

Abstract
Background
The randomized controlled Pediatric Oncology Group study 9233 tested the hypothesis that dose-intensive (DI) chemotherapy would improve event-free survival (EFS) for children <3 years of age with newly diagnosed malignant brain tumors.
Methods
Of 328 enrolled eligible patients, diagnoses were medulloblastoma (n = 112), ependymoma (n = 82), supratentorial primitive neuroectodermal tumor (sPNET, n = 38) and other malignant brain tumors (n = 96), and were randomized to 72 weeks of standard dose chemotherapy (Regimen A, n = 162) or DI chemotherapy (Regimen B, n = 166). Radiation therapy (RT) was recommended for patients with evidence of disease at completion of chemotherapy or who relapsed within 6 months of chemotherapy completion.
Results
Distributions of EFS for Regimens A and B were not significantly different (P = 0.32) with 2- and 10-year rates of 22.8% ± 3.3% and 15.4% ± 3.7%, and 27.1% ± 3.4% and 20.8% ± 3.8%, respectively. Thus, the study hypothesis was rejected. While distributions of EFS and OS were not significantly different between Regimens A and B for patients with medulloblastoma and sPNET, DI chemotherapy resulted in significantly improved EFS distribution (P = .0011) (2-year EFS rates of 42.1% vs. 19.6% with SD chemotherapy), but not OS distribution, for patients with centrally confirmed ependymoma. The degree of surgical resection affected EFS, OS or both for most tumor groups. Approximately 20%, 40% and 20% of patients with medulloblastoma, ependymoma treated with DI chemotherapy, and sPNET, respectively appear to have been cured without RT. Of 11 toxic deaths on study, 10 occurred on the DI chemotherapy arm.
Conclusions
Prolonged dose-intensive chemotherapy given to infants with malignant brain tumors resulted in increased EFS only for patients with ependymoma.
Keywords: brain tumors, chemotherapy, dose-intensive, infants
The groundbreaking study of Duffner et al demonstrated that the use of neoadjuvant chemotherapy could allow successful delay of radiation therapy in 40% of children under the age of 3 years with malignant brain tumors.1 It was hypothesized that this delay allowed a longer period of brain maturation to occur that might have rendered the children less vulnerable to radiation therapy (RT)-induced brain injury. Though unplanned on that study, cases of cure were seen with surgery and chemotherapy only.
In 1992, the Pediatric Oncology Group (POG), now part of the Children's Oncology Group (COG), opened the successor study to prospectively investigate whether more chemotherapy, randomly assigned and delivered through dose-intensification, would result in an improved event-free survival distribution (EFS) compared to standard dosing of the same chemotherapy agents. Patients were also followed for overall survival (OS). For each regimen, secondary objectives of the study were to estimate the EFS rates for children having complete response to surgery and chemotherapy only; to describe acute and chronic toxicities observed, including neuropsychological outcome; and to estimate the secondary EFS of patients treated with RT following disease progression during, or after failing to respond to, chemotherapy. The responses of children with medulloblastoma (MB), ependymoma (EP) and supratentorial primitive neuroectodermal tumors (sPNET) for whom meaningful statistical analyses could be performed are the primary subject of this report. The outcomes of children with less common diagnoses are also described. The results of this study are discussed in context with those of several infant brain tumor studies published in the last 15 years.
Materials and Methods
Patient Eligibility and Registrations
Any child <36 months of age at a participating POG institution with a newly diagnosed and histologically confirmed malignant brain tumor, or intrinsic brainstem glioma diagnosed by clinical and radiographic criteria, was eligible for this study. Written informed consent for enrollment was obtained from parents or guardians of each child according to institutional guidelines. Following tumor resection, patients were evaluated for residual disease and staged for metastases through post-operative MRI or CT, MRI of the spine, CSF cytology, bone scan and bone marrow aspiration. For as many cases as possible, pre- and post-operative images were centrally reviewed. Tumor pathology was centrally reviewed and classified according to World Health Organization criteria of that time. Upon registration, patients were randomized to standard dose (SD; Regimen A) or dose-intensive (DI; Regimen B) chemotherapy and stratified for statistical analyses on the basis of institutional diagnosis and disease status (no disease vs. disease present at the start of chemotherapy). Disease strata included MB, EP, malignant glioma (MG), brainstem glioma (BSG), and sPNET. This last stratum included pineoblastoma, cerebral neuroblastoma and other small, blue, round-cell tumors. Finally, a stratum designated “Other” was used; it included atypical teratoid rhabdoid tumors (ATRT), choroid plexus carcinoma (CPC) and other rare tumor types. Centrally reviewed tumors of low-grade, non-malignant histology were deemed ineligible for statistical analyses.
Chemotherapy
It was recommended that chemotherapy be initiated by 28 post-operative days following maximal safe tumor resection, except in cases of intrinsic pontine glioma. The chemotherapy agents in each regimen were identical and they were given in pairs. Regimen A consisted of Cycles A, A’ and B given every 28 days on an alternating basis of AA'BAA'B … for 72 weeks. Cycle A consisted of cyclophosphamide (65 mg/kg) on day 1 and vincristine (0.065 mg/kg) given on days 1, 8, 15 and 21; Cycle A’ was identical to A except that vincristine was given on days 1 and 8 only. Cycle B consisted of cisplatin (4 mg/kg) on day 1 and etoposide (6.5 mg/kg) on days 3 and 4. Regimen B consisted of Cycles X and Y given every 21 days on an alternating schedule of XXYXXY … . for 72 weeks. Cycle X consisted of cyclophosphamide (65 mg/kg) on days 1 and 2, and vincristine (0.065 mg/kg) on days 1, 8 and 15. Cycle Y consisted of cisplatin (5 mg/kg) on day 1 and etoposide (7.5 mg/kg) on days 3 and 4. (Table 1) Regimen B chemotherapy was 1.8x more DI than Regimen A chemotherapy. G-CSF was used on Regimen B and was optional on Regimen A. Certain laboratory and clinical criteria were required at the start of each chemotherapy cycle to assess for adequate organ function. Chemotherapy doses were adjusted for hematotoxicity, neurotoxicity (vincristine) and renal- or ototoxicity (cisplatin). Chemotherapy was discontinued in those patients who had unacceptable toxicity despite dose modification, and in patients whose disease progressed or recurred. Patients whose tumors were non-metastatic at diagnosis and who were in remission at the end of 72 weeks of therapy discontinued therapy. The doses and schedule of chemotherapy that each patient received were recorded and used to calculate the chemotherapy dose-intensity delivered in each cycle of therapy.
Table 1.
Chemotherapy schedule
| Regimen | Chemotherapy | Doses in mg/kg | Days | DI | RDI |
|---|---|---|---|---|---|
| A | Cyclophosphamide | 65 | A1 | 10.83 | |
| Vincristine | .065 | A1, 8, 15, 22; A'1, 8 | 0.03 | ||
| Cisplatinum | 4 | B1 | 0.33 | ||
| Etoposide | 6.5 | B3, 4 | 1.08 | ||
| B | Cyclophosphamide | 65 | X1, 2 | 28.89 | 2.67 |
| Vincristine | 0.065 | X1, 8, 15 | 0.04 | 1.33 | |
| Cisplatinum | 5 | Y1 | 0.56 | 1.70 | |
| Etoposide | 7.5 | Y3, 4 | 1.67 | 1.55 | |
| Overall | 1.80 |
Chemotherapy on Regimen A was delivered in a pattern of AA'BAA'B … for 72 weeks; the pattern of Regimen B was XXYXXY … was 72 weeks. Dose-intensity (DI) was calculated as mg/kg/week of each agent. Relative DI (RDI) was calculated as the DI of that agent on Regimen B/DI of that agent on Regimen A. Overall RDI of Regimen B was the average of the four individual RDIs of that regimen.
Radiation Therapy
Patients with metastatic disease at diagnosis or who had residual, non-progressive disease at completion of chemotherapy were eligible for radiation therapy (RT) on study 9233. Patients in complete remission at the end of scheduled chemotherapy but who had progressive disease within the subsequent 6 months were eligible for RT on the companion study 9234, as were patients whose disease recurred of progressed on 9233 chemotherapy. Second resection of disease was recommended when feasible prior to the start of RT. The volume of radiation was determined by the histopathologic diagnosis, the presence or absence of metastases at diagnosis, the identification of peri-tumoral edema, and the extent of disease documented at the time of radiation. The dose of radiation was adjusted for these factors and for patient age. Thus, children with tumors unlikely to disseminate, with negative CSF cytology and negative neuraxis imaging, received 48 Gy if <18 months, 50.4 Gy if 18–30 months, and 55.2 Gy if >30 months of age at treatment. The radiation volume included either the posterior fossa with a subsequent cone-down, or the pre-operative or pre-chemotherapy volume, including edema, with a cone-down. Patients with tumors likely to disseminate but with negative CSF cytology and negative neuraxis imaging received craniospinal axis irradiation (CSI) of 27 Gy if <18 months, 30 Gy if 18–30 months, and 34.5 Gy if >30 months of age at the time of treatment. Patients with neuraxis involvement at diagnosis or subsequently received CSI at doses that were 3–4 Gy higher for each age group. Radiation treatment data, including field and dose, were retrospectively reviewed at the Quality Assurance Research Center in Providence, RI.
Statistical Considerations
The primary statistical analysis followed the intent-to-treat premise and all eligible patients who initiated treatment were analyzed as randomized. The study design assumed exponential distributions of EFS and the sample size was sufficient to provide 80% power to detect a 15% absolute improvement in the 2-year EFS rate for patients randomized to Regimen B vs. Regimen A (one-sided tests). Stratified log-rank tests were used to compare the distributions of EFS and OS for patients randomized to Regimens A and B.
Events for EFS were defined as any progression, relapse, second malignancy or death, and EFS was measured from the start of therapy to the earliest event. Patients who did not experience an event were censored at their off-study date. Overall survival was measured from the start of therapy to date of death or date off study for censored patients. Distributions of EFS and OS were estimated using the Kaplan-Meier method for the cohort2 and by treatment, institutional diagnosis, central reviewed diagnosis, gender, metastatic status, degree of resection and age at diagnosis, and compared using stratified log-rank tests. To investigate associations of continuous variables with EFS and OS, Cox proportional hazards models were used. Descriptive statistics for the patient cohort are provided and toxicities are described.
Results
Patient Characteristics
Three-hundred-thirty-eight patients were enrolled on POG 9233. Of those, 8 were ineligible due to diagnosis and 2 patients did not receive therapy on study and were not followed. Thus, analyses were based on 328 eligible patients who were treated on study. Fig. 1 shows the flow of patients through the study. Patient characteristics are shown in Table 2. Of the 328 patients, 186 (56.7%) were male, 72.6% were white, non-Hispanic, the median age was 1.62 years (range 0.01 to 2.97 years), and 15.2% had metastatic (M+) disease at diagnosis. The institutional diagnoses (n; % overall patients; % M+) were MB (112; 34%; 24.1%). EP (82; 25%; 4.9%), sPNET (38; 11.6%; 23.7%), and Other (96; 29.3%; 10.4%). Central review of institutional diagnoses was accomplished for 305 of the 328 patients. Eighty-eight (78.6% of all MB cases, including two posterior fossa PNETs), 76 (92.7%) EP (58 EP NOS and 18 anaplastic type EP), and 10 (26.3%) sPNET were centrally confirmed. One-hundred sixty two (49%) of the patients were registered on Regimen A. Gross total resection (GTR) was achieved in 107 (32.6% of the entire cohort) patients at diagnosis and in 39.3%, 36.6% and 26.3% of patients with MB, EP and sPNET, respectively. Only 2 patients, 1 with MB and 1 with sPNET, were found to have extra-neural metastases, to the bone marrow in each case.
Fig. 1.

Flow of patients through study 9233.
Table 2.
Patient characteristics
| TREATMENT NUMBERS |
All Patients |
|||||||
|---|---|---|---|---|---|---|---|---|
| Regimen-A |
Regimen-B |
|||||||
| n | Row % | n | Row % | n | Row % | |||
| GENDER | MALE | 95 | 51.1 | 91 | 48.9 | 186 | 100.0 | |
| FEMALE | 67 | 47.2 | 75 | 52.8 | 142 | 100.0 | ||
| RACE | WHITE, NON-HISP | 118 | 49.6 | 120 | 50.4 | 238 | 100.0 | |
| BLACK, NON-HISP | 44 | 48.9 | 46 | 51.1 | 90 | 100.0 | ||
| INSTITUTIONAL DIAGNOSIS | EP | 43 | 52.4 | 39 | 47.6 | 82 | 100.0 | |
| MB* | 48 | 42.9 | 64 | 57.1 | 112 | 100.0 | ||
| sPNET | 24 | 63.2 | 14 | 36.8 | 38 | 100.0 | ||
| OTHERS | 47 | 49.0 | 49 | 51.0 | 96 | 100.0 | ||
| M-STAGE | M0 | 137 | 49.3 | 141 | 50.7 | 278 | 100.0 | |
| M+ | 25 | 50.0 | 25 | 50.0 | 50 | 100.0 | ||
| INSTITUTIONAL DIAGNOSIS vs. M-STAGE | EP | M0 | 41 | 52.6 | 37 | 47.4 | 78 | 100.0 |
| M+ | 2 | 50.0 | 2 | 50.0 | 4 | 100.0 | ||
| MB* | M0 | 36 | 42.4 | 49 | 57.6 | 85 | 100.0 | |
| M+ | 12 | 44.4 | 15 | 55.6 | 27 | 100.0 | ||
| sPNET | M0 | 19 | 65.5 | 10 | 34.5 | 29 | 100.0 | |
| M+ | 5 | 55.6 | 4 | 44.4 | 9 | 100.0 | ||
| OTHERS | M0 | 41 | 47.7 | 45 | 52.3 | 86 | 100.0 | |
| M+ | 6 | 60.0 | 4 | 40.0 | 10 | 100.0 | ||
| SURGERY EXTENT | STR/Biopsy | 115 | 52.0 | 106 | 48.0 | 221 | 100.0 | |
| GTR | 47 | 43.9 | 60 | 56.1 | 107 | 100.0 | ||
| INSTITUTIONAL DIAGNOSIS vs. SURGERY EXTENT | EP | STR/Biopsy | 29 | 55.8 | 23 | 44.2 | 52 | 100.0 |
| GTR | 14 | 46.7 | 16 | 53.3 | 30 | 100.0 | ||
| MB* | STR/Biopsy | 29 | 42.6 | 39 | 57.4 | 68 | 100.0 | |
| GTR | 19 | 43.2 | 25 | 56.8 | 44 | 100.0 | ||
| sPNET | STR/Biopsy | 18 | 64.3 | 10 | 35.7 | 28 | 100.0 | |
| GTR | 6 | 60.0 | 4 | 40.0 | 10 | 100.0 | ||
| OTHERS | STR/Biopsy | 39 | 53.4 | 34 | 46.6 | 73 | 100.0 | |
| GTR | 8 | 34.8 | 15 | 65.2 | 23 | 100.0 | ||
| All Patients | 162 | 49.4 | 166 | 50.6 | 328 | 100.0 | ||
Abbreviations used: MB, medulloblastoma; EP, ependymoma, sPNET, supratentorial primitive neuroectodermal tumor; STR, subtotal resection; GTR, gross total resection.
*Including the posterior fossa PNETs.
Outcome of Therapy
For the entire cohort, 2- and 10 year rates of EFS ± standard error (±SE) were 25.0% ± 2.4% and 18.2% ± 2.7%, respectively; corresponding rates of OS were 46.7% ± 2.8% and 29.3% ± 3.2%, respectively. (Fig. 2A) Patients on Regimen A had 2- and 10-year EFS rates of 22.8% ± 3.3% and 15.4% ± 3.7%, respectively; the corresponding rates for patients on Regimen B were 27.1% ± 3.4%, and 20.8% ± 3.8%, respectively. (Fig. 2B) The difference in EFS between the two arms was not significant (P = 0.32). Thus, the hypothesis that DI chemotherapy would increase EFS of infants with malignant brain tumors compared to SD chemotherapy was not proven. Rates of OS at 2- and 10 years for Regimen A patients were 53.7% ± 3.9% and 30.5% ± 4.6%, respectively. For Regimen B patients, these rates were 39.8% ± 3.8% and 28.1% ± 4.5%, respectively. This difference in OS was also not significant (P = 0.25) (Fig. 2C).
Fig. 2.
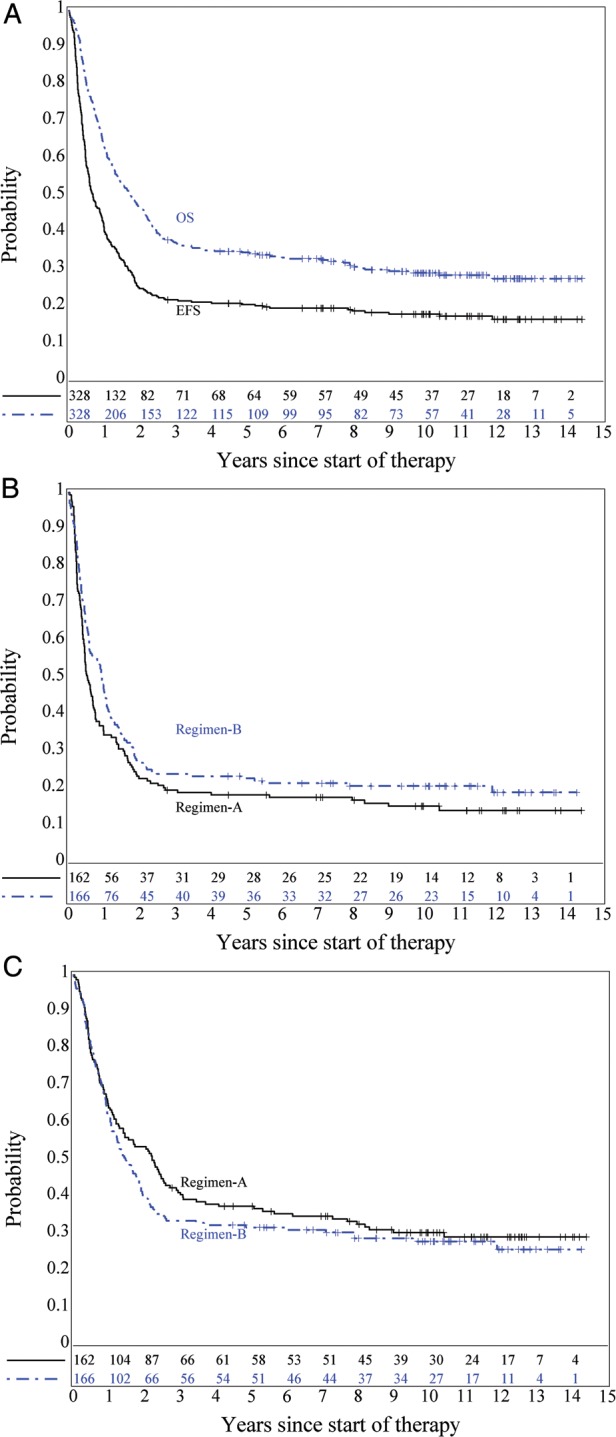
(A) EFS and OS for the entire cohort; (B) EFS by regimen; P = .32; (C) OS by regimen; P = .25.
Further analyses were conducted for the MB, EP and sPNET strata. For patients with institutionally diagnosed EFS and OS distributions did not differ between Regimens A and B (P = 0.96 for EFS, 0.51 for OS; data not shown). Patients with MB were classified based on their M status (M0 vs M+) and by GTR vs sub-total resection (STR) or biopsy. Fig. 3A and B show EFS and OS distributions for these comparisons. EFS was affected by M status: patients with M+ disease had the poorest EFS distribution (P = .0011) and GTR of M0 disease appeared to show no advantage over STR or biopsy for EFS. OS distributions were affected by degree of resection: M0 patients who had undergone GTR of MB had the best OS distribution, which was significantly better (P = .017) than M0 patients who had undergone STR or biopsy who, in turn, had a similar OS distribution as M+ MB patients.
Fig. 3.
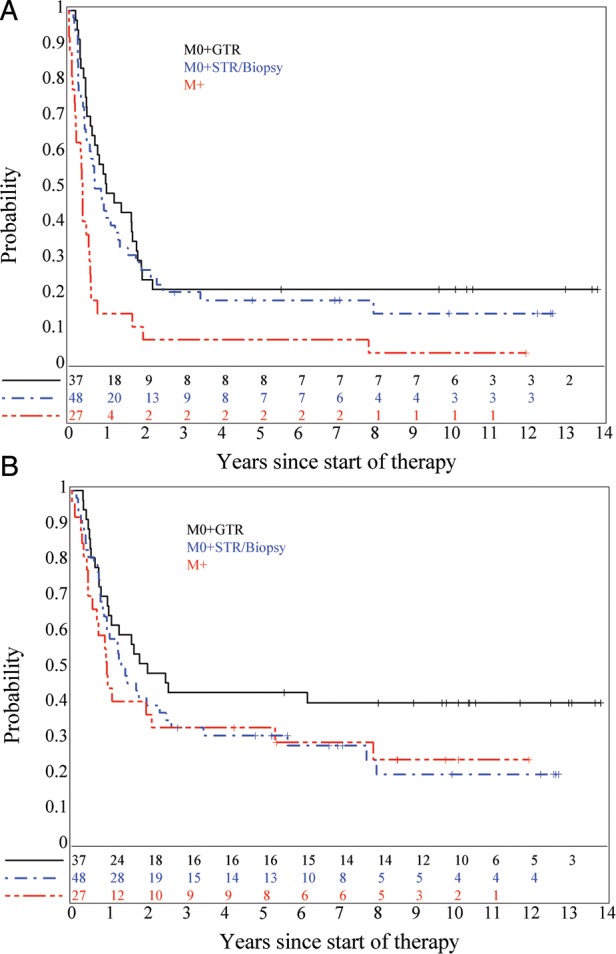
(A) EFS, P = .0011 (M0 + GTR and M0 + STR/biopsy vs. M+) and (B) OS, P = .22, for medulloblastoma patients classified by M-stage and extent of surgical resection. Log-rank P value for OS by surgery extent (GTR vs. STR/Biopsy) is .017.
Treatment of institutionally diagnosed EP patients on DI chemotherapy resulted in significantly improved distribution of EFS (P = .0062) but not of OS (P = .83; Fig. 4A and B). There were only four patients with M+ EP, so those patients were included with patients who underwent STR or biopsy of their tumors for a comparison with EP patients who underwent GTR of disease prior to chemotherapy. For both EFS and OS, GTR was associated with significantly better EFS and OS distributions (P = .008 for EFS [Fig. 4C] and P = .014 for OS).
Fig. 4.
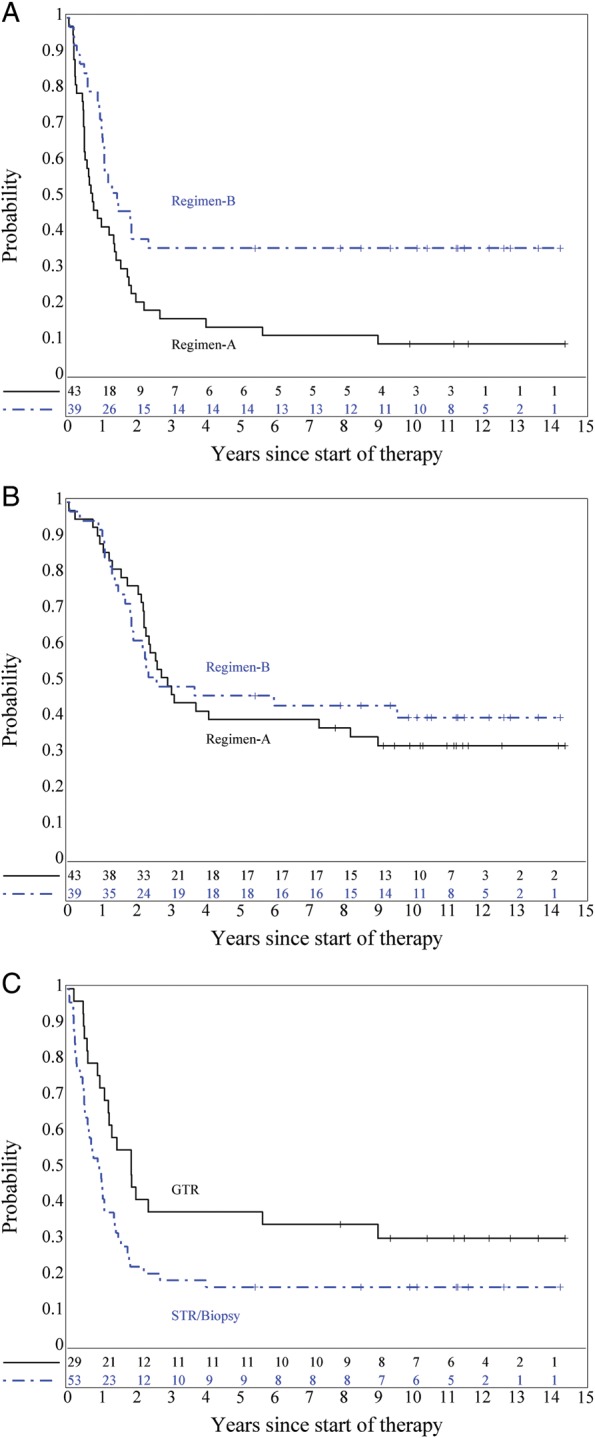
(A) EFS, P = .0062, and (B) OS, P = .83, for institutionally diagnosed ependymoma patients by regimen. (C) EFS for ependymoma patients by extent of surgical resection, P = .008 (GTR vs STR or biopsy).
Similar analyses of patients with sPNET were conducted. Distributions of EFS and OS were not significantly different for Regimen A vs Regimen B, but GTR was associated with significantly improved EFS (P = .026), but not OS (P = .074), distribution as compared to patients with <STR or with M+ disease (Fig. 5A and B).
Fig. 5.
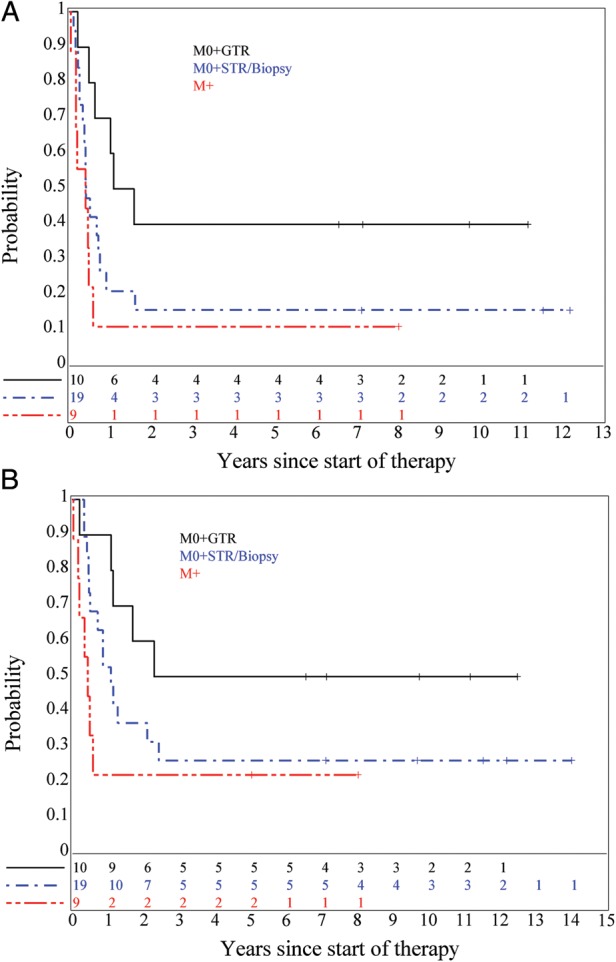
(A) EFS, P = .026 (M0 + GTR vs. M0 + STR or biopsy and M + ), and (B) OS (P = .074) for sPNET patients classified by M-stage and extent of surgical resection.
The primary event on this study was recurrent or progressive disease, and this occurred in 209 of the 328 patients. The prescribed protocol therapy for this was resection of disease, if a significant reduction of tumor bulk was a realistic possibility, followed by RT (see below). EFS rates achieved with surgery and chemotherapy approximated the proportions of patients who did not receive RT. Thus, ∼20%, 40% and 20% of patients with MB, EP treated on DI chemotherapy and sPNET appear to have been cured without having been irradiated.
The associations of age at diagnosis, gender and race were analyzed in the MB, EP and sPNET cohorts, for whom chemotherapy regimen and extent of surgery were used as stratification variables. For these, older age was associated with a more favorable OS for MB patients (P = .034), and male gender was insignificantly associated with a lower OS in the EP cohort (P = .064) (Data not shown.).
Two other diagnostic subgroups of patients were noteworthy. First, during the duration of this study there was increased recognition of ATRT as a distinct entity. While 12 patients carried an institutional diagnosis of ATRT, 36 patients were centrally reviewed as having ATRT. Of these, 7 (20.6%) underwent GTR of disease and 5 (14.7%) had M+ disease at diagnosis. Fourteen (41.2%) were assigned to Regimen A. None of the patients with ATRT survived. The second subgroup was that on the MG stratum, to which 20 patients were registered. Of these, 10 (50%) ultimately survived. The numbers of patients in these two diagnostic subgroups were too low for any meaningful analysis and conclusions. Patients who survived MG, however, underwent complete or less than complete resection of disease, were treated on both chemotherapy regimens and received or did not receive RT; thus, no particular pattern of treatment leading to survival could be discerned. (Data not shown.)
Radiation Therapy
There were 291 patients eligible for RT on study for persistent, recurrent or progressive disease, but only 53 eligible patients were registered on study 9234. Four of these patients were inevaluable for response to RT for reasons of inadequate trial or early death (2 patients, each with “other” tumors), parental refusal for therapy and physician preference for non-protocol RT (1 patient each). Forty-four of the 49 patients began RT with measurable disease and institutions coded responses according to study criteria. Twelve patients achieved a complete response to RT and another had a partial response. Responses were not durable in a large majority of patients; 43 of 49 evaluable irradiated patients ultimately had progressive disease. Twelve patients survive, 3 of these having undergone additional therapy for another recurrence after completion of RT. Because the use of RT was distributed across several diagnostic strata, and across patients treated with standard and DI chemotherapy, meaningful comparisons of results are not possible.
Toxicities of Therapy
Overall, Regimen B was much more toxic than Regimen A. Fatal toxicities were recorded for 11 patients, 10 of whom were on Regimen B. These grade 5 toxicities were recorded for infection (NOS) (n = 1; this patient was on Regimen A), viral pneumonia (n = 3), pulmonary/functional (n = 3), and bacterial pneumonia, varicella, fungal sepsis and viral, generalized in 1 patient each. When recorded, lymphopenia was noted twice as frequently for patients on Regimen B than Regimen A. Infections were generally recorded for two to three times the number of patients on DI chemotherapy. Predictably, ototoxicity was three times as common on Regimen B as on Regimen A. Peripheral neurotoxicity was also nearly three times more common with DI chemotherapy. Without further elaboration, psychosocial toxicities were listed for one and four patients on Regimens A and B, respectively.
Discussion
For the entire study cohort, “more” chemotherapy, delivered through dose-intensification of four active agents, did not result in improved EFS or OS. As such, 9233 constituted a “negative” study, albeit an important one. Study 9233 was built on the results of POG 8633 and the EFS and OS rates achieved on the standard arm of study 9233 were very similar to those reported by Duffner et al for all patients on “Baby POG 1” and for patients with MB, EP and sPNET.1 Intuitively, DI chemotherapy would have resulted in higher rates of EFS and OS. Evidence from medical oncology and from experience in pediatric Ewing sarcoma, osteosarcoma and neuroblastoma suggested that intensification of at least some effective agents would improve patient outcomes.3–7 Given that evidence, the results of this trial were surprising.
The results of several studies contemporary or subsequent to POG 9233 offer context for our results. Direct comparisons are difficult because of differences in definitions of subgroups based on the degree of resection and because of differences in study eligibility, but all of these studies sought to avoid the use of RT during primary therapy. The French Society of Paediatric Oncology used a much less intensive schedule of chemotherapy for patients <5 years of age with MB and achieved PFS rates very similar to those of 9233.8 The Children's Cancer Group (CCG) of North American conducted a trial comparing two regimens of intensive induction chemotherapy lasting only 15 weeks, and followed this with lower intensity maintenance chemotherapy.9 As on 9233, the use of RT was restricted to patients with progressive or metastatic disease. The overall results of the CCG study were similar to those of 9233, but the results for patients with MB appear superior on the CCG study. In the same period of time, the “Head Start” approach utilized an even more DI schedule of induction chemotherapy lasting 15 weeks, followed by a single course of myeloablative chemotherapy with autologous hematopoietic stem cell rescue (AHSCR).10 The 5-year EFS rate of 52% ± 11% for the comparatively low number of patients with non-metastatic MB on the Head Start I and II studies appear superior to the POG and CCG studies. The COG incorporated high-dose chemotherapy with AHSCR into its subsequent clinical trial for infants with malignant brain tumors, the results of which have not yet been published. Meanwhile, the HITSKK92 study from Germany sought to avoid use of CSI by incorporating high-dose systemic and intraventricular methotrexate with vincristine, cyclophosphamide, etoposide and carboplatin.11 The 5-year PFS rate of 82% ± 9% for infants with completely resected M0 or M1 MB is the most promising to date. In that study, desmoplastic histology was strongly prognostic for higher PFS. Overall, while prolonged DI chemotherapy as given on study 9233 did not provide benefit to the entire study population, or to the patients with MB, later trials support further investigation of shorter duration DI chemotherapy achieved with AHSCR or of incorporation of methotrexate into systemic therapy. One therapy for all infant brain tumor diagnoses, or for all types of MB, is no longer a rational or appropriate approach. The results of treatment of infants with non-desmoplastic MB or with incompletely resected or metastatic disease are slowly improving but remain suboptimal.
Another important observation from study 9233 concerns the benefit of DI chemotherapy on EFS for patients with EP. We estimate that 40% of patients treated with DI chemotherapy survive long-term (>10 years) without having received RT. Others have reported that infants with EP can be cured without RT. In the CCG study, 45% of survivors of EP at 5 years had never received RT.9 Grundy et al, for the United Kingdom Children's Cancer Group/International Society of Paediatric Oncology (UKCCSG/SIOP) study CNS9204, reported 42% of survivors not having been treated with RT.12 In that trial, chemotherapy incorporated high-dose methotrexate and RT was reserved for patients with recurrent or progressive disease. It was noted that increased chance of survival was associated with an increased received dose-intensity of chemotherapy. In the SFOP study, ∼23% of patients surviving EP 4 years after diagnosis had not had RT.13 While historically in older children with EP the role of chemotherapy in achieving survival is not clear, the results of 9233 and of other studies suggest that some cases of EP, at least in a subset of infants, are chemotherapy sensitive and possibly curable with surgery and chemotherapy alone.
Subsets of patients with EP for which chemotherapy may be most beneficial and RT not necessary might be identifiable by histology, though published reports are conflicting. Follow-up data submitted from institutions to the study PI (DS) indicated that more than one-half of EP patients (43 of 73) were eventually treated with RT, but less than half of these were treated on study 9233 or 9234. Thus, for the majority of patients with EP who received RT, more detailed information about the RT was not collected. However, the combined institutional and study data indicate that, in our study, similar proportions of patients with grade II EP treated on Regimen B survive without RT (9 of 15) as were treated on Regimen A (5 of 10). The results for patients with grade III EP contrast to this: 11 of 12 patients treated on Regimen A are known to have died, including all 6 who received RT; from Regiment B, 4 of 5 patients who also received RT survive, but only 1 of 6 who did not receive RT survived. These numbers of patients are quite low, but suggest that for those patients with anaplastic histology, survival was more likely following Regimen B chemotherapy with RT, compared to Regimen A chemotherapy with or without RT or Regimen B without RT. These descriptive data suggest a possible interplay of histology and treatment; patients with grade II EP might be more likely to be cured with chemotherapy alone, but patients with grade III EP might require RT to survive. Histology can be subjectively interpreted but likely reflects biology. Subsets of EP patients have also been identified through other cytogenetic, pathologic and molecular features.14,15 As has been developed for medulloblastoma in older children, international collaboration on studies of EP biology might yield valid biological subsets of EP for which distinct therapies might ultimately be designed.16
Not surprisingly, we found gross total resection of disease to be associated with higher rates of EFS and OS for patients with EP, and with higher rates of EFS for patients with MB and sPNET. These are not novel findings, since data show that GTR of most tumor types predicts a higher chance of survival.17 Given this, chemotherapy for infants with brain tumors may facilitate a delayed GTR in patients whose tumors were initially unresectable.18 Chemotherapy may devascularize tumors and establish a more discernable interface between tumor and normal brain. Planned second-look, delayed operation to achieve GTR of disease has been incorporated into ongoing COG infant brain tumor trials.
Finally, infants with ATRT and MG warrant brief discussion. The diagnosis of ATRT was first appreciated in the late 1980s. During the conduct of 9233, it is likely that the diagnosis became better known to pediatric neuropathologists, and that its incidence on 9233 is underestimated. The COG is currently conducting the first prospective study of children of all ages with ATRT, using INI1 analysis to confirm the diagnosis, induction chemotherapy that includes methotrexate, and consolidation therapy that incorporates high-dose chemotherapy with AHSCR. The number of very young children with MG is much lower than MB, EP, sPNET and ATRT. Yet, results of 8633 and 9233 are provocative in showing an apparent cure in 50% of these children, quite unlike the rates achieved in older children and adults, but similar to small series reported by others.19–21 These children, and their treating teams, would benefit highly from an international cooperative trial with correlative analyses of biological characteristics of disease.
In conclusion, the hypothesis that DI chemotherapy would result in higher rates of EFS for very young children with malignant brain tumors was not proven on study 9233. The benefit of DI chemotherapy was reflected only in a significantly higher rate of EFS for patients with EP; for nearly 40% of these patients, RT was not necessary for long-term survival (>10 years). As has been demonstrated by others, brain tumor cure was achieved in some patients following surgery and chemotherapy alone. The arsenal of active chemotherapy agents has changed little in the past two decades; the addition of methotrexate to CNS tumor therapy is relatively new and encouraging, but concerning as well for its potential for adverse long-term effects. High-dose chemotherapy regimens supported by AHSCR are under study. Subsets of patients have been identified, on the basis of degree of tumor resection and tumor histology, to which treatments are being successfully tailored. For the majority of patients, however, curative therapy has not yet been found. The search for such therapy may be aided by further identification of patient- and tumor-derived biologic prognostic factors. Due to the rarity of these tumors, this search will need to be internationally collaborative.
Funding
This study was supported by grant CA098543 from the National Cancer Institute of the National Institutes of Health.
Acknowledgments
The authors acknowledge Dr Henry Friedman for his contribution to the design of this study, Dr James Langston for his review of diagnostic imaging, and the clinical research associates at the former POG institutions for their assistance with data. Results of this study were presented in part at the American Society of Clinical Oncology annual meeting in 2000, and at the International Society of Pediatric Neuro-Oncology annual meetings in 2000 and 2004.
Conflict of interest statement. None declared.
References
- 1.Duffner PK, Horowitz ME, Krischer JP, et al. Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med. 1993;328(24):1725–1731. doi: 10.1056/NEJM199306173282401. [DOI] [PubMed] [Google Scholar]
- 2.Kaplan EL, Meier P. Nonparametric estimations from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 3.Hryniuk WM. Philadelphia: JB Lippincott; 1988. The importance of dose intensity in the outcome of chemotherapy; pp. 121–141. Important advances in oncology. [PubMed] [Google Scholar]
- 4.Hryniuk WM. More is better. J Clin Oncol. 1988;6(9):1365–1367. doi: 10.1200/JCO.1988.6.9.1365. [DOI] [PubMed] [Google Scholar]
- 5.Delepine N, Delepine G, Bacci G, Rosen G, Desbois JC. Influence of methotrexate dose intensity on outcome of patients with high grade osteogenic osteosarcoma. Analysis of the literature. Cancer. 1996;78(10):2127–2135. [PubMed] [Google Scholar]
- 6.Smith MA, Ungerleider RS, Horowitz ME, Simon R. Influence of doxorubicin dose intensity on response and outcome for patients with osteogenic sarcoma and Ewing's sarcoma. J Natl Cancer Inst. 1991;83(20):1460–1470. doi: 10.1093/jnci/83.20.1460. [DOI] [PubMed] [Google Scholar]
- 7.Cheung NV, Heller G. Chemotherapy dose intensity correlates strongly with response, median survival, and median progression-free survival in metastatic neuroblastoma. J Clin Oncol. 1991;9(6):1050–1058. doi: 10.1200/JCO.1991.9.6.1050. [DOI] [PubMed] [Google Scholar]
- 8.Grill J, Sainte-Rose C, Jouvet A, et al. Treatment of medulloblastoma with postoperative chemotherapy alone: an SFOP prospective trial in young children. Lancet Oncol. 2005;6(8):573–580. doi: 10.1016/S1470-2045(05)70252-7. [DOI] [PubMed] [Google Scholar]
- 9.Geyer JR, Sposto R, Jennings M, et al. Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children's Cancer Group. J Clin Oncol. 2005;23(30):7621–7631. doi: 10.1200/JCO.2005.09.095. [DOI] [PubMed] [Google Scholar]
- 10.Dhall G, Grodman H, Ji L, et al. Outcome of children less than three years old at diagnosis with non-metastatic medulloblastoma treated with chemotherapy on the “Head Start” I and II protocols. Pediatr Blood Cancer. 2008;50(6):1169–1175. doi: 10.1002/pbc.21525. [DOI] [PubMed] [Google Scholar]
- 11.Rutkowski S, Bode U, Deinlein F, et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med. 2005;352(10):978–986. doi: 10.1056/NEJMoa042176. [DOI] [PubMed] [Google Scholar]
- 12.Grundy RG, Wilne SA, Weston CL, et al. Primary postoperative chemotherapy without radiotherapy for intracranial ependymoma in children: the UKCCSG/SIOP prospective study. Lancet Oncol. 2007;8(8):696–705. doi: 10.1016/S1470-2045(07)70208-5. [DOI] [PubMed] [Google Scholar]
- 13.Grill J, Le Deley MC, Gambarelli D, et al. Postoperative chemotherapy without irradiation for ependymoma in children under 5 years of age: a multicenter trial of the French Society of Pediatric Oncology. J Clin Oncol. 2001;19(5):1288–1296. doi: 10.1200/JCO.2001.19.5.1288. [DOI] [PubMed] [Google Scholar]
- 14.Godfraind C, Kaczmarska JM, Kocak M, et al. Distinct disease-risk groups in pediatric supratentorial and posterior fossa ependymomas. Acta Neuropathol. 2012;124(2):247–257. doi: 10.1007/s00401-012-0981-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Witt H, Mack SC, Ryzhova M, et al. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell. 2011;20(2):143–157. doi: 10.1016/j.ccr.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123(4):465–472. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lafay-Cousin L, Strother D. Current treatment approaches for infants with malignant central nervous system tumors. Oncologist. 2009;14(4):433–444. doi: 10.1634/theoncologist.2008-0193. [DOI] [PubMed] [Google Scholar]
- 18.Van Poppel M, Klimo P, Jr., Dewire M, et al. Resection of infantile brain tumors after neoadjuvant chemotherapy: the St. Jude experience. J Neurosurg Pediatr. 2011;8(3):251–256. doi: 10.3171/2011.6.PEDS11158. [DOI] [PubMed] [Google Scholar]
- 19.Duffner PK, Horowitz ME, Krischer JP, et al. The treatment of malignant brain tumors in infants and very young children: an update of the Pediatric Oncology Group experience. Neuro Oncol. 1999;1(2):152–161. doi: 10.1093/neuonc/1.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanders RP, Kocak M, Burger PC, Merchant TE, Gajjar A, Broniscer A. High-grade astrocytoma in very young children. Pediatr Blood Cancer. 2007;49(7):888–893. doi: 10.1002/pbc.21272. [DOI] [PubMed] [Google Scholar]
- 21.Grundy RG, Wilne SH, Robinson KJ, et al. Primary postoperative chemotherapy without radiotherapy for treatment of brain tumours other than ependymoma in children under 3 years: results of the first UKCCSG/SIOP CNS 9204 trial. Eur J Cancer. 2010;46(1):120–133. doi: 10.1016/j.ejca.2009.09.013. [DOI] [PubMed] [Google Scholar]