

Abstract
Apolipoprotein A-I (ApoA-I) is the most abundant protein constituent of high-density lipoprotein (HDL). Reduced plasma HDL and ApoA-I levels have been found to be associated with obesity and metabolic syndrome in human beings. However, whether or not ApoA-I has a direct effect on obesity is largely unknown. Here we analysed the anti-obesity effect of ApoA-I using two mouse models, a transgenic mouse with overexpression of ApoA-I and the mice administered with an ApoA-I mimetic peptide D-4F. The mice were induced to develop obesity by feeding with high fat diet. Both ApoA-I overexpression and D-4F treatment could significantly reduce white fat mass and slightly improve insulin sensitivity in the mice. Metabolic analyses revealed that ApoA-I overexpression and D-4F treatment enhanced energy expenditure in the mice. The mRNA level of uncoupling protein (UCP)1 in brown fat tissue was elevated by ApoA-I transgenic mice. ApoA-I and D-4F treatment was able to increase UCP1 mRNA and protein levels as well as to stimulate AMP-activated protein kinase (AMPK) phosphorylation in brown adipocytes in culture. Taken together, our results reveal that ApoA-I has an anti-obesity effect in the mouse and such effect is associated with increases in energy expenditure and UCP1 expression in the brown fat tissue.
Keywords: obesity, apolipoprotein A-I (ApoA-I), brown fat, energy expenditure
Introduction
Obesity is a fast growing social problem that is reaching epidemic expansion worldwide [1]. Primary defects in energy balance that produce obesity and visceral adiposity in particular are sufficient to drive all aspects of metabolic syndrome including insulin resistance, glucose intolerance, hypertension and dyslipidaemia [2]. Obesity is associated with increased levels of circulating free fatty acid (FFA) and other secreted factors that can provoke peripheral insulin resistance in both animals and human beings [3]. Increased availability of FFA derived from adipose tissue also accelerate hepatic synthesis of very low density lipoproteins (VLDL), contributing to dyslipidaemia [4]. Importantly, a majority of studies have demonstrated that even modest weight gain can precipitate the onset of hypertension [5]. Individuals with a central deposition of adipose tissue can experience elevated cardiovascular morbidity and mortality, including stroke, congestive heart failure, myocardial infarction and cardiovascular death [6, 7]. Therefore, to elucidate the underlying mechanisms that link obesity to endothelial dysfunction and cardiovascular diseases has been a focus of studies for the past decades [8].
Apolipoprotein A-I (ApoA-I) is the most abundant protein constituent of high-density lipoprotein (HDL) [9, 10]. Previous studies have demonstrated that the blood level of ApoA-I is more closely correlated with the risk of atherosclerosis than other markers such as HDL-cholesterol [11]. In addition to their anti-atherogenic function, recent studies have indicated that HDL and ApoA-I are inversely correlated to the development of obesity [12]. Obesity has been shown to be associated with a reduced plasma ApoA-I level in the Framingham Offspring Study conducted in 4260 young adult men and women [13]. Another study indicates that the reduced HD level in intra-abdominal obese patients is partly caused by an increased clearance rate of ApoA-I [14]. It was recently reported that ApoA-I gene polymorphisms are risk factors for hypertension and obesity [15]. Study in animals also demonstrated that ApoA-I-deficient mice had an increased fat content [16]. However, it remains unclear at present whether ApoA-I has a direct anti-obesity effect in vivo.
Considering the beneficial effect of ApoA-I in atherosclerosis, recent research have focused on the studies of ApoA-I mimetic peptides. One of these peptides is an ApoA-I mimetic D-4F [17]. Multiple studies have indicated that D-4F have an anti-atherogenic function mimicking ApoA-I [18]. D-4F could increase the formation of pre-β HDL, elevate cholesterol efflux and reduce lipoprotein oxidation in vitro[19, 20]. D-4F also increases antioxidants and improves vascular repair in type 1 diabetic rats [21]. In addition, it has been shown that D-4F could improve vasodilation, oxidative stress, myocardial inflammation and angiogenic potential in a mouse model of scleroderma [22]. D-4F also reduces renal inflammation in low-density lipoprotein (LDL) receptor–null mice [23], ameliorates collagen-induced arthritis in rat [24] and improves the inflammatory properties of HDL in human beings with coronary heart disease [25].
We report here the studies with two mouse models, a transgenic mouse line with overexpression of ApoA-I and the mice administered with an ApoA-I mimetic peptide (D-4F), to investigate the anti-obesity effect of ApoA-I. We found that both ApoA-I overexpression and D-4F treatment could significantly reduce diet-induced obesity (DIO). Together with studies at the cellular and molecular levels, our results demonstrate that ApoA-I has a direct anti-obesity function in the mouse, associated with an elevated uncoupling protein (UCP)1 expression in brown fat and an increased metabolic rate.
Materials and methods
Materials
All tissue culture supplies were obtained from GIBCO BRL (Grand Island, NY, USA). The antibodies for AMP-activated protein kinase (AMPK) and phosphorylated AMPK were from Cell Signaling (Beverly, MA, USA). UCP1 antibody was from Abcam (Cambridge, UK). The antibody for tubulin was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The purified human ApoA-I was from Lee BioSolutions (St. Louis, MO, USA). D-4F was synthesized by GL Biochem (Shanghai, China) as previously described [26]. Norepinephrine was obtained from Sigma-Aldrich (St. Louis, MO, USA).
Animal studies
All animals were maintained and used in accordance with the guidelines of the Institutional Animal Care and Use Committee of the Institute for Nutritional Sciences, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. All mice were housed at a temperature of 22 ± 3°C under a 12-hr dark/light cycle (lights on at 6:30 a.m.) with ad libitum access to food and water. ApoA-I transgenic mice (ApoA-I-Tg) with C57BL/6J background were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) [27]. For DIO in ApoA-I-Tg mice, 6-week-old male ApoA-I-Tg and wild-type littermates were fed with high fat diet (HFD) (Research Diets, Inc., New Brunswick, NJ, USA) for 3 months. For D-4F injected DIO mice, 8-week-old C57BL/6J male mice (purchased from Slaccas, Shanghai, China) were fed with HFD for a total of 16 weeks. After HFD feeding for 12 weeks, the mice were injected with either D-4F (1 mg/kg body weight/day) or PBS (as control) continuously for 4 weeks. The body weight of animals was monitored weekly. The body fat content was determined by Nuclear Magnetic Resonance (NMR) 1 day before mice were killed using a Minispec mq10 NMR Analyzer (Bruker Optics, Billerica, MA, USA). An insulin tolerance test was performed in 3 hrs (10:00–13:00) fasted male mice. Glucose concentrations were measured in blood collected by venous bleeding from tail vein, immediately before and 30, 60 and 120 min. after i.p. injection with human insulin (Lilly France S.A.S, Fegersheim, France) at 0.5 units/kg using the FreeStyle blood glucose monitoring system (FreeStyle, TheraSense, Alameda, CA, USA). All mice were killed after overnight fasting for 16 hrs before measurement of the epididymal fat pad and retroperitoneal fat pad.
Analysis of serum parameters
The serum total cholesterol, triglyceride, HDL cholesterol and LDL cholesterol were determined by kits from Sysmex (Shanghai, China). The serum FFA was determined by a kit from Roche Diagnostics Corporation (Indianapolis, IN, USA). The serum insulin level was determined by a radioimmunoassay (BNIBT, Beijing, China).
Studies of metabolic profile of the mouse
After the mouse was acclaimed to a powdered high-fat diet, the metabolic profile of the animal was measured, including food intake, oxygen consumption, carbon dioxide production, respiratory exchange ratio (RER) and locomotive movement in metabolic cages (Columbus Instruments, Columbus, OH, USA). The calculated metabolic rate (Weir equation) is expressed per gram body weight [28].
Real-time quantitative RT-PCR analysis
The mice were fasted overnight before killing and tissue separation. The brown fat tissue was removed and snap-frozen immediately in liquid nitrogen for subsequent RNA extraction. Real-time quantitative PCR was performed with ABI Prism 7500 sequence detection system following the manufacturer’s recommendations. The gene encoding β-actin was used for internal normalization. The primers used for the genes were designed by GenScript Real-time PCR (TaqMan) Primer Design online (https://www.genscript.com/ssl-bin/app/primer).
Brown fat precursor cell isolation and culture
Brown fat precursor cells were isolated from 6–8-week-old male C57BL/6J mice as previously described [29]. The cell preparation was made from about 10 mice. The isolated precursor cells were pooled and planted into two 12-well plates with a density of ∼1.2 × 105 cells/cm2. The cells were cultivated in a culture medium consisting of Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% newborn calf serum (PAA Laboratories GmbH, Pasching, Austria), 4 nM insulin (Sigma-Aldrich), 10 mM Hepes, with 50 IU of penicillin, 50 pg of streptomycin and 25 pg of sodium ascorbate per millilitre. The cells were cultured at 37°C in water-saturated atmosphere with 8% CO2. The medium was completely changed with fresh pre-warmed medium on days 1, 3, 6 and 9.
Western blotting analysis
For Western blotting analysis, the cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris-HCl, pH 7.4) containing phosphatase inhibitors and a protease inhibitor cocktail (Sigma-Aldrich). The lysate was subjected to SDS-PAGE, transferred to poly(vinylidene fluoride) membranes and incubated with the primary antibodies, followed by horseradish peroxidase-conjugated secondary antibody (Amersham, Little Chalfont, Bucks, UK). The bound antibody was visualized using enhanced chemiluminescence reagents (Pierce, Rockford, IL, USA).
Results
ApoA-I transgenic mice are less obese than wild-type animals after HFD feeding
Our previous studies have demonstrated that the body fat content was significantly increased in ApoA-I null mice [16]. To further examine whether ApoA-I plays a role in the development of obesity, we generated DIO in the mice. ApoA-I-Tg and wild-type mice were fed with HFD for 3 months. Both groups of mice had the same body weight gain (Fig. 1A) and food intake (Fig. 1B). However, the body fat content was significantly lower in ApoA-I-Tg mice (18.10 ± 0.04% of fat/body) than the wild-type mice (24.63 ± 0.04%) (Fig. 1C). The weight of epididymal fat pad and retroperitoneal fat pad was also significantly lower in ApoA-I-Tg mice than in the wild-type mice (Fig. 1D). Consistent with a previous report [27], ApoA-I-Tg mice had increased levels of circulating cholesterol and HDL-cholesterol (Table 1). However, the ApoA-I-Tg mice had significantly lower FFA level than the wild-type animals (Table 1). Collectively, these data indicate that overexpression of ApoA-I in the ApoA-I-Tg mice had reduced obesity upon HFD feeding in comparison with the wild-type mice, providing evidence that ApoA-I had an anti-obesity effect in DIO mice.
Fig 1.
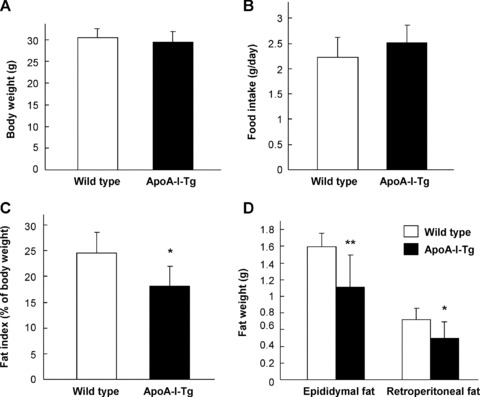
Reduction of fat mass in ApoA-I-Tg mice fed with HFD. Six-week-old male ApoA-I-Tg (n= 6) and wild-type mice (n= 7) were fed with HFD for 3 months. The body weight (A), food intake (B), body fat content (C) and the weight of epididymal fat pad and retroperitoneal fat pad (D) were measured. The data are shown in mean ± S.D. * indicates P < 0.05 and ** for P < 0.01 by Student’s t-test between the two groups.
Table 1.
Serum parameters in ApoA-I-Tg and D-4F-treated mice after HFD feeding
| TCHO (mM) | TG (mM) | HDL-C (mM) | LDL-C (mM) | FFA (mM) | Glucose (mM) | Insulin (μIU/ml) | |
|---|---|---|---|---|---|---|---|
| Wild-type (n= 6) | 4.46(0.50) | 0.76(0.20) | 2.85(0.32) | 0.96(0.25) | 1.15(0.15) | 10.25(1.30) | 10.36(1.56) |
| ApoA-I Tg (n= 6) | 8.36(2.31)** | 0.75(0.26) | 5.31(1.04)** | 2.04(1.39) | 0.79(0.13)** | 9.20(1.62) | 10.23(2.49) |
| Control (n= 5) | 5.73(0.38) | 0.54(0.18) | 3.27(0.18) | 1.66(0.25) | 0.63(0.15) | 9.54(0.97) | 8.74(1.22) |
| D-4F (n= 6) | 5.62(0.94) | 0.59(0.20) | 3.27(0.43) | 1.64(0.49) | 0.61(0.07) | 10.08(1.64) | 11.22(3.37) |
All parameters were measured in mice fasted overnight except for blood glucose that was examined after fasting for 3 hrs. The data are shown as mean (S.D.). ‘n’ indicates the number of mouse used in each group. ** indicates P < 0.01 as a comparison between the ApoA-I-Tg mice and the wild-type mice.
ApoA-I mimetic peptide D-4F reduces HFD-induced obesity
To further investigate the anti-obesity effect of ApoA-I, we treated DIO mice with an ApoA-I mimetic peptide D-4F [17]. D-4F is an 18-amino acid peptide with a sequence (DWFKAFYDKVAEKFKEAF) with limited homology to ApoA-I protein. D-4F has the capacity to form a class A amphipathic helix similar to that found in ApoA-I to mimic many of the lipid binding properties of ApoA-I. Injection of D-4F (1 mg/kg/day) for 1 month had no effect on body weight (Fig. 2A) and food intake (Fig. 2B) as compared with PBS-injected control group. Similar to the ApoA-I-Tg mice, D-4F-injected mice showed a slight decrease in body fat content in comparison with the control animals (Fig. 2C). In addition, the weight of epididymal fat pad was significantly lower in D-4F-treated mice than the control animals (Fig. 2D). As expected, injection of D-4F had no effect on lipid profile as compared with the control group (Table 1). Taken together, these observations indicate that D-4F could protect mice from DIO, similar to the findings in the ApoA-I-Tg mice.
Fig 2.
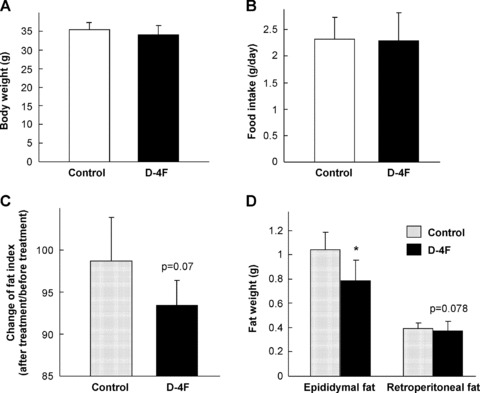
Decreased fat mass in HFD-fed mice after treatment with D-4F. Eight-week-old C57BL/6J male mice were fed with HFD for 12 weeks, and then injected with either D-4F (n= 6) or PBS (n= 5) continuously for 4 weeks. The body weight (A), food intake (B), percentage of body fat content (C) and the weight of epididymal and retroperitoneal fat pads (D) were measured. The data are shown in mean ± S.D. * indicates P < 0.05 by Student’s t-test between the two groups.
Effects of ApoA-I overexpression and D-4F on insulin sensitivity
As central deposition of adipose tissue often leads to insulin resistance [30], we suggested that the reduced fat mass in ApoA-I-Tg and D-4F-treated DIO mice might be associated with increased insulin sensitivity. Insulin tolerance test was performed with these mice (Fig. 3). It appeared that both ApoA-I-Tg mice and D-4F-treated mice, in comparison with the control animals, had a slight but significant improvement of insulin resistance. However, the fasting blood glucose and insulin levels were not altered in these animals (Table 1). These data, therefore, indicate that ApoA-I overexpression or D-4F administration could slightly improve insulin sensitivity, consistent with their anti-obesity effect as observed (Figs 1 and 2).
Fig 3.
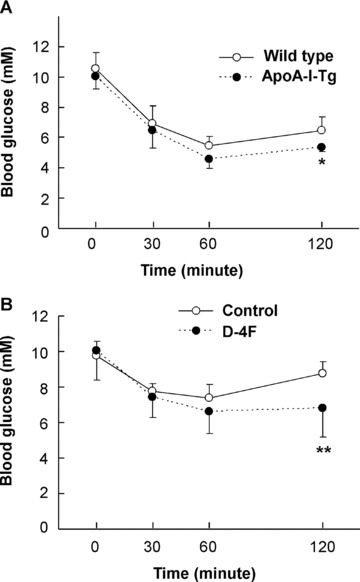
Improved insulin sensitivity in ApoA-I-Tg mice and D-4F-treated DIO mice. Insulin tolerance test was performed with the ApoA-I-Tg and wild-type mice (A), and D-4F-treated and control animals (B). The blood glucose levels were determined at the indicated time-points following the intraperitoneal administration of insulin (n= 5 for ApoA-I-Tg mice, n= 6 for wild-type mice; n= 6 for D-4F-injected group, n= 5 for PBS-injected group). * indicates P < 0.05 and ** for P < 0.01 as comparison between the two experimental groups by Student’s t-test.
ApoA-I and D-4F enhance energy expenditure in the mouse
The reduction of fat accumulation in ApoA-I-Tg and D-4F-treated mice versus the control animals upon HFD feeding was not associated with a reduction of food intake (Figs 1 and 2), indicating a possible shift in total energy balance in these mice. To address this issue, we investigated the metabolic profile of the animals using metabolic cages. The ApoA-I-Tg mice had a significantly increased RER (VCO2/VO2) and metabolic rate in general in both the light and dark cycles as compared with the wild-type controls (Fig. 4A and B), with no difference in locomotive movement (data not shown). Furthermore, D-4F treatment in DIO mice also had a trend of increase in RER and metabolic rate especially in the dark cycle (Fig. 5A and B), without changes in locomotive movement as compared with the control group (data not shown). Collectively, these results suggest that both ApoA-I overexpression and D-4F treatment can enhance energy expenditure in the mouse.
Fig 4.
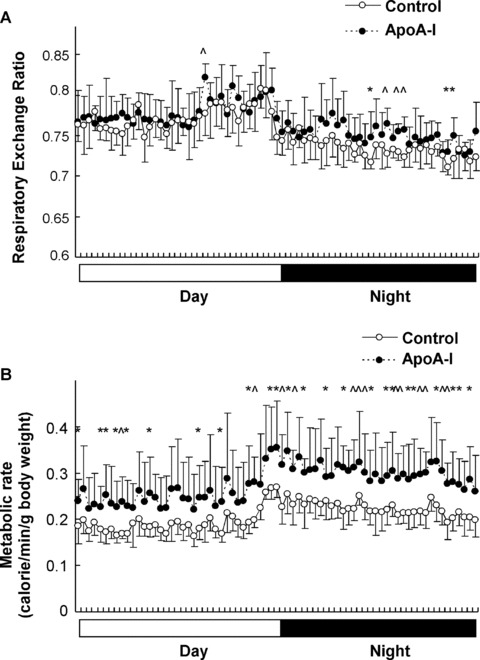
Increased energy expenditure in ApoA-I-Tg mice. Respiratory exchange ratio (A) and metabolic rates (B) were measured in ApoA-I-Tg and wild-type mice fed with HFD for 3 months (n= 7 for ApoA-I-Tg, n= 7 for wild-type mice). The data are shown in mean ± S.D. * indicates P < 0.05 and ∧ indicates P < 0.01 by Student’s t-test between the two groups.
Fig 5.
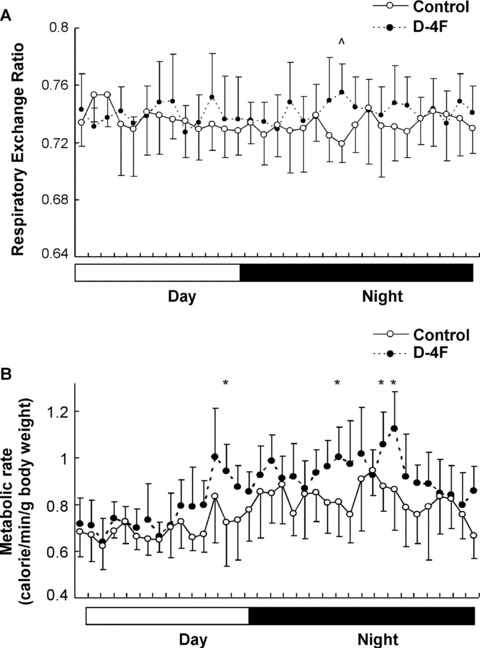
Elevated energy expenditure in D-4F-treated DIO mice. Respiratory exchange ratio (A) and metabolic rates (B) were measured in D-4F- or PBS-injected mice (n= 6 for D-4F group mice and n= 5 for the control group). The data are shown in mean ± S.D. * indicates P < 0.05 and ∧ indicates P < 0.01 by Student’s t-test between the two groups.
ApoA-I and D-4F up-regulate UCP-1 expression in brown fat tissue
Uncoupling respiration that occurs in brown adipocytes can promote energy expenditure and decrease the fat content of the body [31]. We next assessed the mRNA levels of UCPs, PRD1-BF1-RIZ1 homologous domain containing 16 (PRDM16) that controls the determination of brown fat fate [32] and peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) that is a critical transcriptional regulator that controls expression of mitochondria proteins [33], in the brown adipose tissue. In comparison with the wild-type mice, ApoA-I-Tg mice had a significant increase in UCP1 mRNA level in the brown adipose tissue (Fig. 6A). On the other hand, D-4F treatment could also elevate UCP1 expression in the brown fat pad in comparison with the control animals (Fig. 6B). However, UCP2, UCP3, PRDM16 and PGC-1α were not significantly altered in ApoA-I-Tg and D-4F-treated mice. We also analysed the protein level of UCP1 in the brown fat tissue. As shown in Fig. 6C, UCP1 protein level was significantly higher in ApoA-I-Tg mice than in the wild-type mice, although D-4F had no obvious effect on UCP1 protein level (Fig. 6D). These observations, collectively, indicate that ApoA-I may enhance energy expenditure by up-regulation of UCP1 expression in brown fat tissue.
Fig 6.
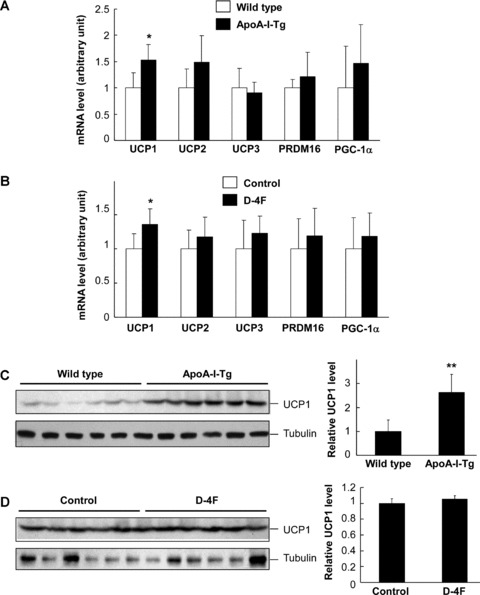
Analysis of gene expression in brown fat tissue of ApoA-I-Tg and D-4F treated DIO mice. The mRNA levels of UCP1, UCP2, UCP3, PRDM16 and PGC-1α were determined by real-time RT-PCR in brown fat tissue of ApoA-I-Tg and wild-type mice (A) and D-4F-treated and control mice (B). The UCP1 protein level was determined by Western blotting using brown fat tissues of ApoA-I-Tg and wild-type mice (C) and D-4F-treated mice and the control mice (D). The brown fat tissue was isolated from overnight-fasted mice. The data were normalized with the level of β-actin for mRNA and tubulin for protein. The right panels for (C) and (D) are semi-quantitative data based on densitometry analysis. The data are shown in mean ± S.D. * indicates P < 0.05 and ** for P < 0.01 as comparison between the two experimental groups in each experiment by Student’s t-test.
ApoA-I and D-4F up-regulate UCP1 expression and stimulate AMPK phosphorylation in brown adipocytes in culture
To further examine whether ApoA-I directly regulates UCP1 expression in brown adipocytes, we isolated brown fat precursor cells that were differentiated into brown adipocytes in vitro. As a positive control, treatment of the brown adipocytes with norepinephrine could markedly increase the expression of UCP1 in these cells at the both the mRNA and protein levels (Fig. 7A and B). Importantly, treatment of the cells with ApoA-I or D-4F also led to significant increase of both UCP1 mRNA and protein in these cells (Fig. 7A and B). These data, therefore, suggest that UCP1 is one of the target genes regulated by ApoA-I and D-4F in brown fat, and such regulation may contribute to the increase of energy expenditure observed in the ApoA-I-Tg and D-4F-treated mice. In addition, we analysed whether ApoA-I and D-4F could stimulate AMPK phosphorylation in brown fat cells. Treatment of brown adipocytes with either ApoA-I or D4F could indeed activate AMPK to certain degrees (Fig. 7C), similar to the findings in endothelial cells and myocytes [16, 34].
Fig 7.
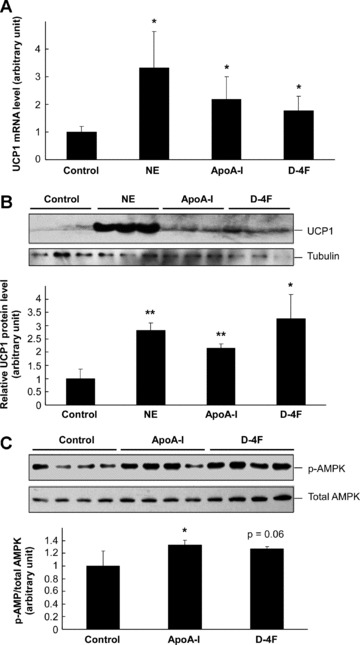
Up-regulation of UCP1 in primary brown adipocytes by ApoA-I and D-4F. Brown fat precursor cells were isolated from 6–8-week-old male C57BL/6J mice and cultured for 9 days. The differentiated brown adipocytes were treated with 0.1 μM norepinephrine, 20 μg/ml ApoA-I, 20 μg/ml D-4F for 24 hrs as indicated. The mRNA level of UCP1 was determined by real-time RT-PCR and normalized with the level of β-actin mRNA (A). The protein level of UCP1 was determined by Western blotting and normalized with the protein level of tubulin (B). The differentiated brown adipocytes were serum-starved overnight and then treated with 20 μg/ml ApoA-I, 20 μg/ml D-4F for 10 min. before Western blotting to detect phosphorylated and total AMPK (C). The lower panels for (B) and (C) are semi-quantitative data based on densitometry analysis. The data are shown in mean ± S.D. * indicates P < 0.05 and ** for P < 0.01 as comparison with the control group by Student’s t-test.
Discussion
As the most abundant protein constituent of HDL, ApoA-I has been a focus of studies in the past to elucidate the anti-atherogenic effects of HDL [9]. However, a few recent studies have linked ApoA-I to glucose and energy metabolism, revealing that ApoA-I can stimulate AMPK in different cells [16, 34].These reports indicate that in addition to its atheroprotective effect, ApoA-I may play a role in other metabolic processes. Here we report the anti-obesity effect of ApoA-I by using two mice models. Both the ApoA-I-Tg mice and administration of ApoA-I mimetic peptide D-4F could ameliorate fat mass accumulation upon HFD feeding. The decrease in obesity is associated with improved insulin sensitivity, enhanced energy expenditure and increased UCP1 expression in brown fat tissue. Our in vitro study revealed that ApoA-I and D-4F directly regulate UCP1 expression in primary brown adipocytes. These results, collectively, reveal for the first time that ApoA-I has an anti-obesity effect through enhancement of energy expenditure likely mediated by an up-regulation of UCP1 expression in the brown fat tissue.
The brown adipose tissue is a special form of adipose tissue found in human infants as well as in adult rodents, small mammals and hibernators [35]. The importance of brown fat tissues in human beings are highlighted by recent discoveries that brown fat can be detected in adult human beings [36, 37]. Most importantly, the amount of brown adipose tissue is inversely correlated with body-mass index [38]. Brown adipocytes differ from white adipocytes by the presence of numerous mitochondria which exhibit a spontaneous uncoupling of respiration. The unique function of brown adipocytes in energy expenditure is mediated by specific membrane transporter termed UCPs including UCP1, UCP2 and UCP3. UCP1 is unique to brown adipocytes and it is the only UCP that possesses the respiration-uncoupling activity and the thermogenic activity in the brown fat tissue. UCP1-associated uncoupling respiration that occurs in brown adipocytes can promote energy expenditure, providing a means to reduce obesity [31]. In our study, both ApoA-I overexpression in ApoA-I-Tg mice and D-4F treatment lead to reduced adiposity and increased metabolic rate. Furthermore, our results suggest that ApoA-I and D-4F can up-regulate UCP1 expression both in vivo and in vitro. Therefore, we postulate that the anti-obesity effect of ApoA-I is, at least partially, mediated by a direct function of ApoA-I on UCP1 expression and energy expenditure in brown fat tissue.
D-4F is an 18-amino acid peptide that has the capacity to form a class A amphipathic helix similar to those found in ApoA-I and mimics many of the lipid binding properties of ApoA-I [39]. A majority of studies have revealed the atheroprotective effects of D-4F both in vivo and in vitro[17]. Consistent with our result, injection of L-4F, a peptide with the same amino acid sequence as D-4F but synthesized from L-amino acids, with a dose of 2 mg/kg/day in ob/ob mice for 6 weeks showed reduced adiposity and improved insulin sensitivity [40]. In our study, D-4F acts differently from ApoA-I in terms of the lipid profile in serum. ApoA-I-Tg mice have increased HDL cholesterol and decreased FFA level. However, injection of D-4F has no effect on the serum lipid profile as compared with the control animals. Nevertheless, D-4F has a similar effect as ApoA-I in the reduction of adiposity, increase of energy expenditure and up-regulation of UCP1 expression in the brown fat. Therefore, we postulate that in addition to its anti-atherogenic effect, D-4F has a therapeutical potential to treat obesity. Although this idea still awaits vigorous testing in human beings in the future.
AMPK is considered to be a cellular energy sensor that regulates glucose and lipid metabolism by phosphorylating and regulating key proteins that control cellular metabolism [41]. ApoA-I has been found to stimulate the phosphorylation of AMPK in endothelial cells and C2C12 myocytes [16, 34]. In our study, ApoA-I also activates AMPK in brown fat cells. As injection of AMPK agonist AICAR could enhance energy expenditure with up-regulation of UCP1 in mice [42], activation of AMPK is possibly one of the underlying molecular mechanisms accounting for ApoA-I- and D-4F-induced UCP1 up-regulation in brown fat.
Taken together, our study reveals a new paradigm to demonstrate the protective effect of ApoA-I in obesity. ApoA-I could ameliorate high fat DIO by enhancing energy expenditure associated with up-regulation of UCP1 in brown fat. Considering its dual effects of anti-atherosclerosis and anti-obesity, ApoA-I appears to be an ideal choice for the therapy of obesity and obesity-associated metabolic syndrome in human beings.
Acknowledgments
This work was supported by research grants from Chinese Academy of Sciences (Knowledge Innovation Program KSCX1-YW-02), National Natural Science Foundation of China (30830037) and the Ministry of Science and Technology of China (2007CB947100 and 2006CB503900) to Y.C.
References
- 1.James PT, Rigby N, Leach R. The obesity epidemic, metabolic syndrome and future prevention strategies. Eur J Cardiovasc Prev Rehabil. 2004;11:3–8. doi: 10.1097/01.hjr.0000114707.27531.48. [DOI] [PubMed] [Google Scholar]
- 2.Moller DE, Kaufman KD. Metabolic syndrome: a clinical and molecular perspective. Annu Rev Med. 2005;56:45–62. doi: 10.1146/annurev.med.56.082103.104751. [DOI] [PubMed] [Google Scholar]
- 3.Boden G. Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes. 1997;46:3–10. [PubMed] [Google Scholar]
- 4.Ginsberg HN. Insulin resistance and cardiovascular disease. J Clin Invest. 2000;106:453–8. doi: 10.1172/JCI10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Must A, Spadano J, Coakley EH, et al. The disease burden associated with overweight and obesity. Jama. 1999;282:1523–9. doi: 10.1001/jama.282.16.1523. [DOI] [PubMed] [Google Scholar]
- 6.Kenchaiah S, Evans JC, Levy D, et al. Obesity and the risk of heart failure. N Engl J Med. 2002;347:305–13. doi: 10.1056/NEJMoa020245. [DOI] [PubMed] [Google Scholar]
- 7.Lakka TA, Lakka HM, Salonen R, et al. Abdominal obesity is associated with accelerated progression of carotid atherosclerosis in men. Atherosclerosis. 2001;154:497–504. doi: 10.1016/s0021-9150(00)00514-1. [DOI] [PubMed] [Google Scholar]
- 8.Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–80. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- 9.Assmann G, Nofer JR. Atheroprotective effects of high-density lipoproteins. Annu Rev Med. 2003;54:321–41. doi: 10.1146/annurev.med.54.101601.152409. [DOI] [PubMed] [Google Scholar]
- 10.Linsel-Nitschke P, Tall AR. HDL as a target in the treatment of atherosclerotic cardiovascular disease. Nat Rev Drug Discov. 2005;4:193–205. doi: 10.1038/nrd1658. [DOI] [PubMed] [Google Scholar]
- 11.Maciejko JJ, Holmes DR, Kottke BA, et al. Apolipoprotein A-I as a marker of angiographically assessed coronary-artery disease. N Engl J Med. 1983;309:385–9. doi: 10.1056/NEJM198308183090701. [DOI] [PubMed] [Google Scholar]
- 12.Rashid S, Genest J. Effect of obesity on high-density lipoprotein metabolism. Obesity. 2007;15:2875–88. doi: 10.1038/oby.2007.342. [DOI] [PubMed] [Google Scholar]
- 13.Garrison RJ, Wilson PW, Castelli WP, et al. Obesity and lipoprotein cholesterol in the Framingham offspring study. Metabolism. 1980;29:1053–60. doi: 10.1016/0026-0495(80)90216-4. [DOI] [PubMed] [Google Scholar]
- 14.Vajo Z, Terry JG, Brinton EA. Increased intra-abdominal fat may lower HDL levels by increasing the fractional catabolic rate of Lp A-I in postmenopausal women. Atherosclerosis. 2002;160:495–501. doi: 10.1016/s0021-9150(01)00610-4. [DOI] [PubMed] [Google Scholar]
- 15.Chen ES, Mazzotti DR, Furuya TK, et al. Apolipoprotein A1 gene polymorphisms as risk factors for hypertension and obesity. Clin Exp Med. 2009;9:319–25. doi: 10.1007/s10238-009-0051-3. [DOI] [PubMed] [Google Scholar]
- 16.Han R, Lai R, Ding Q, et al. Apolipoprotein A-I stimulates AMP-activated protein kinase and improves glucose metabolism. Diabetologia. 2007;50:1960–8. doi: 10.1007/s00125-007-0752-7. [DOI] [PubMed] [Google Scholar]
- 17.Navab M, Anantharamaiah GM, Reddy ST, et al. Apolipoprotein A-I mimetic peptides. Arterioscler Thromb Vasc Biol. 2005;25:1325–31. doi: 10.1161/01.ATV.0000165694.39518.95. [DOI] [PubMed] [Google Scholar]
- 18.Van Lenten BJ, Wagner AC, Anantharamaiah GM, et al. Apolipoprotein A-I mimetic peptides. Curr Atheroscler Rep. 2009;11:52–7. doi: 10.1007/s11883-009-0008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Navab M, Hama SY, Anantharamaiah GM, et al. Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: steps 2 and 3. J Lipid Res. 2000;41:1495–508. [PubMed] [Google Scholar]
- 20.Troutt JS, Alborn WE, Mosior MK, et al. An apolipoprotein A-I mimetic dose-dependently increases the formation of prebeta1 HDL in human plasma. J Lipid Res. 2008;49:581–7. doi: 10.1194/jlr.M700385-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Peterson SJ, Husney D, Kruger AL, et al. Long-term treatment with the apolipoprotein A1 mimetic peptide increases antioxidants and vascular repair in type I diabetic rats. J Pharmacol Exp Ther. 2007;322:514–20. doi: 10.1124/jpet.107.119479. [DOI] [PubMed] [Google Scholar]
- 22.Weihrauch D, Xu H, Shi Y, et al. Effects of D-4F on vasodilation, oxidative stress, angiostatin, myocardial inflammation, and angiogenic potential in tight-skin mice. Am J Physiol Heart Circ Physiol. 2007;293:H1432–41. doi: 10.1152/ajpheart.00038.2007. [DOI] [PubMed] [Google Scholar]
- 23.Buga GM, Frank JS, Mottino GA, et al. D-4F reduces EO6 immunoreactivity, SREBP-1c mRNA levels, and renal inflammation in LDL receptor-null mice fed a Western diet. J Lipid Res. 2008;49:192–205. doi: 10.1194/jlr.M700433-JLR200. [DOI] [PubMed] [Google Scholar]
- 24.Charles-Schoeman C, Banquerigo ML, Hama S, et al. Treatment with an apolipoprotein A-1 mimetic peptide in combination with pravastatin inhibits collagen-induced arthritis. Clin Immunol. 2008;127:234–44. doi: 10.1016/j.clim.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 25.Bloedon LT, Dunbar R, Duffy D, et al. Safety, pharmacokinetics, and pharmacodynamics of oral apoA-I mimetic peptide D-4F in high-risk cardiovascular patients. J Lipid Res. 2008;49:1344–52. doi: 10.1194/jlr.P800003-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Navab M, Anantharamaiah GM, Hama S, et al. Oral administration of an Apo A-I mimetic Peptide synthesized from D-amino acids dramatically reduces atherosclerosis in mice independent of plasma cholesterol. Circulation. 2002;105:290–2. doi: 10.1161/hc0302.103711. [DOI] [PubMed] [Google Scholar]
- 27.Rubin EM, Ishida BY, Clift SM, et al. Expression of human apolipoprotein A-I in transgenic mice results in reduced plasma levels of murine apolipoprotein A-I and the appearance of two new high density lipoprotein size subclasses. Proc Natl Acad Sci USA. 1991;88:434–8. doi: 10.1073/pnas.88.2.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weir JB. New methods for calculating metabolic rate with special reference to protein metabolism. J Physiol. 1949;109:1–9. doi: 10.1113/jphysiol.1949.sp004363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bronnikov G, Houstek J, Nedergaard J. Beta-adrenergic, cAMP-mediated stimulation of proliferation of brown fat cells in primary culture. Mediation via beta 1 but not via beta 3 adrenoceptors. J Biol Chem. 1992;267:2006–13. [PubMed] [Google Scholar]
- 30.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–6. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 31.Ricquier D. Respiration uncoupling and metabolism in the control of energy expenditure. Proc Nutr Soc. 2005;64:47–52. doi: 10.1079/pns2004408. [DOI] [PubMed] [Google Scholar]
- 32.Seale P, Kajimura S, Yang W, et al. Transcriptional control of brown fat determination by PRDM16. Cell Metab. 2007;6:38–54. doi: 10.1016/j.cmet.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu Z, Puigserver P, Andersson U, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–24. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 34.Drew BG, Fidge NH, Gallon-Beaumier G, et al. High-density lipoprotein and apolipoprotein AI increase endothelial NO synthase activity by protein association and multisite phosphorylation. Proc Natl Acad Sci USA. 2004;101:6999–7004. doi: 10.1073/pnas.0306266101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 36.van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, et al. Cold-activated brown adipose tissue in healthy men. N Engl J Med. 2009;360:1500–8. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 37.Virtanen KA, Lidell ME, Orava J, et al. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360:1518–25. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- 38.Cypess AM, Lehman S, Williams G, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–17. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anantharamaiah GM. Synthetic peptide analogs of apolipoproteins. Methods Enzymol. 1986;128:627–47. doi: 10.1016/0076-6879(86)28096-9. [DOI] [PubMed] [Google Scholar]
- 40.Peterson SJ, Drummond G, Kim DH, et al. L-4F treatment reduces adiposity, increases adiponectin levels, and improves insulin sensitivity in obese mice. J Lipid Res. 2008;49:1658–69. doi: 10.1194/jlr.M800046-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Villena JA, Viollet B, Andreelli F, et al. Induced adiposity and adipocyte hypertrophy in mice lacking the AMP-activated protein kinase-alpha2 subunit. Diabetes. 2004;53:2242–9. doi: 10.2337/diabetes.53.9.2242. [DOI] [PubMed] [Google Scholar]
- 42.Narkar VA, Downes M, Yu RT, et al. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134:405–15. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]