercise bout muscle biopsies were obtained from vastus lateralis muscles of both legs. Thus, a total of eight (2 × 4) biopsies were obtained from each subject. All biopsies were obtained from different incisions, separated by at least 5 cm. Muscle biopsies were obtained under local anaesthesia (2–4 ml of 2% lidocaine) and were immediately stored at −80°C.
Incubation of intact mouse skeletal muscle in vitro
Extensor digitorum longus (EDL) and soleus muscles were quickly removed from anaesthetized (intraperitoneal injection of pentobarbital; 6 mg/100 g body weight–1) fed male mice aged 16–20 weeks and incubated in Krebs-Henseleit buffer (117 mm NaCl, 4.7 mm KCl, 2.5 mm CaCl2, 1.2 mm KH2PO4, 1.2 mm MgSO4, and 24.6 mm NaHCO3 with addition of 8 mm mannitol, 2 mm pyruvate, and 0.1% BSA) at 30°C, oxygenated with a gas containing 95% O2 and 5% CO2; Multi Myograph system, DMT, Denmark). To assess the effects of contraction on AMPK signalling and TBC1D1 and TBC1D4 phosphorylation, muscles from α2 AMPK KO/KD mice (Mu et al. 2001; Viollet et al. 2003) and corresponding wild-type (WT) littermates (n = 8–12) were incubated for 40 min. Subsequently, contraction was induced by electrical stimulation with a 10 s train (100 Hz, 0.2 ms impulse, ∼30 V) per minute for 10 min. Contralateral muscles were incubated for the same duration under resting condition. After incubation, muscles were harvested, washed in ice-cold Krebs-Henseleit buffer, blotted on filter paper, and quickly frozen with aluminium tongs pre-cooled in liquid nitrogen and stored at −80°C. We chose to determine TBC1D1 phosphorylation patterns in response to contraction in EDL muscle and TBC1D4 phosphorylation patterns in response to contraction in soleus muscles. This was in part due to the restricted number of animals available for these analyses, but also because of our previous findings showing that TBC1D1 is more abundant in EDL compared with soleus and that TBC1D4 is more abundant in soleus compared with EDL (Pehmøller et al. 2009).
Mouse treadmill running protocol
Five days prior to the experimental day, male C57BL/6J WT mice (n = 24, 12–14 weeks of age) were familiarised to treadmill running for four consecutive days (10–15 min at 8–18 m min−1). Mice rested 1 day prior to the experiment. On the day of the experiment mice (n = 12) were run on a motorized treadmill (Exer 3/6, Columbus Instruments, OH, USA) for 1 h at 18 m min−1 with an incline of 5 deg. Control mice (n = 12) were rested on a dummy treadmill (no motor) that was placed next to the motorized treadmill so that control mice were exposed to the same environment as the exercised mice. Immediately after exercise mice were killed by cervical dislocation and soleus and EDL muscles were rapidly dissected and quickly frozen in liquid nitrogen. Samples were stored at −80°C until processed as described below.
Blood chemistry
Plasma glucose and lactate levels were determined using an ABL 615 (Radiometer Medical A/S, Denmark). Plasma insulin levels were determined by ELISA (EIA1825, DRG International, Inc., Mountainside, NJ, USA).
Glycogen
Muscle glycogen was measured as glucosyl units after acid hydrolysis. The analysis was performed by fluorometry using freeze-dried, dissected muscle specimens (Passonneau et al. 1967).
Muscle sample preparation
A piece of each human muscle sample was freeze-dried and dissected free of visible fat and connective tissue under a microscope. Human and mouse muscle samples were homogenized in ice-cold buffer (50 mm Hepes, 150 mm NaCl, 20 mm Na4P2O7, 20 mm β-glycerophosphate, 10 mm NaF, 2 mm Na3VO4, 2 mm EDTA, 1% Nonidet P-40, 10% glycerol, 2 mm phenylmethylsulfonyl fluoride (PMSF), 10 μg ml−1 leupeptin, 10 μg ml−1 aprotinin and 3 mm benzamidine). Homogenates were rotated end-over-end (4°C, 1 h) and were subjected to centrifugation (16,000 g, 20 min, 4°C). Supernatants were harvested and protein concentrations were determined (BCA no. 23225, Pierce, Rockford, IL, USA).
Antibodies
The phosphorylation status of TBC1D1/TBC1D4 was measured using a phospho-Akt-substrate (PAS) antibody (no. 9611, Cell Signalling Technology, MA, USA), and site- and phospho-specific antibodies against TBC1D4 (S318, S341, S588, T642, S666, S704 and S751; Geraghty et al. 2007; Treebak et al. 2010) and TBC1D1 (S237, T596, S660, and S700; Vichaiwong et al. 2010), as previously described and validated. All phosphorylation sites refer to the human sequence for TBC1D1 (UniProt entry Q86TI0) and TBC1D4 (UniProt entry O60343), except for S660 and S700 on TBC1D1 which refer to the mouse sequence (UniProt entry Q60949). Supplemental Table S1 provides an overview of the surrounding amino acids of the different phospho-sites. Total TBC1D4 protein was determined using an antibody specific for human TBC1D4 (ab24469, Abcam plc, Cambridge, UK) and total TBC1D1 protein was determined with an antibody specific for human TBC1D1 as described previously (Chen et al. 2008). Phosphorylation of T172 on AMPK (no. 2531, Cell Signalling Technology), S221 on acetyl-CoA carboxylase (ACC; no. 07-303, Upstate), Akt on T308 (no. 06-678 Upstate), and on S473 (no. 9271, Cell Signalling Technology) was measured using the site- and phospho-specific antibodies indicated. Total Akt was measured using antibody no. 9272 from Cell Signalling Technology, and AMPK α1 and α2 subunits were detected using antibodies kindly provided by Professor D. Grahame Hardie (University of Dundee, UK).
SDS-PAGE and Western blot analyses
Western blot analyses were performed as previously described (Treebak et al. 2007). Membranes probed with the phospho-specific antibodies for TBC1D4/TBC1D1 were stripped in a buffer containing 100 mm 2-mercaptoethanol, 2% SDS, and 62.5 mm Tris-HCl (pH 6.7) and re-probed with antibodies recognizing total TBC1D4 and TBC1D1 protein. The signal coming from the phospho-specific antibodies was related to total TBC1D4/TBC1D1 in the sample. Human muscle samples from the two exercise trials (i.e. fasted and fed) were run on the same gel during Western blot analyses, allowing for direct comparisons between the two trials.
AMPK activity assay
AMPK activity was measured on muscle lysates by immunoprecipitation (IP) of the γ3 subunit (isolating the α2/β2/γ3 complex from human and mouse lysate) followed by IP of the α2 (isolating the α2/β2/γ1 from human lysate, and the α2/β1/γ1 and α2/β2/γ1 from mouse lysate) and lastly the α1 subunit (isolating the α1/β2/γ1 from human lysate, and the α1/β1/γ1 and α1/β2/γ1 from mouse lysate) as described previously (Birk & Wojtaszewski, 2006; Treebak et al. 2009a).
14-3-3 overlays
14-3-3 overlays were carried out as previously described (Pozuelo Rubio et al. 2004; Pehmøller et al. 2009). Briefly, endogenous TBC1D4 and TBC1D1 protein was immunoprecipitated from 150 μg of skeletal muscle lysates, electrophoretically separated on Invitrogen Nu-PAGE gels, and subsequently transferred to polyvinylidene difluoride membranes (Millipore, Denmark). Membranes were subsequently probed with digoxigenin-labelled 14-3-3 at 4°C overnight followed by incubation with a HRP-conjugated anti-digoxigenin antibody (Roche, UK). Immuno-complexes were visualised and quantified using a Kodak Image Station (2000MM, Kodak, Denmark) and normalised to TBC1D4/TBC1D1 protein.
Statistics
Statistical analyses were performed in SigmaPlot 11.0 (Systat software, Inc., Germany). Unpaired t tests and two-, or three-way ANOVAs with repeated measures were used to analyse the data. When sets of data contained more than two factors, data obtained from the samples taken before the exercise bout commenced were analysed to test whether there were differences between the fasted and the fed trial and between the rested and the exercised leg, respectively. In all cases, except for the TBC1D4 pT642 and α1/β2/γ1 AMPK activity data, there were no differences between the trials or legs before the exercise. Thus, within each trial, the delta values (e.g. rested leg post exercise minus rested leg before exercise) were calculated and the resulting set of data was analysed by two-way repeated measures ANOVA. Although presented in the same figure panel (Fig. 7E), the TBC1D4 pT642 data from the fasted and the fed trials have been analysed separately by a two-way ANOVA with repeated measures. The α1/β2/γ1 AMPK activity data were analysed with a three-way ANOVA with repeated measures (Fig. 3C). In the case of interaction effects, the Tukey test was used post-hoc in all analyses. Correlation analyses were performed by calculating Pearson's product moment correlation coefficient. Statistical significance was accepted at P < 0.05.
Figure 7.
Phosphorylation signatures of TBC1D4 in human skeletal muscle in response to exercise and physiological insulin stimulation
Figure 3.
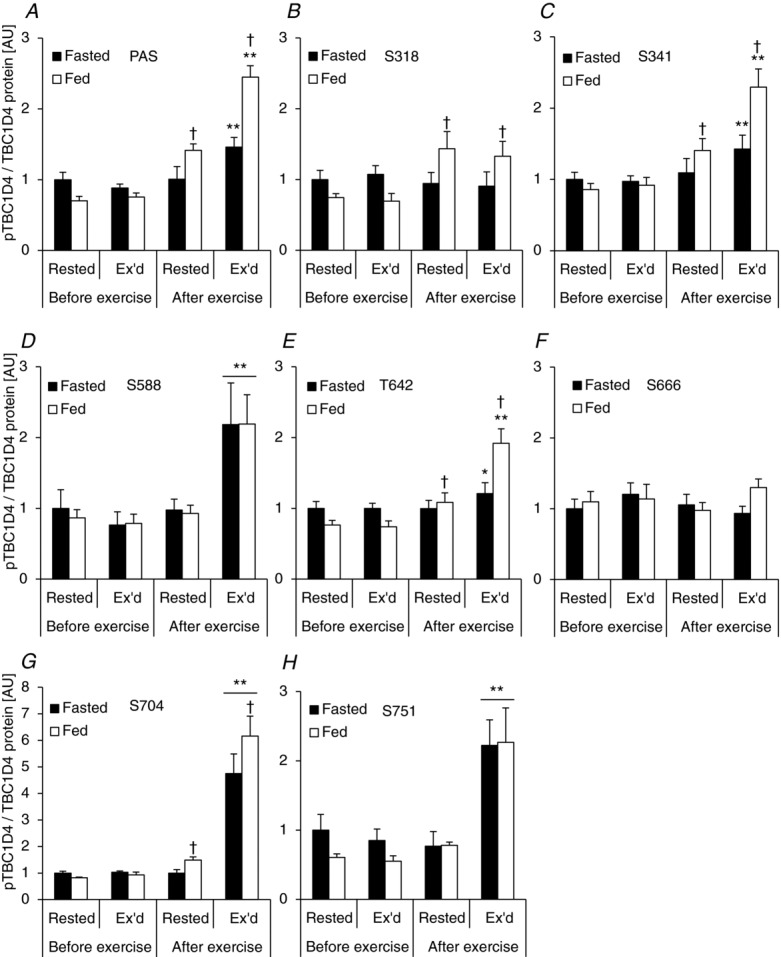
Acute exercise increases AMPK trimer-specific activity in human skeletal muscle
Results
Blood chemistry and muscle glycogen
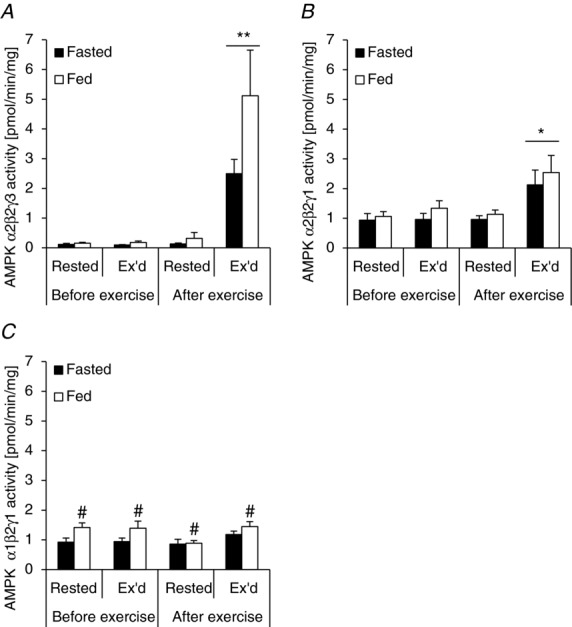
We set out to investigate the specific phosphorylation patterns on TBC1D1 and TBC1D4 in response to an acute bout of one-legged knee extensor exercise, acute physiological ‘insulin’ stimulation induced by a small carbohydrate rich meal, and the two stimuli combined. Using the one-legged knee extensor exercise model in the fasted and fed condition combined with biopsies from both legs in each trial, muscle samples were obtained representing four different experimental conditions. One leg was rested (non-stimulated), one leg was rested and insulin-stimulated, one leg was exercised, and one leg was exercised and insulin-stimulated. When the subjects were fed a meal just prior to (i.e. 15 min) the exercise bout, plasma glucose levels were significantly elevated compared with the fasted trial (Fig. 1A). At baseline, plasma insulin levels were not different between the two trials, but after the meal, plasma insulin levels were >6-fold higher than during the fasted trial (Fig. 1B). During the course of the exercise period in the fasted state, plasma insulin levels did not change significantly. However, in the fed state, plasma insulin levels decreased significantly after 30 and 60 min but remained elevated compared with the fasted trial (Fig. 1B). Plasma lactate increased to a similar extent in response to the exercise bout in the fasted and fed trials (Fig. 1C), and muscle glycogen was similarly reduced in the exercised leg in the two trials (Fig. 1D).
Figure 1.
Plasma levels of glucose, lactate and insulin as well as muscle glycogen content
Activation of potential upstream TBC1D1/4 kinases
AMPK and Akt are putative upstream kinases of TBC1D1/4 (Kane et al. 2002; Treebak et al. 2006; Kramer et al. 2006a; Pehmøller et al. 2009; Vichaiwong et al. 2010). Furthermore, these kinases are known to be activated by insulin and exercise. Phosphorylation of AMPK on T172 is essential for its activation (Hawley et al. 1996) and S221 on ACC is a well-established AMPK target (Ha et al. 1994). AMPK and ACC were phosphorylated in response to exercise in the exercised leg only, and the increase was similar in the fasted and fed condition (Fig. 2A and B). Full activation of Akt requires phosphorylation of T308 and S473 (Alessi et al. 1996). It has previously been shown that Akt phosphorylation can be induced by submaximal insulin levels (Karlsson et al. 2006) and exercise (Sakamoto et al. 2004; Deshmukh et al. 2006; Treebak et al. 2007) in human skeletal muscle. To our knowledge no-one has investigated whether the two stimuli together have additive effects on Akt phosphorylation in human skeletal muscle. In the fasted state, phosphorylation of T308 did not increase significantly with exercise (Fig. 2C). In the fed state, on the other hand, T308 phosphorylation was significantly increased in both the rested and exercised leg. Interestingly, we observed an interaction effect indicating a synergistic effect on T308 when exercising in the fed state (Fig. 2C). Contrary to T308, phosphorylation on S473 increased significantly with exercise in the fasted state (Fig. 2D). Furthermore, S473 phosphorylation was increased in the rested leg in the fed state, and there was an additive effect on S473 phosphorylation when the two stimuli were combined (Fig. 2D).
Figure 2.
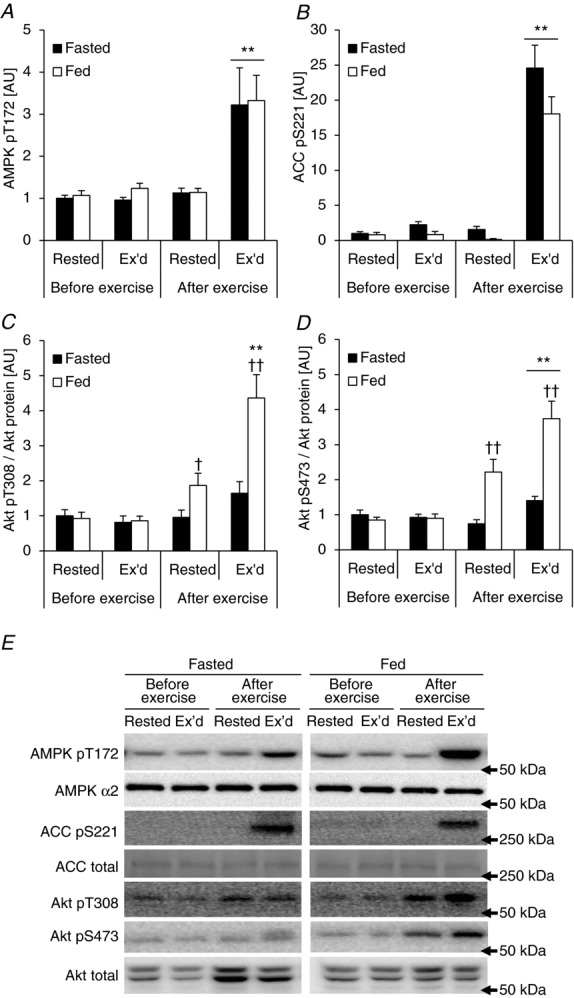
Acute exercise and physiological insulin stimulation affect AMPK and Akt signalling in human skeletal muscle
Specific AMPK trimers are activated in response to one-legged knee extensor exercise
Phosphorylation of AMPK at T172 correlates well with total AMPK kinase activity in human skeletal muscle during whole body exercise (Wojtaszewski et al. 2002; Birk & Wojtaszewski, 2006). However, it is not known whether specific AMPK trimer complexes are activated in response to one-legged knee extensor exercise. Furthermore, in order for us to assess the association between specific upstream AMPK kinase complexes and the phosphorylation of TBC1D1 and TBC1D4, we measured the activity of the three different AMPK trimer complexes found in human skeletal muscle (Wojtaszewski et al. 2005). Activity of both AMPK α2/β2/γ1 and α2/β2/γ3 trimers increased in the exercised leg only and there was no statistical difference between the fasted and fed condition (Fig. 3A and B). The AMPK α1/β2/γ1 complex did not respond to exercise, but the activity was higher in the fed compared with the fasted trial (Fig. 3C).
TBC1D1 phosphorylation signatures in human skeletal muscle
Site-specific phosphorylation of TBC1D1 in human skeletal muscle in response to exercise has been studied recently (Frøsig et al. 2010; Jessen et al. 2011), but no prior study has investigated the effect of physiological insulin stimulation or the interaction between exercise and physiological insulin stimulation. Here we found that exercise, but not physiological insulin, increased phosphorylation of S237, a site regulated by AMPK α2 in response to muscle contraction ex vivo in mice (Pehmøller et al. 2009; Frøsig et al. 2010; Fig. 4A). On the other hand, T596 phosphorylation was regulated by both exercise (∼70%) and physiological insulin (∼35%), and the two stimuli combined gave an additive response (Fig. 4B). TBC1D1 has two other phosphorylation sites (S660 and S700; residues refer to the mouse sequence of TBC1D1; Jessen et al. 2011) that have been predicted to be phosphorylated by AMPK (Taylor et al. 2008). This was recently confirmed for S660 (Vichaiwong et al. 2010). In addition, S660 has been shown to be phosphorylated in response to exercise in human skeletal muscle (Jessen et al. 2011). Here we show that both S660 and S700 are phosphorylated exclusively by exercise and not by physiological insulin in human skeletal muscle (Fig. 4C and D).
Figure 4.
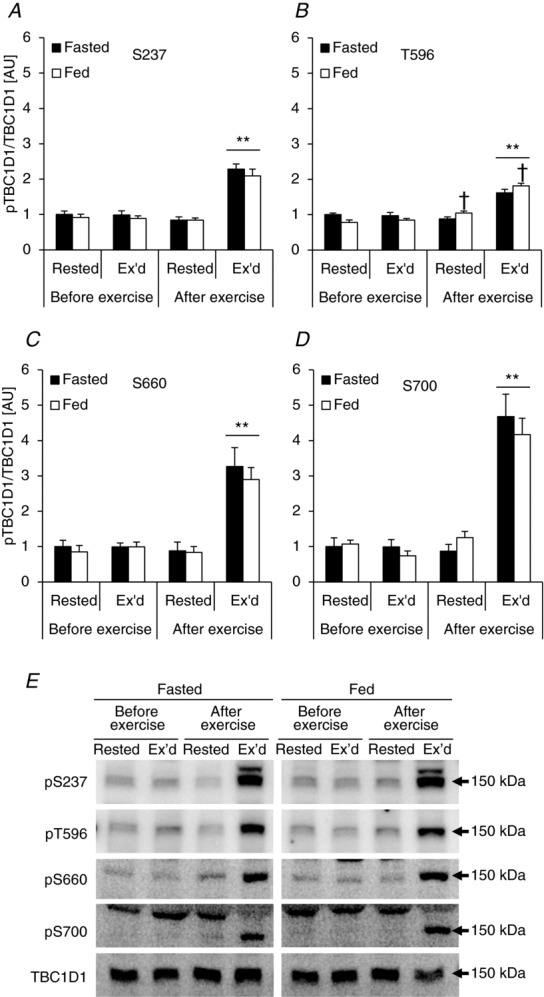
Phosphorylation signatures of TBC1D1 in human skeletal muscle in response to exercise and physiological insulin stimulation
Muscle contraction induces phosphorylation of TBC1D1 on specific sites in an AMPK-dependent manner
To investigate the potential upstream kinase regulating the different phosphorylation sites on TBC1D1 in response to exercise in humans we employed a genetic approach. We have previously investigated whether phosphorylation of TBC1D1 in intact skeletal muscle in response to contraction ex vivo is AMPK dependent (Pehmøller et al. 2009; Frøsig et al. 2010; Vichaiwong et al. 2010). By incubating and electrically stimulating EDL muscle to contract we confirm that phosphorylation of S237, T596, and S660 are increased with contraction and that this occurs in an AMPK-dependent manner (Fig. 5A–C). In addition, we show that the increased phosphorylation of S700 in response to contraction is also dependent on AMPK (Fig. 5D). Thus, it is possible that in human skeletal muscle, the kinase responsible for phosphorylation of S237, T596, S660, and S700 during exercise is indeed AMPK. In order to test whether specific AMPK trimer complexes could be responsible for phosphorylating TBC1D1, we measured AMPK activity in EDL muscles from WT and α2 AMPK knockout mice. Both AMPK γ1- and γ3-containing trimers were robustly activated in response to contraction in the WT mice (Fig. 6), and we were therefore not able in this way to identify a specific AMPK trimer as the responsible TBC1D1 kinase.
Figure 5.
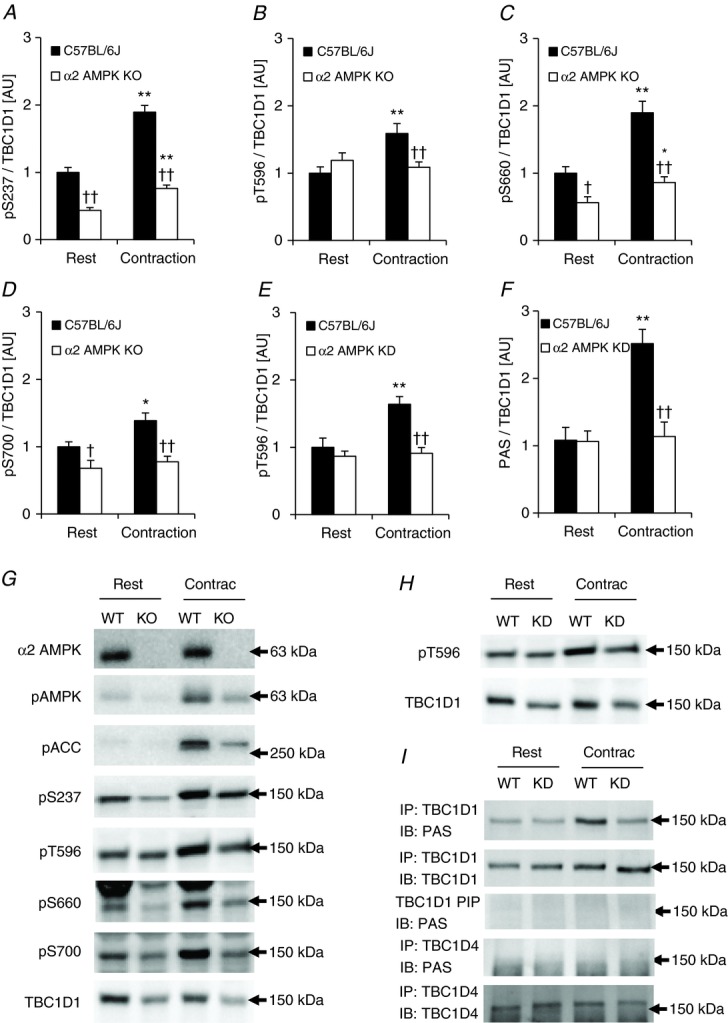
TBC1D1 phosphorylation signature in response to in vitro contraction in mouse skeletal muscle
Figure 6.
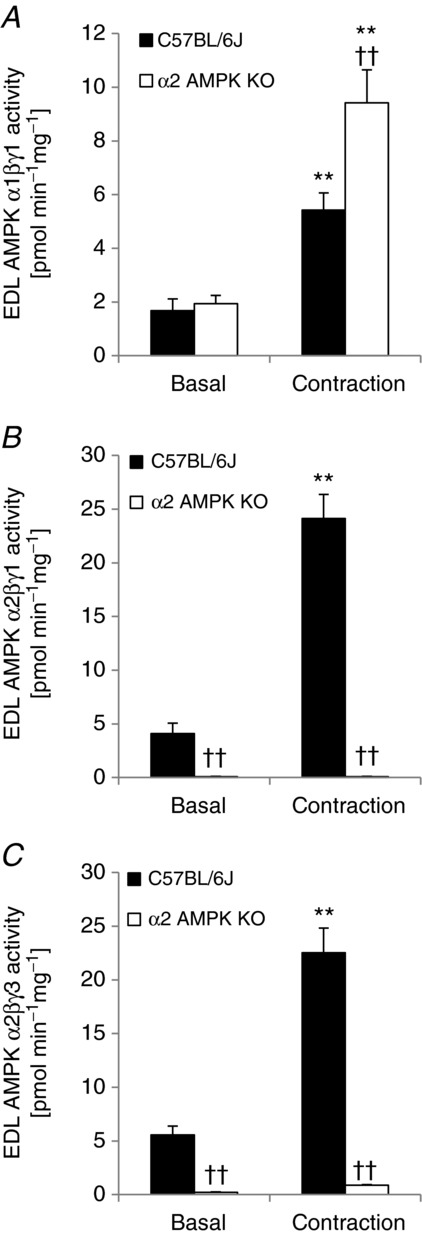
In vitro contraction of mouse skeletal muscle activates γ1 and γ3 AMPK trimers
Using the PAS antibody, we have previously reported that TBC1D4 was phosphorylated during contraction in EDL muscle and that this increased phosphorylation was ablated in the α2 AMPK KO and KD mice (Treebak et al. 2006). We and others have subsequently shown that the main PAS signal in EDL muscle derives from TBC1D1 (Taylor et al. 2008; Pehmøller et al. 2009; Ducommun et al. 2012), and data from HEK293 and 3T3-L1 adipocytes suggest that the PAS antibody primarily detects T596 on TBC1D1. Here we reanalysed the samples from our previous α2 AMPK KD/WT contraction experiment (Treebak et al. 2006) and evaluated both T596 and PAS phosphorylation. T596 phosphorylation was measured on the individual lysates (n = 8–10), whereas PAS phosphorylation was measured on immunoprecipitated TBC1D1 from pools of lysates (n = 4–5). In accordance with data from the α2 AMPK KO/WT samples (Fig. 5B), contraction increased phosphorylation of T596 in muscle from WT mice, but not in the α2 AMPK KD muscles (Fig. 5E and H). Furthermore, contraction increased PAS phosphorylation in TBC1D1 immunoprecipitates, but only in WT mice (Fig. 5F and top panel in 5I). We also performed Western blot analyses to examine the supernatants from the TBC1D1 immunoprecipitates and found that the PAS signal around 150–160 kDa was completely absent (Fig. 5I). Finally, we immunoprecipitated TBC1D4 using the same samples as described in the above and analysed these immunocomplexes for PAS phosphorylation. We were unable to detect any phosphorylation of TBC1D4 in these samples using the PAS antibody (Fig. 5I, fourth panel from the top). Thus, in EDL muscle, contraction-induced PAS phosphorylation is AMPK dependent and seems to involve only TBC1D1 and not TBC1D4.
TBC1D4 phosphorylation signatures in human skeletal muscle
TBC1D4 contains nine confirmed phosphorylation sites (i.e. S318, S341, T568, S570, S588, T642, S666, S704, and S751; Geraghty et al. 2007; Treebak et al. 2010), and we investigated the phosphorylation signatures of TBC1D4 induced by exercise and physiological insulin, separately or combined on seven of these sites. We have not been able to obtain good quality Western blots with antibodies against T568 and S570. Overall, TBC1D4 contains sites that only respond to insulin (S318), that only respond to exercise (S588 and S751), that respond to both insulin and exercise (S341, T642, and S704), and that do not respond to either stimuli (S666). We have previously shown that phosphorylation of TBC1D4 in human skeletal muscle in response to both exercise and insulin can be detected by the PAS antibody (Treebak et al. 2007; Højlund et al. 2008) and there is strong evidence that the PAS antibody
primarily detects pT642 in cell-based models (Sano et al. 2003; Ramm et al. 2006; Geraghty et al. 2007). In line with this observation, we found that both PAS and T642 phosphorylation increased with exercise and insulin, and that the two stimuli combined induced a synergistic effect on the phosphorylation level (Figs 7A and E and 8). The phosphorylation patterns of S341 and S704 were similar to that of T642 (Figs 7C and G and 8) in that these sites responded significantly to both insulin and exercise. Furthermore, there was an additive (S704) and even a synergistic (S341) response when insulin and exercise were combined. S704 phosphorylation was found earlier to respond to both contraction and maximal insulin stimulation in rodent skeletal muscle (Treebak et al. 2010), and to insulin during hyperinsulinaemic euglycaemic clamp conditions in human skeletal muscle (Pehmøller et al. 2012). Here we show that this also occurs in human skeletal muscle under physiological conditions. We have previously found that both S588 and S751 can be phosphorylated in response to high insulin concentrations during a hyperinsulinaemic euglycaemic clamp (Treebak et al. 2009b; Vind et al. 2011; Biensø et al. 2012; Pehmøller et al. 2012). In the present study, however, S588 and S751 were only phosphorylated after exercise and did not respond to a physiological insulin stimulus (Figs 7D and H and 8). Finally, S666 did not respond to either exercise or insulin (Figs 7F and 8) and S318 responded only to insulin (Figs 7B and 8).
Figure 8.
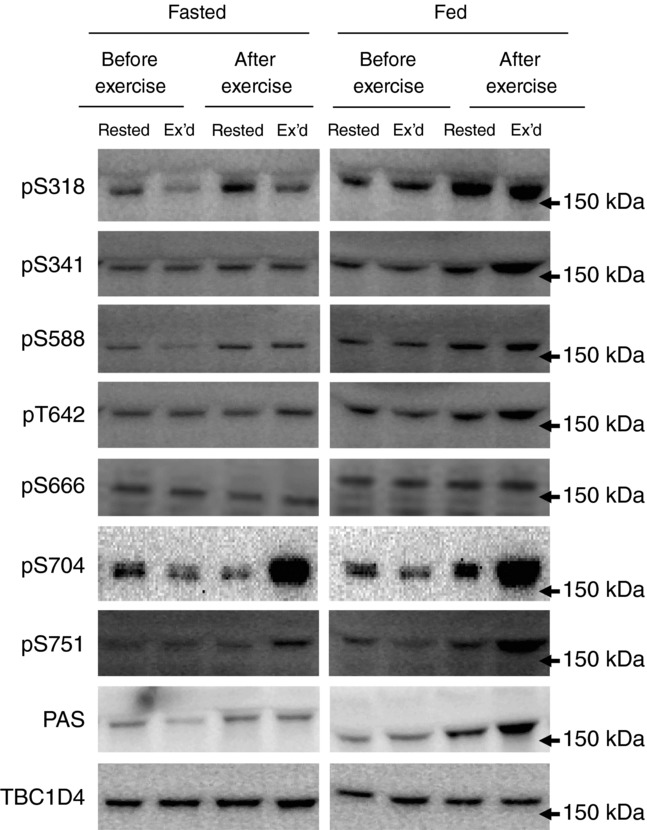
Representative Western blots for the data presented in Fig. 7 with molecular weight marker indications
AMPK trimer activity correlates with phosphorylation level on specific TBC1D1 and TBC1D4 sites in response to exercise in humans
To further explore the possibility that AMPK is regulating phosphorylation of specific sites on TBC1D1 and TBC1D4 in human skeletal muscle, we performed correlation analyses between specific AMPK trimer activities and phosphorylation levels of those phosphorylation sites on TBC1D1 and TBC1D4 that were phosphorylated in response to exercise (i.e. in the exercised leg after exercise in the fasted state). On TBC1D1 all four sites investigated were phosphorylated in response to exercise, and there was a significant correlation between α2/β2/γ3 AMPK trimer activity and phosphorylation of S237 (P < 0.05) and T596 (P < 0.01; Fig. 9A and B). Although phosphorylation of S660 and S700 correlated with borderline significance with α2/β2/γ3 AMPK activity (P = 0.12; R2 = 0.49; n = 7 for S660; P = 0.09; R2 = 0.55; n = 7 for S700; data not shown) these analyses may be biased by the distribution of the data and the low number of subjects. Interestingly, of the three existing human AMPK trimers, exercise-induced TBC1D1 phosphorylation at S237 and T596 correlated only with activity associated with the α2/β2/γ3 trimer. On the other hand, only α2/β2/γ1 trimer activity correlated with specific exercise-induced phosphorylation sites on TBC1D4. We found that several TBC1D4 sites were phosphorylated in response to exercise (i.e. S341, S588, T642, S704, and S751), but only S341 and S704 correlated significantly with α2/β2/γ1 trimer activity (Fig. 9C and D). Phosphorylation of S588 and S751 that was highly induced by exercise correlated only with borderline significance (P = 0.12; R2 = 0.41; n = 7 for S588; P = 0.1; R2 = 0.45; n = 7 for S751; data not shown). We designed this study in a paired fashion which enabled us to correlate the increase in phosphorylation with the increase in AMPK trimer activity in response to exercise (i.e. delta vs. delta correlation). When changes in TBC1D1 phosphorylation were correlated with changes in activity of the AMPK trimer complexes, this slightly improved the correlation between S700 and AMPK α2/β2/γ3 activity (P = 0.07; R2 = 0.58; n = 7; data not shown), but the only significant correlation remaining was between S237 phosphorylation and α2/β2/γ3 trimer activity (P < 0.05; R2 = 0.60; n = 7; data not shown). In addition, changes in TBC1D4 phosphorylation at S341 (P < 0.01; R2 = 0.77; n = 7; data not shown) and S704 (P < 0.01; R2 = 0.86; n = 7; data not shown) remained significantly correlated with changes in α2/β2/γ1 AMPK complex activity. In short, correlation analyses support the idea that TBC1D1 (S237) and TBC1D4 (S341, S704) are phosphorylated by AMPK in human skeletal muscle during exercise.
Figure 9.
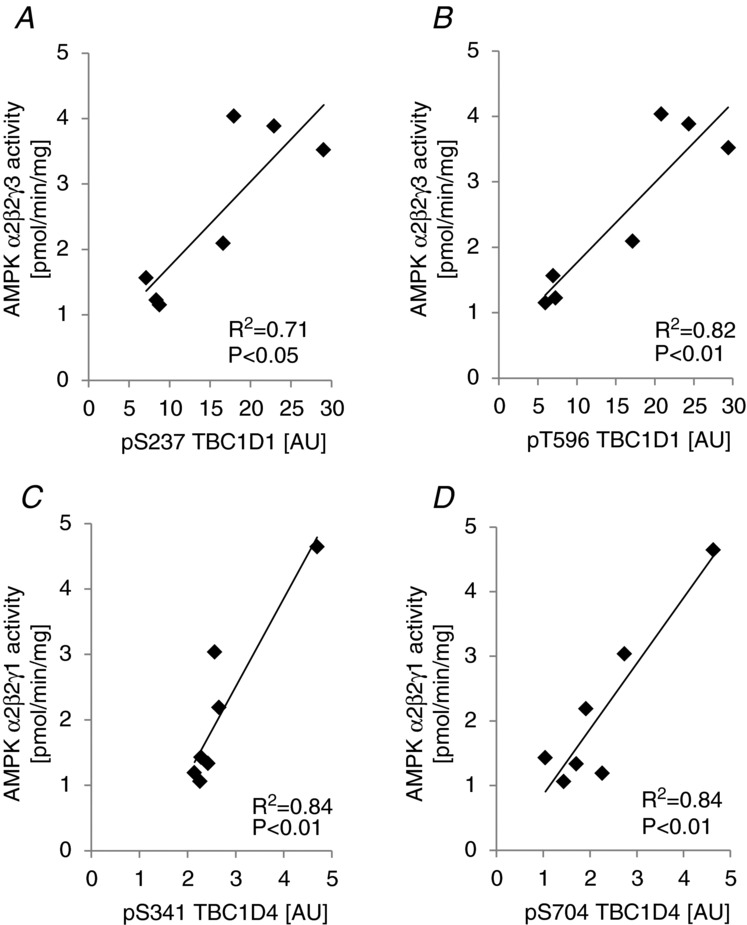
AMPK trimer activity correlates with phosphorylation of TBC1D1/4 on specific sites in human skeletal muscle
Muscle contraction induces phosphorylation of TBC1D4 on specific sites in an AMPK-dependent manner
Exercise in humans led to increased phosphorylation of S341, S588, T642, S704, and S751 on TBC1D4 (Fig. 7). To explore the idea that AMPK is the upstream kinase mediating this effect we incubated soleus muscles from WT and α2 AMPK KO mice and electrically stimulated them to contract. As shown before (Treebak et al. 2010), phosphorylation of S704 was increased 2-fold in response to contraction in WT mice and lack of AMPK led to an overall reduced level of phosphorylation (main effect of genotype; Fig. 10C). Interestingly, none of the other specific sites were phosphorylated by contraction and there was even a significant reduction in phosphorylation on sites T642 and S751 (Fig. 10B and D). Phosphorylation of S588 was generally lower in the knockout mice, but did not respond to contraction (Fig. 10A). Using a low percentage polyacrylamide gel (4.5%), and performing Western blot analyses with the PAS antibody, we were able to separate two bands, one running at 150 kDa and one running at 160 kDa. We have previously found that these bands correspond to TBC1D1 and TBC1D4, respectively (Taylor et al. 2008; Pehmøller et al. 2009). We found that in vitro contraction significantly increased phosphorylation of the 150 kDa band in both WT and α2 AMPK KO animals (Fig. 10E). Although the increase in phosphorylation was not statistically different between the two genotypes, there was a generally lower level of phosphorylation in the α2 AMPK KO mice. This difference between genotypes was also present for the 160 kDa band, but phosphorylation decreased significantly in response to contraction (Fig. 10F). Thus, it appears that in soleus the PAS antibody detects both TBC1D1 and TBC1D4, and that these proteins are differentially regulated during muscle contraction.
Figure 10.
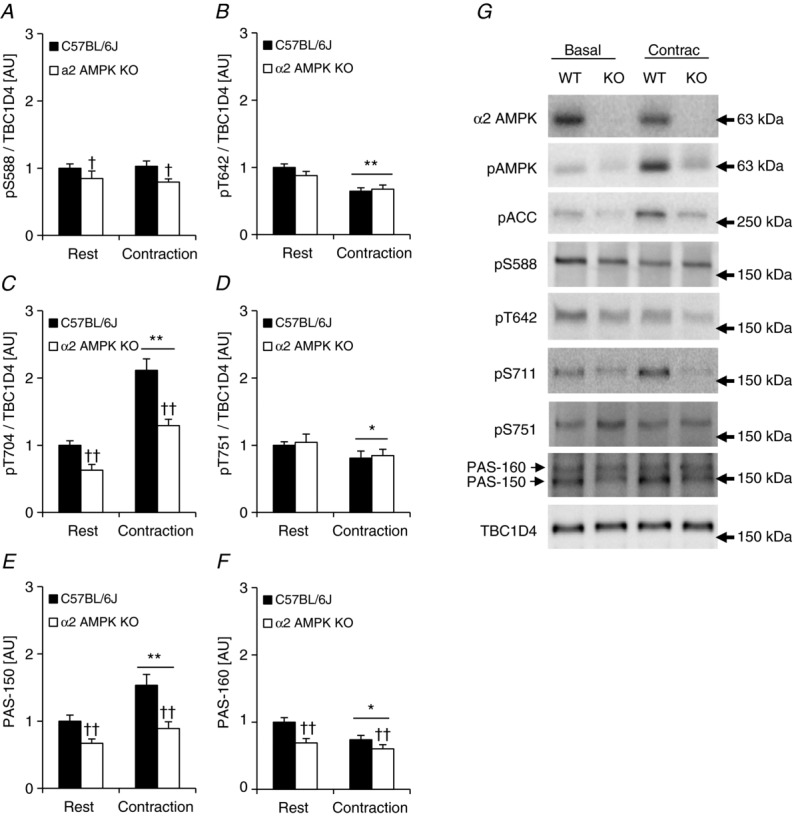
TBC1D4 phosphorylation signatures in response to in vitro contraction in mouse skeletal muscle
In vitro contraction of mouse soleus muscle activates γ1 and γ3 AMPK trimers
In mouse skeletal muscle we have previously identified five different AMPK complexes of which the four AMPK γ1-containing complexes in soleus muscle constitute the majority (>95%) of AMPK complexes (Treebak et al. 2009a). Considering the finding that activity of AMPK γ1-containing complexes associates with phosphorylation of TBC1D4 on specific sites (i.e. S341 and S704) in human skeletal muscle, one possible explanation for the lack of TBC1D4 phosphorylation in mouse soleus muscle in response to contraction could be that activation of specific γ1-containing AMPK complexes does not occur in response to the experimental protocols we have used. We have previously shown that in response to both endurance type exercise (∼70% 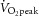 ) and to sprint type exercise in human skeletal muscle, the AMPK α2/β2/γ3 complex is activated earlier and more potently compared with the α2/β2/γ1 complex (Treebak et al. 2007). In fact, we found unchanged and even reduced activation of AMPK α2/β2/γ1 in response to various types of sprint exercises ranging from 30 s to 20 min (Birk & Wojtaszewski, 2006; Treebak et al. 2007). In our hands, the stimulation protocol used in the present study to contract soleus and EDL muscles in vitro induces fatigue very rapidly. Thus, it is possible that conditions in our contraction experiments do not allow for AMPK γ1-mediated phosphorylation of TBC1D4 in mouse skeletal muscle. To investigate this we measured AMPK α1β(1/2)γ1, α2β(1/2)γ1, and α2/β2/γ3 trimer-specific activity in soleus from α2 AMPK KD mice (Fig. 11). In this assay we did not distinguish between the different β AMPK trimer complexes. As shown in Fig. 11B and C, in vitro contraction of soleus activated γ1- and γ3-containing AMPK trimers in WT mice. In addition, α1-containing AMPK trimers were also activated in the WT mice and as expected in the AMPK KD mice (Fig. 11A). Thus, lack of TBC1D4 phosphorylation on specific sites in soleus muscle in response to in vitro contraction is not due to lack of AMPK activation of specific trimers.
) and to sprint type exercise in human skeletal muscle, the AMPK α2/β2/γ3 complex is activated earlier and more potently compared with the α2/β2/γ1 complex (Treebak et al. 2007). In fact, we found unchanged and even reduced activation of AMPK α2/β2/γ1 in response to various types of sprint exercises ranging from 30 s to 20 min (Birk & Wojtaszewski, 2006; Treebak et al. 2007). In our hands, the stimulation protocol used in the present study to contract soleus and EDL muscles in vitro induces fatigue very rapidly. Thus, it is possible that conditions in our contraction experiments do not allow for AMPK γ1-mediated phosphorylation of TBC1D4 in mouse skeletal muscle. To investigate this we measured AMPK α1β(1/2)γ1, α2β(1/2)γ1, and α2/β2/γ3 trimer-specific activity in soleus from α2 AMPK KD mice (Fig. 11). In this assay we did not distinguish between the different β AMPK trimer complexes. As shown in Fig. 11B and C, in vitro contraction of soleus activated γ1- and γ3-containing AMPK trimers in WT mice. In addition, α1-containing AMPK trimers were also activated in the WT mice and as expected in the AMPK KD mice (Fig. 11A). Thus, lack of TBC1D4 phosphorylation on specific sites in soleus muscle in response to in vitro contraction is not due to lack of AMPK activation of specific trimers.
Figure 11.
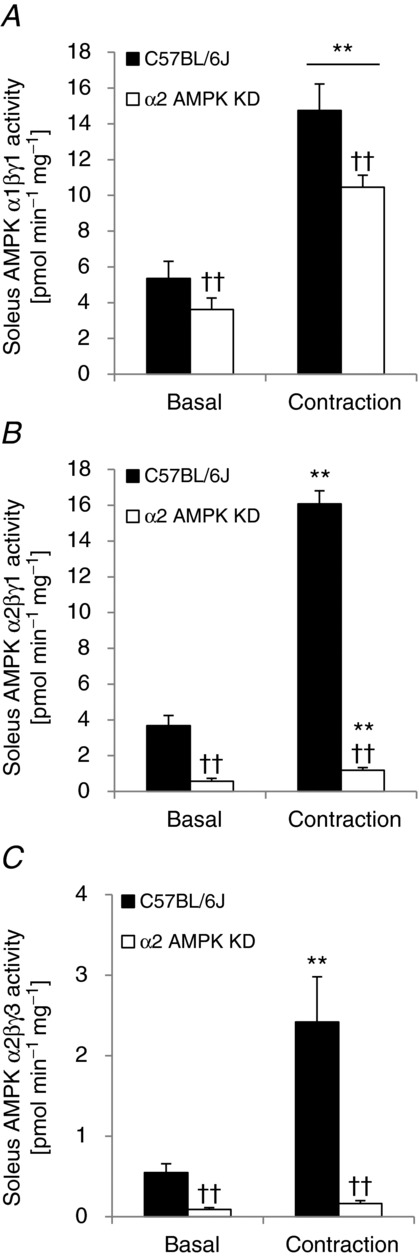
In vitro contraction of mouse skeletal muscle activates γ1 and γ3 AMPK trimers
Treadmill running induces muscle-specific changes in TBC1D1 and TBC1D4 phosphorylation patterns in mouse skeletal muscle
To test whether the decreased TBC1D4 phosphorylation of T642 and S751 in response to muscle contraction (Fig. 10B and D) was due to an in vitro rather than in vivo experimental approach, we ran male C57BL/6J mice (n = 12) on a treadmill (1 h; 18 m min−1; 5 deg slope), dissected out soleus and EDL muscles immediately after exercise, and compared the phosphorylation level of TBC1D4 and TBC1D1 on specific sites with rested control mice. This exercise protocol activated AMPK as indicated by significantly increased phosphorylation of T172 of AMPK and of S212 of ACC, but only in EDL muscles (Fig. 12A and B). In soleus muscle TBC1D4 phosphorylation was either unchanged (Fig. 13B and D) or significantly decreased (Fig. 13A, C and E). We speculated that this decrease in TBC1D4 phosphorylation level with exercise could be due to putatively reduced insulin levels compared with the rested control mice. We measured Akt phosphorylation in soleus and EDL muscles and found significantly reduced pT308 and pS473 levels in both muscles with exercise (Fig. 12C and D). In addition, in the exercised soleus muscles there were significant (P < 0.01; n = 6) positive correlations between Akt phosphorylation status of S473, and phosphorylation of TBC1D4 on sites T642 and S751 (R2 = 0.85 for Akt S473 vs. T642; R2 = 0.82 for Akt S473 vs. S751). There were additional borderline significant correlations between Akt T308 and S473 phosphorylation and specific phosphorylation sites on TBC1D4 (data not shown). Thus, it is possible that Akt is responsible for maintaining basal TBC1D4 phosphorylation levels on specific sites in mouse skeletal muscle. However, since the conditions under which we incubate muscles in vitro are not influenced by circulating insulin levels, contractions per se may still, independently of AMPK activation, lead to alterations in activity of upstream TBC1D4 kinases or phosphatases. A distinct possibility to explain the lack of TBC1D4 phosphorylation in both soleus and EDL with exercise could also be lack of muscle recruitment. In these samples, we did not measure glycogen levels. However, in a separate experiment where mice ran for 30 min at various intensities with 5 deg incline, we observed significant reductions (∼25%) in glycogen levels in both soleus and EDL already at 14 m min−1 (data not shown). Therefore, we believe that both muscles were recruited during the current exercise bout. In accordance with the TBC1D1 phosphorylation data obtained in the incubated EDL muscles, phosphorylation of S237, S660, and S700 increased in response to exercise (Fig. 13F, H and I). It is possible that the lack of T596 phosphorylation in response to exercise in this muscle could be due activation/deactivation of opposing upstream kinases (Fig. 13G). To support this idea, it has previously been shown that both Akt and AMPK are upstream kinases of TBC1D1 on this site (Pehmøller et al. 2009; Vichaiwong et al. 2010).
Figure 12.
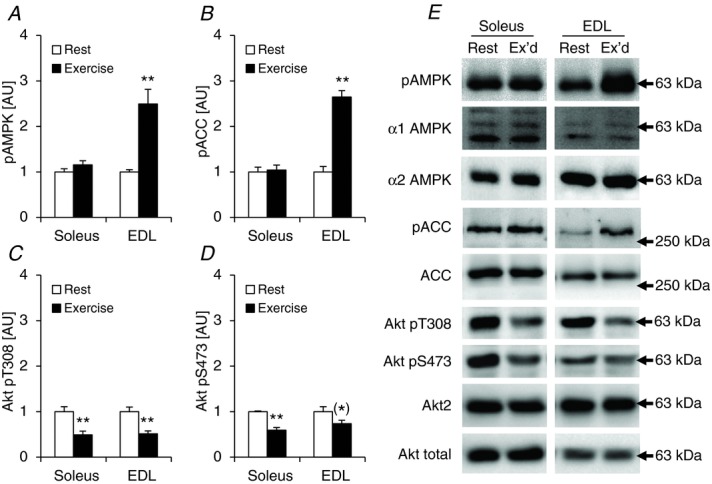
Exercise-induced phosphorylation of AMPK, ACC and Akt in mouse soleus and EDL muscles
Figure 13.
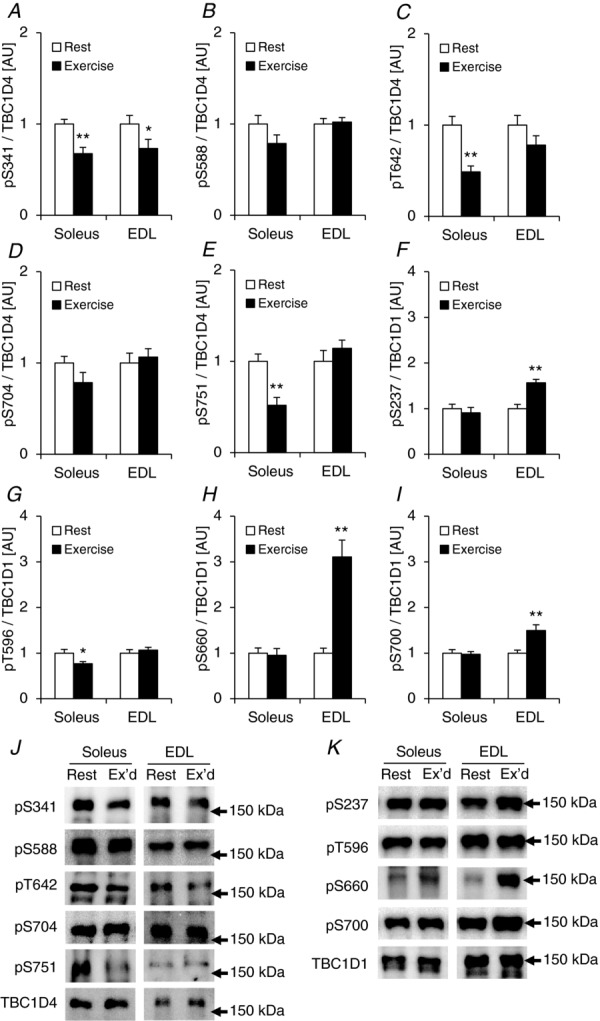
Exercise-induced phosphorylation signatures of TBC1D4 and TBC1D1 in mouse soleus and EDL muscles
14-3-3 binding capacity to TBC1D1 and TBC1D4
We have previously shown that acute cycle ergometer exercise in human subjects and in vitro contraction of mouse EDL muscle increases TBC1D1 S237 phosphorylation and that this is associated with increased 14-3-3 binding capacity towards TBC1D1 in vitro (Pehmøller et al. 2009; Frøsig et al. 2010). In addition, it has been shown in L6 myotubes that pharmacological activation of AMPK results in S237 phosphorylation and increased 14-3-3 binding capacity towards TBC1D1 (Chen et al. 2008), and that insulin stimulation promotes T596 phosphorylation but without changes in 14-3-3 binding capacity in both L6 cells (Chen et al. 2008) and intact skeletal muscle (Pehmøller et al. 2009). Although we found a significant increase in S237 phosphorylation in the current study using the one-legged knee extensor exercise model we were not able to detect any changes in 14-3-3 binding capacity in response to either exercise or physiological insulin (Fig. 14A).
Figure 14.
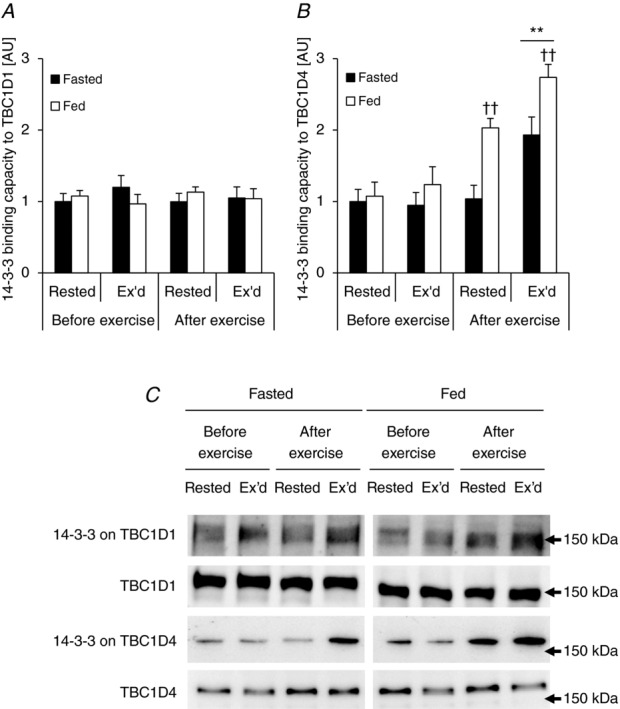
Exercise and physiological insulin stimulation results in differential 14-3-3 binding capacity to TBC1D1 and TBC1D4
Others have shown that binding capacity of 14-3-3 to TBC1D4 increases in human skeletal muscle in response to acute aerobic exercise (Howlett et al. 2008), and we and others have shown that 14-3-3 binding capacity to TBC1D4 in human skeletal muscle can be induced by raising insulin levels to high physiological levels during hyperinsulinaemic clamp conditions (Howlett et al. 2008; Treebak et al. 2009b). In the present study we investigated the effect of exercise and physiological levels of insulin on 14-3-3 binding capacity to TBC1D4. Both stimuli alone induce significant increases in 14-3-3 binding capacity and there was a complete additive effect when the two stimuli were combined (Fig. 14B). Data from several in vitro cell culture models suggest that phosphorylation of S341 and/or T642 is necessary for 14-3-3 to bind to TBC1D4 (Ramm et al. 2006; Geraghty et al. 2007). In this current study the phosphorylation patterns of S341 and T642 are similar to the data obtained from the TBC1D4 14-3-3 overlay (compare Fig. 7C and E with Fig. 14B). However, correlations between 14-3-3 binding capacity and phosphorylation on either S341 or T642 in response to either stimulus were not significant (data not shown).
Phosphorylation level of Akt correlates with phosphorylation level of specific TBC1D1 and TBC1D4 sites in response to insulin in humans
We showed that Akt was phosphorylated on T308 and S473 in human skeletal muscle in response to physiological insulin (i.e. in the rested leg after exercise in the fed state; Fig. 12C and D). In addition, we showed that several sites on TBC1D1/4 were phosphorylated in this condition (i.e. T596, S318, S341, T642, and S704; Figs 14 and 7). Except for S704, Akt has been proposed as an upstream kinase regulating these sites due to their location within the preferred Akt recognition sequence (RXRXXS/T; Sano et al. 2003; Roach et al. 2007). However, of these sites, recombinant Akt has been shown to phosphorylate only T596, S318, and T642 (Geraghty et al. 2007; Chen et al. 2008). Thus, we performed correlation analyses to examine whether any of the sites responding to physiological insulin stimulation could potentially be phosphorylated by Akt in human skeletal muscle. Akt2 has been shown to be responsible for the majority of the increase in Akt phosphorylation upon insulin stimulation in human skeletal muscle (Vind et al. 2012), and since Akt2 has been shown to be necessary for insulin to induce phosphorylation of T642 (Kramer et al. 2006a), it was surprising that phosphorylation of T642 did not correlate significantly with the phosphorylation level of any of the two Akt phosphorylation sites (P > 0.05; R2 = 0.19 for Akt T308 vs. T642; P > 0.05; R2 = 0.45 for Akt S473 vs. T642; n = 8). All other sites correlated significantly with the phosphorylation level of both T308 (Fig. 15) and S473 (Fig. 16). To further explore the pT642 data in relation to Akt phosphorylation, we correlated the delta changes of T642 phosphorylation to the delta changes of Akt T308 and S473 phosphorylation. However, we were still not able to obtain significant correlations (Supplemental Figs S1D and S2D). In addition, we correlated the delta changes in phosphorylation of all of the other TBC1D1/4 sites regulated by physiological insulin stimulation with delta changes in Akt phosphorylation. All delta correlations were either of similar strength or stronger compared with the non-delta correlations (Supplemental Figs S1 and S2). The PAS antibody has been shown to primarily detect phosphorylation of T642 (Sano et al. 2003; Ramm et al. 2006; Geraghty et al. 2007), and while phosphorylation of T642 did not correlate with the phosphorylation level of Akt, phosphorylation measured with the PAS antibody strongly correlated with Akt phosphorylation (Figs 15E, 16E and Supplemental Figs S1F, and S2F). Since all detectable PAS signal in human skeletal muscle can be accounted for in a TBC1D4 IP (Højlund et al. 2008), it is therefore possible that the PAS antibody detects other TBC1D4 sites than T642 in human skeletal muscle.
Figure 15.
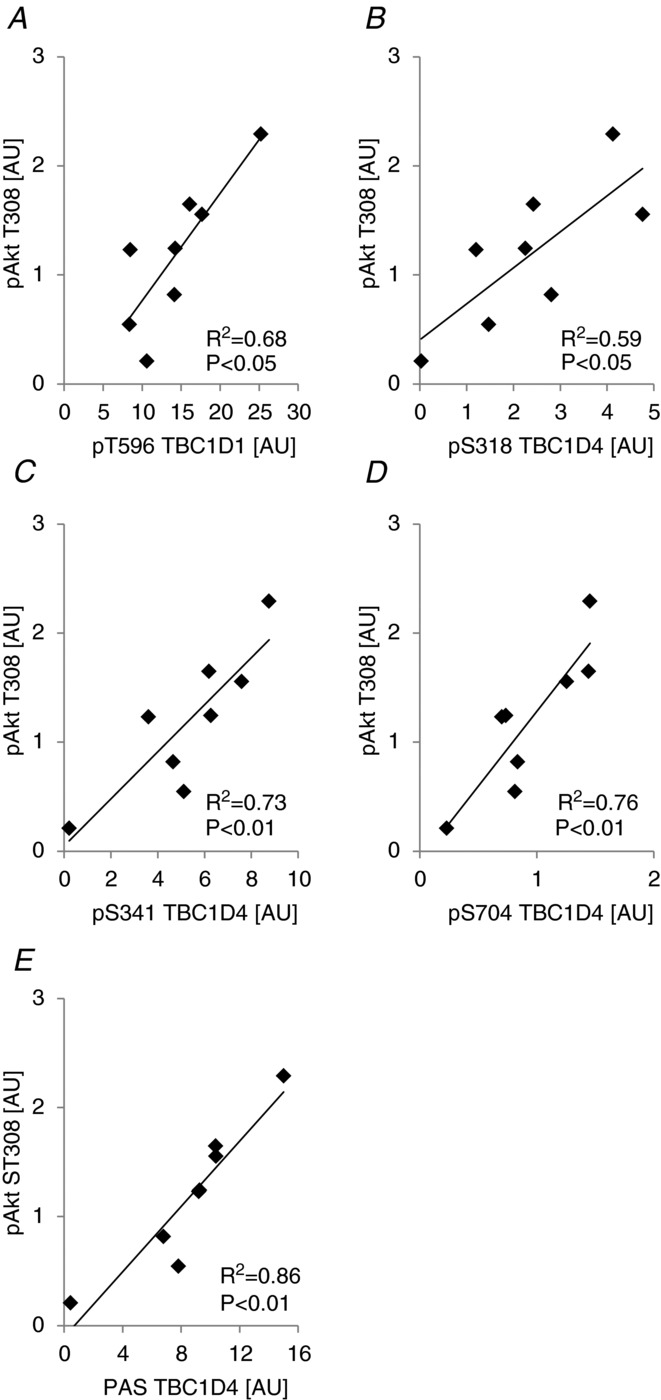
Akt phosphorylation status on T308 correlates with phosphorylation of TBC1D1/4 on specific sites in human skeletal muscle
Figure 16.
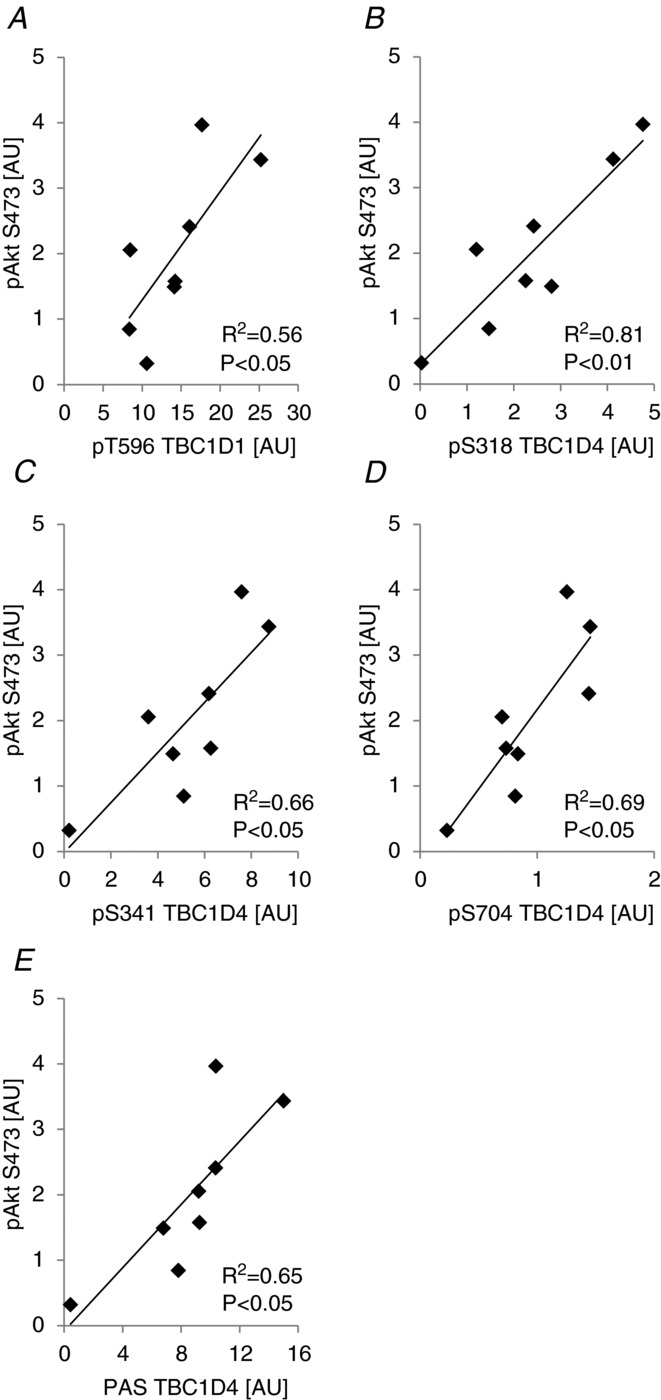
Akt phosphorylation status on S473 correlates with phosphorylation of TBC1D1/4 on specific sites in human skeletal muscle
Discussion
Phosphorylation patterns on TBC1D1 and TBC1D4 for different stimuli constitute important information regarding how these proteins are regulated and what sites may be important for specific stimuli to exert their effects through these proteins. Most studies that have investigated phosphorylation patterns on TBC1D1 and TBC1D4 have used rodent or cell culture-based models and stimulus conditions that are not physiological, and it is possible that results from these studies do not directly translate to what is seen in human skeletal muscle under physiological conditions. In the present study, TBC1D1 phosphorylation patterns between species were similar for the sites investigated. On the other hand, muscle contractions induced by in vivo exercise resulted in very different phosphorylation signatures on TBC1D4 in skeletal muscle from mice compared with humans. TBC1D4 was phosphorylated on a variety of residues (S341, S588, T642, S704, and S751) in human muscle during exercise, whereas several residues on TBC1D4 were either unchanged or underwent significant dephosphorylation during ex vivo contraction or during in vivo exercise. Along the same lines, a recent study reported increased S588 phosphorylation but unchanged S318, S341, and S751 phosphorylation after in situ contraction of the mouse gastrocnemius muscle (Ducommun et al. 2012). In the present study, we did not attempt to match exercise protocols used for the human volunteers and the cohort of mice in terms of intensity, duration, or mode. Furthermore, fibre type differences, and differences in substrate utilization during exercise may be contributing factors to explain the species difference on TBC1D4 phosphorylation patterns observed. However, the differential phosphorylation patterns of TBC1D4 among species highlight the potential risk associated with uncritical use of animal data to explain physiological phenomena in humans.
Using the one-legged knee extensor exercise model and having the same subjects complete an acute bout of exercise in the fasted and fed condition allowed us to utilize paired statistics for a more powerful evaluation of our research question. The meal ingested elicited regulation by phosphorylation of both TBC1D1 and TBC1D4, although in general the increases in phosphorylation were smaller compared with findings during hyperinsulinaemic euglycaemic clamp conditions (Treebak et al. 2009b; Pehmøller et al. 2012; Vendelbo et al. 2012). Moreover, the phosphorylation signatures on TBC1D1 and TBC1D4 in response to the meal were different to what we have previously reported during hyperinsulinaemic euglycaemic clamps (Treebak et al. 2009b; Pehmøller et al. 2012; Vendelbo et al. 2012), as we were not able to see regulation of S588 and S751 on TBC1D4 in the current study. Also, in the present study we found that those sites regulated by both exercise in the fasted state and in response to the meal (i.e. T596, S341, T642, and S704) were all regulated in an additive manner by exercise in the fed state. 14-3-3 binding to TBC1D4 has been suggested to be necessary for insulin-mediated GLUT4 trafficking in adipocytes (Ramm et al. 2006). Here we showed that in vitro 14-3-3 binding capacity to TBC1D4 was up-regulated by exercise per se, by the meal per se, and synergistically by exercise in the fed state. This may suggest that signalling arising from insulin and contraction converges on TBC1D4. The additive effects on phosphorylation and 14-3-3 binding may lead to elevated glucose transporter translocation to the plasma membrane, and may along with the elevated extracellular glucose concentration mediate the expected increased glucose uptake during exercise in the fed state compared to the fasted state (DeFronzo et al. 1981). Although this may also be true for the TBC1D1, we were unable to detect regulation of the 14-3-3 binding capacity on this protein in either of the conditions. This contrasts our own observations during more intense exercise (Frøsig et al. 2010).
TBC1D1 has four confirmed phosphorylation sites and while S237, S660, and S700 were phosphorylated in response to exercise only, T596 was phosphorylated in response to both postprandial increase in insulin and exercise. Furthermore, the two stimuli together gave an additive effect on T596 phosphorylation, suggesting that both exercise- and insulin-stimulated kinases regulate this site. Our findings are partly in line with recent studies showing increased S237 and S660 phosphorylation in response to exercise in human skeletal muscle (Frøsig et al. 2010; Jessen et al. 2011). Exercise-induced TBC1D1 phosphorylation on S237 and T596 correlated significantly with activity of α2/β2/γ3 AMPK trimer complexes indicating that this AMPK complex could be the in vivo upstream kinase for these sites. However, when correlation analyses on delta values were performed, the only significant correlation remaining was between S237 and α2/β2/γ3 AMPK activity. We have previously shown that recombinant AMPK can phosphorylate S237 in vitro (Frøsig et al. 2010), further supporting the idea that AMPK is a direct upstream TBC1D1 kinase. We found that Akt phosphorylation of S473, but not T308, was significantly increased in the exercised leg indicating that Akt may have been activated in the exercised leg. This could have contributed to the positive correlation between phosphorylation level of T596 and α2/β2/γ3 AMPK activity. In EDL muscle from α2 AMPK KO mice we confirmed that S237, T596, and S660 on TBC1D1 are regulated exclusively by α2 AMPK in response to contractions (Pehmøller et al. 2009; Vichaiwong et al. 2010), and we have identified S700 as another site regulated by AMPK in contracting skeletal muscle.
TBC1D4 has nine confirmed phosphorylation sites and we were able to measure the phosphorylation status of seven of these. Since S341, S588, T642, S704, and S751 all were exercise-responsive sites, we correlated phosphorylation level with AMPK activity. We have previously shown that recombinant AMPK can phosphorylate S704 in vitro (Treebak et al. 2010). Here we showed that S341 and S704 correlated significantly with activity of α2/β2/γ1 AMPK complexes, suggesting that this trimer may be responsible for phosphorylating these sites in vivo. Interestingly, and in contrast to TBC1D1, α2/β2/γ3 AMPK trimer activity did not correlate with phosphorylation level of any of the exercise-responsive TBC1D4 sites. We have previously found that cycle ergometer exercise induced PAS phosphorylation in human muscle and that this correlated with activation of α2/β2/γ1 AMPK complexes (Treebak et al. 2007); however, in the present study where the exercise mode was different, we did not find this correlation.
The idea that different AMPK trimers target specific Rab-GAPs is intriguing and suggests that AMPK and TBC1D1/4 could be localized to distinct compartments in the cell, and that TBC1D1 and TBC1D4 may play distinct roles in inhibiting vesicle mobilization. It is possible, for instance, that each Rab-GAP controls different Rab-GTPases responsible for separate steps in the mobilization of GLUT4 containing vesicles. To understand the function of TBC1D1 and TBC1D4 phosphorylation in response to AMPK activation, one would need specific AMPK activators that target specific AMPK heterotrimers. Although, A-769662 has been shown to target specific AMPK trimers in vitro (Cool et al. 2006; Scott et al. 2008), off-target effects in intact skeletal muscle have been reported (Treebak et al. 2009a). The fact that phosphorylation of S588 and S751 did not correlate significantly with AMPK α2/β2/γ1 activity could be due to other kinases than AMPK mediating the phosphorylation of these sites. A recent study has shown that phosphorylation of S588 and T642 in response to insulin occurs on different pools of TBC1D4 in 3T3-L1 adipocytes and that protein kinase Cζ (PKCζ) is a likely upstream kinase for S588 (Ng et al. 2010). Furthermore, PKCζ is activated in response to exercise in human skeletal muscle (Richter et al. 2004; Rose et al. 2004), supporting the idea that this kinase could potentially target TBC1D4 in vivo.
Overexpression of the TBC1D4 mutant in which four key residues (S318, S588, T642, and S751) have been mutated to alanine (4P mutant) has been shown to decrease contraction-stimulated glucose uptake in mouse skeletal muscle (Kramer et al. 2006b). However, phosphorylation of T642 does not appear to be required for this effect of contraction (Ducommun et al. 2012), and since TBC1D4 phosphorylation has been reported to be unchanged or even reduced on three of the four sites in the 4P mutant (present study and Ducommun et al. 2012), it is possible that TBC1D4 does not mediate contraction-stimulated glucose uptake in muscle. On the other hand, phosphorylation of S588, one of the sites mutated in the 4P mutant, was reported to increase in response to in situ contraction of the gastrocnemius muscle (Ducommun et al. 2012). Whether phosphorylation of S588 alone is sufficient to increase glucose uptake, or whether aggregate mutation of multiple sites on TBC1D4 is necessary to disrupt function and glucose uptake is not known. Future studies using transgenic or knock-in mouse models are required to answer these questions.
The correlation analyses performed on the raw pTBC1D1/4 and pAkt data from the rested leg after exercise in the fed state (insulin-stimulated) together with the analyses performed on delta values (i.e. rested leg in the fed trial after exercise minus rested leg in the fed trial before exercise) showed significant correlations between both Akt sites (T308/S473) and T596, S318, S341, S704, and PAS. T596, S318, and S341 are all within the Akt consensus sequence and it is likely that Akt is responsible for phosphorylating these sites in response to insulin. Indeed, phosphorylation of T596 in response to insulin has been shown to be ablated in Akt2 knockout mice (Vichaiwong et al. 2010). As previously shown, phosphorylation of S704 in response to insulin is completely ablated if mouse skeletal muscles are pre-incubated with the PI3 kinase inhibitor wortmannin (Treebak et al. 2010). In addition, the S704 phosphorylation was similar between WT and Akt2 knockout mice in response to a maximal dose of insulin, and recombinant Akt1/2 and PKCζ did not phosphorylate S704 in vitro (Treebak et al. 2010). Thus, although we found strong correlations between Akt phosphorylation and S704 in human skeletal muscle in the present study we do not know whether this relationship is causal. It would be interesting to study the effect on S704 phosphorylation in Akt2 knockout animals in response to a submaximal/physiological insulin dose, as muscles from these mice do not respond the same way to a maximal compared with a submaximal insulin doses in terms of glucose uptake (Cho et al. 2001).
The strength of the current study is that we have investigated the phosphorylation signatures of TBC1D1 and TBC1D4 in human skeletal muscle in response to physiological stimuli in a paired manner. We have applied transgenic animal models to further substantiate our findings from human skeletal muscle. However, data from these experiments indicate that it is not always possible to transfer findings across species, underlining the necessity to study intact muscle from human beings. Our findings show that in human skeletal muscle TBC1D1 and TBC1D4 are points of convergence of signalling events activated during postprandial physiological increase in insulin and acute exercise, and that the proposed upstream kinases AMPK and Akt may mediate this effect. Our working hypothesis is that TBC1D1 and TBC1D4 may be involved in the insulin-sensitizing effect of exercise, and perhaps other insulin-sensitizing agents (Pehmøller et al. 2012).
Acknowledgments
We would like to acknowledge skilled technical assistance from Betina Bolmgren, Per Schack Larsen, Steve Risis, Christine Janssens, Irene Bech Nielsen, and Radhika Vadalasetty. Founder AMPK α2 knockout and AMPK α2 KD mice were provided by Dr Benoit Viollet and Professor Morris Birnbaum, respectively.
Glossary
- ACC
acetyl-CoA carboxylase
- Akt
serine/threonine-specific protein kinase
- AMPK
AMP-activated protein kinase
- ANOVA
analysis of variance
- AU
arbitrary units
- EDL
extensor digitorum longus
- GLUT4
glucose transporter 4
- IB
immunoblot
- IP
immunoprecipitation
- KD
kinase dead
- KO
knockout
- PAS
phospho-Akt substrate
- Rab-GAP
Rab-GTPase activating protein
- TBC1D1/4
TBC1 domain family member 1/4
- WT
wild-type
- 14-3-3
highly conserved regulatory eukaryotic proteins binding to phosphorylated sites of target proteins
Competing interests
The authors report no competing interests
Author contributions
The experiments in the present manuscript were conducted in Section of Molecular Physiology, the August Krogh Centre, Department of Nutrition, Exercise and Sports, University of Copenhagen and in Section of Integrated Physiology at the Novo Nordisk Foundation Center for Basic Metabolic Research, University of Copenhagen. Conception and design of the experiments: J.T.T., J.F.P.W. Collection, analysis and interpretation of data: J.T.T., C.P., J.M.K., R.K., J.B., P.S., E.A.R., L.J.G., J.F.P.W. Drafting the article or revising it critically for important intellectual content: J.T.T., P.S., L.J.G., E.A.R., J.F.P.W. All authors read and approved the final version of the manuscript and all authors listed qualify for authorship.
Funding
This study was supported by grants from the Danish Medical Research Council, The Novo Nordisk Research Foundation, The Lundbeck Foundation, and Danish Ministry of Food, Agriculture and Fisheries, an integrated project funded by the European Union (LSHMCT-2004-005272), and NIH R0142238 (LJG). This work was carried out as part of the research program of the UNIK: Food, Fitness and Pharma for Health and Disease (www.foodfitnesspharma.ku.dk). The UNIK project is supported by the Danish Ministry of Science, Technology and Innovation. Jonas T. Treebak was supported by a postdoctoral fellowship from The Danish Agency for Science, Technology and Innovation, and by the Novo Nordisk Foundation Center for Basic Metabolic Research. The Novo Nordisk Foundation Center for Basic Metabolic Research is an independent Research Center, based at the University of Copenhagen, Denmark and partially funded by an unconditional donation from the Novo Nordisk Foundation (http://metabol.ku.dk).
Authors’ present addresses
Jonas T. Treebak: The Novo Nordisk Foundation Center for Basic Metabolic Research, Section of Integrative Physiology, University of Copenhagen, DK-2200 Copenhagen, Denmark.
Jonas M. Kristensen: Department of Endocrinology, Odense University Hospital, Institute of Clinical Research, Section of Molecular Diabetes and Metabolism, University of Southern Denmark, DK-5000 Odense, Denmark.
References
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- An D, Toyoda T, Taylor EB, Yu H, Fujii N, Hirshman MF, Goodyear LJ. TBC1D1 regulates insulin- and contraction-induced glucose transport in mouse skeletal muscle. Diabetes. 2010;59:1358–1365. doi: 10.2337/db09-1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen P, Adams RP, Sjogaard G, Thorboe A, Saltin B. Dynamic knee extension as model for study of isolated exercising muscle in humans. J Appl Physiol. 1985;59:1647–1653. doi: 10.1152/jappl.1985.59.5.1647. [DOI] [PubMed] [Google Scholar]
- Arias EB, Kim J, Funai K, Cartee GD. Prior exercise increases phosphorylation of Akt substrate of 160 kDa (AS160) in rat skeletal muscle. Am J Physiol Endocrinol Metab. 2007;292:E1191–E1200. doi: 10.1152/ajpendo.00602.2006. [DOI] [PubMed] [Google Scholar]
- Biensø RS, Ringholm S, Kiilerich K, Aachmann-Andersen NJ, Krogh-Madsen R, Guerra B, Plomgaard P, van HG, Treebak JT, Saltin B, Lundby C, Calbet JA, Pilegaard H, Wojtaszewski JF. GLUT4 and glycogen synthase are key players in bed rest-induced insulin resistance. Diabetes. 2012;61:1090–1099. doi: 10.2337/db11-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birk JB, Wojtaszewski JFP. Predominant α2/β2/γ3 AMPK activation during exercise in human skeletal muscle. J Physiol. 2006;577:1021–1032. doi: 10.1113/jphysiol.2006.120972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzakri K, Zachrisson A, Al-Khalili L, Zhang BB, Koistinen HA, Krook A, Zierath JR. siRNA-based gene silencing reveals specialized roles of IRS-1/Akt2 and IRS-2/Akt1 in glucose and lipid metabolism in human skeletal muscle. Cell Metab. 2006;4:89–96. doi: 10.1016/j.cmet.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Cartee GD, Douen AG, Ramlal T, Klip A, Holloszy JO. Stimulation of glucose transport in skeletal muscle by hypoxia. J Appl Physiol. 1991;70:1593–1600. doi: 10.1152/jappl.1991.70.4.1593. [DOI] [PubMed] [Google Scholar]
- Chen S, Murphy J, Toth R, Campbell DG, Morrice NA, MacKintosh C. Complementary regulation of TBC1D1 and AS160 by growth factors, insulin and AMPK activators. Biochem J. 2008;409:449–459. doi: 10.1042/BJ20071114. [DOI] [PubMed] [Google Scholar]
- Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, III, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBβ. Science. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- Consitt LA, Van Meter J, Newton CA, Collier DN, Dar MS, Wojtaszewski FP, Jr, Treebak JT, Tanner CJ, Houmard JA. Impairments in site-specific AS160 phosphorylation and effects of exercise training. Diabetes. 2013;62:3437–3747. doi: 10.2337/db13-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, Zhao G, Marsh K, Kym P, Jung P, Camp HS, Frevert E. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metabolism. 2006;3:403–416. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, Ferrannini E, Sato Y, Felig P, Wahren J. Synergistic interaction between exercise and insulin on peripheral glucose uptake. J Clin Invest. 1981;68:1468–1474. doi: 10.1172/JCI110399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh A, Coffey VG, Zhong Z, Chibalin AV, Hawley JA, Zierath JR. Exercise-induced phosphorylation of the novel Akt substrates AS160 and Filamin A in human skeletal muscle. Diabetes. 2006;55:1776–1782. doi: 10.2337/db05-1419. [DOI] [PubMed] [Google Scholar]
- Douen AG, Ramlal T, Klip A, Young DA, Cartee GD, Holloszy JO. Exercise-induced increase in glucose transporters in plasma membranes of rat skeletal muscle. Endocrinology. 1989;124:449–454. doi: 10.1210/endo-124-1-449. [DOI] [PubMed] [Google Scholar]
- Ducommun S, Wang HY, Sakamoto K, MacKintosh C, Chen S. Thr649Ala-AS160 knockin mutation does not impair contraction/AICAR-induced glucose transport in mouse muscle. Am J Physiol Endocrinol Metab. 2012;302:E1036–E1043. doi: 10.1152/ajpendo.00379.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrichsen M, Mortensen B, Pehmoller C, Birk JB, Wojtaszewski JF. Exercise-induced AMPK activity in skeletal muscle: Role in glucose uptake and insulin sensitivity. Mol Cell Endocrinol. 2012;366:204–214. doi: 10.1016/j.mce.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Frøsig C, Pehmoller C, Birk JB, Richter EA, Wojtaszewski JF. Exercise-induced TBC1D1 Ser237 phosphorylation and 14-3-3 protein binding capacity in human skeletal muscle. J Physiol. 2010;588:4539–4548. doi: 10.1113/jphysiol.2010.194811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frøsig C, Rose AJ, Treebak JT, Kiens B, Richter EA, Wojtaszewski JFP. Effects of endurance exercise training on insulin signaling in human skeletal muscle: interactions at the level of phosphatidylinositol 3-kinase, Akt, and AS160. Diabetes. 2007;56:2093–2102. doi: 10.2337/db06-1698. [DOI] [PubMed] [Google Scholar]
- Funai K, Schweitzer GG, Sharma N, Kanzaki M, Cartee GD. Increased AS160 phosphorylation, but not TBC1D1 phosphorylation, with increased post-exercise insulin sensitivity in rat skeletal muscle. Am J Physiol Endocrinol Metab. 2009;297:E242–E251. doi: 10.1152/ajpendo.00194.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraghty K, Chen S, Harthill JE, Ibrahim AF, Toth R, Morrice NA, Vandermoere F, Moorhead GB, Hardie DG, MacKintosh C. Regulation of multisite phosphorylation and 14-3-3 binding of AS160 in response to insulin-like growth factor 1, EGF, PMA and AICAR. Biochem J. 2007;407:231–241. doi: 10.1042/BJ20070649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha J, Daniel S, Broyles SS, Kim KH. Critical phosphorylation sites for acetyl-CoA carboxylase activity. J Biol Chem. 1994;269:22162–22168. [PubMed] [Google Scholar]
- Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996;271:27879–27887. doi: 10.1074/jbc.271.44.27879. [DOI] [PubMed] [Google Scholar]
- Hill MM, Clark SF, Tucker DF, Birnbaum MJ, James DE, Macaulay SL. A role for protein kinase Bbeta/Akt2 in insulin-stimulated GLUT4 translocation in adipocytes. Mol Cell Biol. 1999;19:7771–7781. doi: 10.1128/mcb.19.11.7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeg LD, Sjoberg KA, Jeppesen J, Jensen TE, Frosig C, Birk JB, Bisiani B, Hiscock N, Pilegaard H, Wojtaszewski JF, Richter EA, Kiens B. Lipid-induced insulin resistance affects women less than men and is not accompanied by inflammation or impaired proximal insulin signaling. Diabetes. 2011;60:64–73. doi: 10.2337/db10-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman NJ, Elmendorf JS. Signaling, cytoskeletal and membrane mechanisms regulating GLUT4 exocytosis. Trends Endocrinol Metab. 2011;22:110–116. doi: 10.1016/j.tem.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Højlund K, Glintborg D, Andersen NR, Birk JB, Treebak JT, Frosig C, Beck-Nielsen H, Wojtaszewski JFP. Impaired insulin-stimulated phosphorylation of Akt and AS160 in skeletal muscle of women with polycystic ovary syndrome is reversed by pioglitazone treatment. Diabetes. 2008;57:357–366. doi: 10.2337/db07-0706. [DOI] [PubMed] [Google Scholar]
- Howlett K, Mathews A, Garnham A, Sakamoto K. The effect of exercise and insulin on AS160 phosphorylation and 14-3-3 binding capacity in human skeletal muscle. Am J Physiol Endocrinol Metab. 2008;294:E401–E407. doi: 10.1152/ajpendo.00542.2007. [DOI] [PubMed] [Google Scholar]
- Howlett KF, Sakamoto K, Garnham A, Cameron-Smith D, Hargreaves M. Resistance exercise and insulin regulate AS160 and interaction with 14-3-3 in human skeletal muscle. Diabetes. 2007;56:1608–1614. doi: 10.2337/db06-1398. [DOI] [PubMed] [Google Scholar]
- Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab. 2007;5:237–252. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Ishikura S, Koshkina A, Klip A. Small G proteins in insulin action: Rab and Rho families at the crossroads of signal transduction and GLUT4 vesicle traffic. Acta Physiol (Oxf) 2008;192:61–74. doi: 10.1111/j.1748-1716.2007.01778.x. [DOI] [PubMed] [Google Scholar]
- Jensen TE, Richter EA. Regulation of glucose and glycogen metabolism during and after exercise. J Physiol. 2012;590:1069–1076. doi: 10.1113/jphysiol.2011.224972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen N, An D, Lihn AS, Nygren J, Hirshman MF, Thorell A, Goodyear LJ. Exercise increases TBC1D1 phosphorylation in human skeletal muscle. Am J Physiol Endocrinol Metab. 2011;301:E164–E171. doi: 10.1152/ajpendo.00042.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane S, Sano H, Liu SCH, Asara JM, Lane WS, Garner CC, Lienhard GE. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J Biol Chem. 2002;277:22115–22118. doi: 10.1074/jbc.C200198200. [DOI] [PubMed] [Google Scholar]
- Karlsson HK, Hallsten K, Bjornholm M, Tsuchida H, Chibalin AV, Virtanen KA, Heinonen OJ, Lonnqvist F, Nuutila P, Zierath JR. Effects of metformin and rosiglitazone treatment on insulin signaling and glucose uptake in patients with newly diagnosed type 2 diabetes: a randomized controlled study. Diabetes. 2005a;54:1459–1467. doi: 10.2337/diabetes.54.5.1459. [DOI] [PubMed] [Google Scholar]
- Karlsson HK, Zierath JR, Kane S, Krook A, Lienhard GE, Wallberg-Henriksson H. Insulin-stimulated phosphorylation of the Akt substrate AS160 is impaired in skeletal muscle of type 2 diabetic subjects. Diabetes. 2005b;54:1692–1697. doi: 10.2337/diabetes.54.6.1692. [DOI] [PubMed] [Google Scholar]
- Karlsson HKR, Ahlsen M, Zierath JR, Wallberg-Henriksson H, Koistinen HA. Insulin signaling and glucose transport in skeletal muscle from first-degree relatives of type 2 diabetic patients. Diabetes. 2006;55:1283–1288. doi: 10.2337/db05-0853. [DOI] [PubMed] [Google Scholar]
- Klip A, Ramlal T, Young DA, Holloszy JO. Insulin-induced translocation of glucose transporters in rat hindlimb muscles. FEBS Lett. 1987;224:224–230. doi: 10.1016/0014-5793(87)80452-0. [DOI] [PubMed] [Google Scholar]
- Kramer HF, Witczak CA, Fujii N, Jessen N, Taylor EB, Arnolds DE, Sakamoto K, Hirshman MF, Goodyear LJ. Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes. 2006a;55:2067–2076. doi: 10.2337/db06-0150. [DOI] [PubMed] [Google Scholar]
- Kramer HF, Witczak CA, Taylor EB, Fujii N, Hirshman MF, Goodyear LJ. AS160 regulates insulin- and contraction-stimulated glucose uptake in mouse skeletal muscle. J Biol Chem. 2006b;281:31478–85. doi: 10.1074/jbc.M605461200. [DOI] [PubMed] [Google Scholar]
- Kristiansen S, Hargreaves M, Richter EA. Exercise-induced increase in glucose transport, GLUT-4, and VAMP-2 in plasma membrane from human muscle. Am J Physiol Endocrinol Metab. 1996;270:E197–E201. doi: 10.1152/ajpendo.1996.270.1.E197. [DOI] [PubMed] [Google Scholar]
- Mu J, Brozinick JT, Jr, Valladares O, Bucan M, Birnbaum MJ. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Molecular Cell. 2001;7:1085–1094. doi: 10.1016/s1097-2765(01)00251-9. [DOI] [PubMed] [Google Scholar]
- Nagase T, Ishikawa K, Miyajima N, Tanaka A, Kotani H, Nomura N, Ohara O. Prediction of the coding sequences of unidentified human genes. IX. The complete sequences of 100 new cDNA clones from brain which can code for large proteins in vitro. DNA Res. 1998;5:31–39. doi: 10.1093/dnares/5.1.31. [DOI] [PubMed] [Google Scholar]
- Ng Y, Ramm G, Burchfield JG, Coster AC, Stockli J, James DE. Cluster analysis of insulin action in adipocytes reveals a key role for Akt at the plasma membrane. J Biol Chem. 2010;285:2245–2257. doi: 10.1074/jbc.M109.060236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Gorman DJ, Karlsson HK, McQuaid S, Yousif O, Rahman Y, Gasparro D, Glund S, Chibalin AV, Zierath JR, Nolan JJ. Exercise training increases insulin-stimulated glucose disposal and GLUT4 (SLC2A4) protein content in patients with type 2 diabetes. Diabetologia. 2006;49:2983–2992. doi: 10.1007/s00125-006-0457-3. [DOI] [PubMed] [Google Scholar]
- Passonneau JV, Gatfield PD, Schulz DW, Lowry OH. An enzymic method for measurement of glycogen. Anal Biochem. 1967;19:315–326. doi: 10.1016/0003-2697(67)90167-4. [DOI] [PubMed] [Google Scholar]
- Pehmøller C, Brandt N, Birk JB, Høeg LD, Sjøberg KA, Goodyear LJ, Kiens B, Richter EA, Wojtaszewski JF. Exercise alleviates lipid-induced insulin resistance in human skeletal muscle-signaling interaction at the level of TBC1 domain family member 4. Diabetes. 2012;61:2743–2752. doi: 10.2337/db11-1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehmøller C, Treebak JT, Birk JB, Chen S, MacKintosh C, Hardie DG, Richter EA, Wojtaszewski JF. Genetic disruption of AMPK signaling abolishes both contraction- and insulin-stimulated TBC1D1 phosphorylation and 14-3-3 binding in mouse skeletal muscle. Am J Physiol Endocrinol Metab. 2009;297:E665–E675. doi: 10.1152/ajpendo.00115.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploug T, van DB, Ai H, Cushman SW, Ralston E. Analysis of GLUT4 distribution in whole skeletal muscle fibers: identification of distinct storage compartments that are recruited by insulin and muscle contractions. J Cell Biol. 1998;142:1429–1446. doi: 10.1083/jcb.142.6.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozuelo Rubio M, Geraghty KM, Wong BHC, Wood NT, Campbell DG, Morrice N, MacKintosh C. 14-3-3-affinity purification of over 200 human phosphoproteins reveals new links to regulation of cellular metabolism, proliferation and trafficking. Biochem J. 2004;379:395–408. doi: 10.1042/BJ20031797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramm G, Larance M, Guilhaus M, James DE. A role for 14-3-3 in insulin-stimulated GLUT4 translocation through its interaction with the RabGAP AS160. J Biol Chem. 2006;281:29174–29180. doi: 10.1074/jbc.M603274200. [DOI] [PubMed] [Google Scholar]
- Richter EA, Mikines KJ, Galbo H, Kiens B. Effect of exercise on insulin action in human skeletal muscle. J Appl Physiol. 1989;66:876–885. doi: 10.1152/jappl.1989.66.2.876. [DOI] [PubMed] [Google Scholar]
- Richter EA, Vistisen B, Maarbjerg SJ, Sajan M, Farese RV, Kiens B. Differential effect of bicycling exercise intensity on activity and phosphorylation of atypical protein kinase C and extracellular signal-regulated protein kinase in skeletal muscle. J Physiol. 2004;560:909–918. doi: 10.1113/jphysiol.2004.071373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach WG, Chavez JA, Miinea CP, Lienhard GE. Substrate specificity and effect on GLUT4 translocation of the Rab GTPase-activating protein Tbc1d1. Biochem J. 2007;403:353–358. doi: 10.1042/BJ20061798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AJ, Michell BJ, Kemp BE, Hargreaves M. Effect of exercise on protein kinase C activity and localization in human skeletal muscle. J Physiol. 2004;561:861–870. doi: 10.1113/jphysiol.2004.075549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, Arnolds DEW, Ekberg I, Thorell A, Goodyear LJ. Exercise regulates Akt and glycogen synthase kinase-3 activities in human skeletal muscle. Biochem Biophysl Res Commun. 2004;319:419–425. doi: 10.1016/j.bbrc.2004.05.020. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Holman GD. Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am J Physiol Endocrinol Metab. 2008;295:E29–E37. doi: 10.1152/ajpendo.90331.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–14602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- Scott JW, van Denderen BJ, Jorgensen SB, Honeyman JE, Steinberg GR, Oakhill JS, Iseli TJ, Koay A, Gooley PR, Stapleton D, Kemp BE. Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chem Biol. 2008;15:1220–1230. doi: 10.1016/j.chembiol.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Slot JW, Geuze HJ, Gigengack S, Lienhard GE, James DE. Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J Cell Biol. 1991;113:123–135. doi: 10.1083/jcb.113.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor EB, An D, Kramer HF, Yu H, Fujii NL, Roeckl KSC, Bowles N, Hirshman MF, Xie J, Feener EP, Goodyear LJ. Discovery of TBC1D1 as an insulin-, AICAR-, and contraction-stimulated signaling nexus in mouse skeletal muscle. J Biol Chem. 2008;283:9787–9796. doi: 10.1074/jbc.M708839200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treebak JT, Birk JB, Hansen BF, Olsen GS, Wojtaszewski JF. A-769662 activates AMPK β1-containing complexes but induces glucose uptake through a PI3 kinase-dependent pathway in mouse skeletal muscle. Am J Physiol Cell Physiol. 2009a;297:C1041–C1052. doi: 10.1152/ajpcell.00051.2009. [DOI] [PubMed] [Google Scholar]
- Treebak JT, Birk JB, Rose AJ, Kiens B, Richter EA, Wojtaszewski JF. AS160 phosphorylation is associated with activation of α2β2γ1 but not α2β2γ3 AMPK trimeric complex in skeletal muscle during exercise in humans. Am J Physiol Endocrinol Metab. 2007;292:E715–E722. doi: 10.1152/ajpendo.00380.2006. [DOI] [PubMed] [Google Scholar]
- Treebak JT, Frøsig C, Pehmøller C, Chen S, Maarbjerg SJ, Brandt N, MacKintosh C, Zierath JR, Hardie DG, Kiens B, Richter EA, Pilegaard H, Wojtaszewski JF. Potential role of TBC1D4 in enhanced post-exercise insulin action in human skeletal muscle. Diabetologia. 2009b;52:891–900. doi: 10.1007/s00125-009-1294-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treebak JT, Glund S, Deshmukh A, Klein DK, Long YC, Jensen TE, Jorgensen SB, Viollet B, Andersson L, Neumann D, Wallimann T, Richter EA, Chibalin AV, Zierath JR, Wojtaszewski JFP. AMPK-mediated AS160 phosphorylation in skeletal muscle is dependent on AMPK catalytic and regulatory subunits. Diabetes. 2006;55:2051–2058. doi: 10.2337/db06-0175. [DOI] [PubMed] [Google Scholar]
- Treebak JT, Taylor EB, Witczak CA, An D, Toyoda T, Koh HJ, Xie J, Feener EP, Wojtaszewski JF, Hirshman MF, Goodyear LJ. Identification of a novel phosphorylation site on TBC1D4 regulated by AMP-activated protein kinase in skeletal muscle. Am J Physiol Cell Physiol. 2010;298:C377–C385. doi: 10.1152/ajpcell.00297.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendelbo MH, Clasen BF, Treebak JT, Moller L, Krusenstjerna-Hafstrom T, Madsen M, Nielsen TS, Stodkilde-Jorgensen H, Pedersen SB, Jorgensen JO, Goodyear LJ, Wojtaszewski JF, Moller N, Jessen N. Insulin resistance after a 72-h fast is associated with impaired AS160 phosphorylation and accumulation of lipid and glycogen in human skeletal muscle. Am J Physiol Endocrinol Metab. 2012;302:E190–E200. doi: 10.1152/ajpendo.00207.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vichaiwong K, Purohit S, An D, Toyoda T, Jessen N, Hirshman MF, Goodyear LJ. Contraction regulates site-specific phosphorylation of TBC1D1 in skeletal muscle. Biochem J. 2010;431:311–320. doi: 10.1042/BJ20101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vind BF, Birk JB, Vienberg SG, Andersen B, Beck-Nielsen H, Wojtaszewski JF, Hojlund K. Hyperglycaemia normalises insulin action on glucose metabolism but not the impaired activation of AKT and glycogen synthase in the skeletal muscle of patients with type 2 diabetes. Diabetologia. 2012;55:1435–1445. doi: 10.1007/s00125-012-2482-8. [DOI] [PubMed] [Google Scholar]
- Vind BF, Pehmøller C, Treebak JT, Birk JB, Hey-Mogensen M, Beck-Nielsen H, Zierath JR, Wojtaszewski JF, Højlund K. Impaired insulin-induced site-specific phosphorylation of TBC1 domain family, member 4 (TBC1D4) in skeletal muscle of type 2 diabetes patients is restored by endurance exercise-training. Diabetologia. 2011;54:157–167. doi: 10.1007/s00125-010-1924-4. [DOI] [PubMed] [Google Scholar]
- Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, Gomas E, Nicolas G, Wojtaszewski JFP, Kahn A, Carling D, Schuit FC, Birnbaum MJ, Richter EA, Burcelin R, Vaulont S. The AMP-activated protein kinase α2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszewski JFP, Birk JB, Frosig C, Holten M, Pilegaard H, Dela F. 5′AMP activated protein kinase expression in human skeletal muscle: effects of strength training and type 2 diabetes. J Physiol. 2005;564:563–573. doi: 10.1113/jphysiol.2005.082669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszewski JFP, Mourtzakis M, Hillig T, Saltin B, Pilegaard H. Dissociation of AMPK activity and ACCβ phosphorylation in human muscle during prolonged exercise. Biochem Biophys Res Commun. 2002;298:309–316. doi: 10.1016/s0006-291x(02)02465-8. [DOI] [PubMed] [Google Scholar]