

Malignant peripheral nerve sheath tumors (MPNSTs) are rare, but deadly, soft tissue sarcomas that occur more commonly and at a younger age in individuals affected with the neurofibromatosis type 1 (NF1) inherited cancer predisposition syndrome. Currently, the only curative treatment for MPNSTs is surgery, and treatment of advanced disease is limited to cytotoxic chemotherapy with generally little effect on overall survival. Moreover, there is a pressing need for targeted therapeutic options for MPNSTs arising in individuals with NF1 and in the general population.
Mutations in the NF1 gene have been reported in both NF1-related and sporadic MPNSTs,1 such that loss of NF1 protein (neurofibromin) function leads to increased Ras signaling.2–4 Given that the BRAF kinase molecule is also a known downstream Ras effector and that BRAF mutations are found in numerous nervous system cancers,5,6 we hypothesized that BRAF mutations may identify a molecularly-distinct subset of MPNST.
To explore this hypothesis, we employed immunohistochemistry using a BRAFV600E mutation-specific antibody on formalin-fixed/paraffin-embedded samples representing 62 MPNSTs and reviewed the associated clinical data to determine NF1 status, overall survival, and time to metastasis. Representative images are shown in Fig. 1 (left panels). Confirmatory BRAFV600E sequencing was not performed on these tumors, based on our previous experience revealing complete concordance between sequencing and immunohistochemistry results in ganglioglioma7 as well as in other laboratories using different tumor types.8,9
Fig. 1.
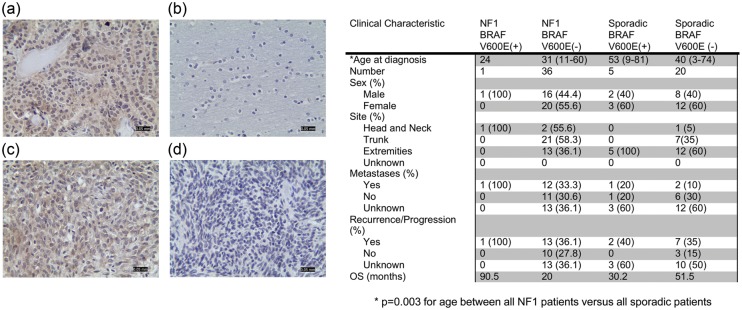
Left. BRAFV600E immunostaining in (a) human papillary thyroid cancer, positive control; (b) normal human brain, negative control; (c) one representative BRAFV600E-immunopositive MPNST; and (d) one representative BRAFV600E-immunonegative MPNST. Magnification, 200x; Scale bar, 50 microns. Right. summary of clinical, demographic, and survival data for individuals with MPNST.
First, we found that individuals with NF1 developed MPNSTs ∼10–15 years earlier than their sporadic counterparts (P < .003; Fig. 1; right panel), which was similar to previous reports.10–12 Second, BRAFV600E mutation was observed in 20% (5/25) of sporadic MPNSTs. In these BRAFV600E-positive MPNSTs, 90%–100% of the tumor cells were BRAFV600E-immunoreactive, suggesting that this mutation could be a primary driver of malignancy and not merely a mutation found in a subset of tumor cells. The prevalence of BRAFV600E mutation in our series of 62 MPNSTs was slightly higher than previously reported in a smaller series of tumors (1 of 13 sporadic MPNSTs13) and was likewise higher than observed in sporadic glioblastoma.14 In this regard, analysis of available The Cancer Genome Atlas (TCGA) data using the C-BIO website application (http://cbio.mskcc.org) revealed that BRAFV600E mutations predominated in thyroid cancer (182/323 tumors; ∼56%) and melanoma (81/228 tumors; ∼36%) but were far less common in all other cancers examined (<9%). Interestingly, melanocytes and Schwann cells have neural crest origins, suggesting a susceptible cell of origin for BRAF mutation in the genesis of these cancers. Third, BRAFV600E mutation was not observed in benign neurofibromas (0/11 tumors), implicating BRAF mutation in malignant progression rather than in neurofibroma tumorigenesis. Fourth, only one NF1-associated MPNST harbored a BRAFV600E mutation (1/37; 2.7%). There was a statistically significant difference in the percentage of BRAFV600E mutations in NF1-associated MPNSTs relative to their sporadic counterparts (P = .035). While these molecular events might be considered mutually exclusive, BRAF molecular alterations have also been reported in another NF1-associated nervous system tumor (pilocytic astrocytoma).15 Fifth, BRAFV600E mutation does not appear to confer any statistically significant differences in overall patient survival or time to metastasis when patients were censored by either date of death or date of last follow-up. This may reflect the small sample size, the rarity of these tumors, and the fact that nearly one-third of the patients were lost to follow-up (and assumed to be living as they could not be found in the Social Security Death Index). While the prognostic value has yet to be established, this study identifies BRAF mutation as a significant molecular alteration in MPNST, potentially offering another therapeutic target for a deadly cancer with no currently efficacious treatment options.
Methods
Cases with the diagnosis of MPNST were collected from UCSF (1990–2012) and Washington University (1990–2005). Clinical data were retrieved from electronic medical records, and all cases with available material were reviewed by at least one of two experienced neuropathologists for diagnostic confirmation (S.D., A.P.). Nerve sheath tumors with equivocal features of malignancy were excluded. The most representative tumor blocks were selected in each case, and microarray blocks were constructed containing two cores of each cancer tissue from the most representative area of tumors, away from necrosis.
Immunohistochemistry was performed, as previously described using a BRAFV600E-specific antibody (clone VE1; Spring Bioscience; 1:100 dilution).7 Tumors were scored as immunopositive if they displayed strong and diffuse cytoplasmic expression. Overall survival data were generated by Kaplan-Meier analysis and log-rank test using Graphpad Prism Version 5.03. A Student' t test was employed to determine the age differences between NF1 and sporadic MPNST patients. A Fisher exact test was employed to determine the difference in frequency of the BRAFV600E mutation in NF1 versus sporadic MPNST patients.
Funding
None declared.
Conflict of interest statement. None declared.
References
- 1.Gupta G, Mammis A, Maniker A. Malignant peripheral nerve sheath tumors. Neurosurg Clin N Am. 2008;19:533–543. doi: 10.1016/j.nec.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 2.Williams VC, Lucas J, Babcock MA, et al. Neurofibromatosis type 1 revisited. Pediatrics. 2009;123:124–133. doi: 10.1542/peds.2007-3204. [DOI] [PubMed] [Google Scholar]
- 3.Basu TN, Gutmann DH, Fletcher JA, et al. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature. 1992;356:713–715. doi: 10.1038/356713a0. [DOI] [PubMed] [Google Scholar]
- 4.DeClue JE, Papageorge AG, Fletcher JA, et al. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell. 1992;69:265–273. doi: 10.1016/0092-8674(92)90407-4. [DOI] [PubMed] [Google Scholar]
- 5.Dienstmann R, Tabernero J. BRAF as a target for cancer therapy. Anticancer Agents Med Chem. 2011;11:285–295. doi: 10.2174/187152011795347469. [DOI] [PubMed] [Google Scholar]
- 6.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 7.Dahiya S, Haydon DH, Alvarado D, et al. BRAF(V600E) mutation is a negative prognosticator in pediatric ganglioglioma. Acta Neuropathol. 2013;125:901–910. doi: 10.1007/s00401-013-1120-y. [DOI] [PubMed] [Google Scholar]
- 8.Colomba E, Helias-Rodzewicz Z, Von Deimling A, et al. Detection of BRAF p.V600E mutations in melanomas: comparison of four methods argues for sequential use of immunohistochemistry and pyrosequencing. J Mol Diagn. 2013;15:94–100. doi: 10.1016/j.jmoldx.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 9.Capper D, Preusser M, Habel A, et al. Assessment of BRAF V600E mutation status by immunohistochemistry with a mutation-specific monoclonal antibody. Acta Neuropathol. 2011;122:11–19. doi: 10.1007/s00401-011-0841-z. [DOI] [PubMed] [Google Scholar]
- 10.Ducatman BS, Scheithauer BW, Piepgras DG, et al. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57:2006–2021. doi: 10.1002/1097-0142(19860515)57:10<2006::aid-cncr2820571022>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 11.Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002;62:1573–1577. [PubMed] [Google Scholar]
- 12.Evans DG, Baser ME, McGaughran J, et al. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39:311–314. doi: 10.1136/jmg.39.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Serrano C, Simonetti S, Hernandez-Losa J, et al. BRAF V600E and KRAS G12S mutations in peripheral nerve sheath tumours. Histopathology. 2013;62:499–504. doi: 10.1111/his.12021. [DOI] [PubMed] [Google Scholar]
- 14.Dahiya S, Haydon DH, Leonard JR, et al. BRAF-V600E mutation in pediatric and adult glioblastoma. Neuro Oncol. 2014;16:318–319. doi: 10.1093/neuonc/not146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriguez FJ, Ligon AH, Horkayne-Szakaly I, et al. BRAF duplications and MAPK pathway activation are frequent in gliomas of the optic nerve proper. J Neuropathol Exp Neurol. 2012;71:789–794. doi: 10.1097/NEN.0b013e3182656ef8. [DOI] [PMC free article] [PubMed] [Google Scholar]