

Abstract
Herein, we report that insulin-activated extracellular signal-regulated kinase (ERK) is translocated to the nuclear envelope by caveolin-2 (cav-2) and associates with lamin A/C in the inner nuclear membrane in response to insulin. We identified that the Ser154–Val155–Ser156 domain on the C-terminal of cav-2 is essential for insulin-induced phosphorylation and nuclear targeting of ERK and cav-2. In human embryonic kidney 293T cells, ERK was not activated and translocated to the nucleus by insulin in comparison to insulin-like growth factor-1 (IGF-1). However, insulin-stimulated activation of ERK was induced by exogenous addition of cav-2. The activated ERK associated and translocated with the cav-2 to the nucleus. In turn, cav-2 promoted phospho-ERK interaction with lamin A/C in the inner nuclear membrane. In contrast, ERK, but not cav-2, was phosphorylated and translocated to the nucleus by IGF-1. The nuclear targeted phospho-ERK failed to localize in the nuclear envelope in response to IGF-1. Together, our data demonstrate that translocation of phospho-ERK to the nuclear envelope is mediated by Ser154–Val155–Ser156 domain of cav-2 and this event is an insulin-specific action.
Keywords: caveolin-2, phospho-ERK, lamin A/C, nuclear translocation, insulin, IGF-1
Introduction
Caveolin-1 (cav-1) and cav-2 have been studied exclusively as major structural components of plasma membrane caveolae [1–4]. Recently, functions of cav-1 outside of caveolae in the plasma membrane have been reported [4–6]. Distinctively from the caveolae, cav-1 oligomers can form functional microdomains and interact with proteins and lipids to regulate raft dynamics and receptor signalling [5–7]. Cav-1 has also been shown to be present in the cytoplasm, focal adhesions, mitochondria, lipid droplets and nucleus, suggesting its function in a variety of cell signalling and mechanotransduction processes to regulate tumour cell migration and metastasis [5–10]. Relative to cav-1, it is reported that cav-2 is not essential for caveolae biogenesis [11, 12]. The functional role of cav-2 in lipid microdomains distinct from caveolae has not been explored.
Recent works in our laboratory show that cav-2 is translocated to the nucleus in response to insulin. The nuclear localized cav-2 regulates transcriptional factors [c-Jun and signal transducers and activators of transcription 3 (STAT3)], cell cycle regulator (cyclinD1) and DNA synthesis in extracellular signal-regulated kinase (ERK)-mediated mitogenesis of insulin signalling [13–15]. Phospho-ERK has been shown to re-localize into the nucleus in a nuclear localization sequence (NLS)-independent manner [16] and interact with lamin A/C at the nuclear envelope [17]. Lamin A/C, intermediate filament proteins at the inner nuclear membrane play important roles in the control of gene expression and DNA replication via their interaction with transcriptional factors, cell cycle regulators and histones [17–19]. We demonstrated that tyrosines 19 and 27 in the N-terminal of cav-2 are necessary for insulin-induced nuclear targeting of phospho-ERK [14, 15]. Cav-2 does not contain a classical NLS, and no specific nuclear import receptor for cav-2 has been reported. Thus, little is known of cav-2 intracellular trafficking and the molecular machinery that regulates the nuclear translocation of cav-2. Further, insulin-induced cellular mitogenesis regulated by cross-talk among cav-2, ERK and lamin A/C in the nucleus has not been investigated.
Insulin and insulin-like growth factor-1 (IGF-1) are closely related peptide hormones that are essential for metabolic homeostasis and regulate cell growth and differentiation [20–24]. Insulin and IGF-1 exert their biological effects by binding to their respective transmembrane receptors. Insulin and IGF-1 receptors (IR and IGF-1R, respectively) sharing the structural homology are composed of two extracellular α subunits containing the ligand-binding domain and two transmembrane β-subunits possessing the ligand-stimulated intrinsic tyrosine kinase activity [22, 25, 26]. Each receptor is capable of cross-binding insulin and IGF-1 but binds its own ligand with a 100–1000-fold higher affinity than that of the heterologous peptide [27, 28]. Both IR and IGF-1R phosphorylate on tyrosine residues of the same substrates such as insulin receptor substrate (IRS) [29, 30], Gab-1 [31] and Shc [32], and activate the Ras-Raf-ERK [33, 34] and phosphoinositide 3-kinase / Akt [35] pathways. Despite close similarities in their receptor structure and signal pathways, many studies have revealed that insulin and IGF-1 have distinct biological functions. Insulin functions predominantly as a regulator of metabolic signalling, whereas IGF-1 controls primarily cell growth, survival and differentiation [20, 22, 23]. We have been investigating insulin action in cellular mitogenesis and have identified a positive regulator, cav-2, of ERK-mediated mitogenic signalling in response to insulin [13–15]. However, how differently cav-2-dependent cellular mitogenesis is regulated in response to insulin versus IGF-1 has not been investigated.
In this study, we identify a novel domain, Ser154–Val155–Ser156 in the C-terminal of cav-2 that is required for the nuclear targeting of phospho-ERK in insulin signalling. We demonstrate insulin-specific cav-2-depedent activation of ERK, its interaction with cav-2 and their co-localization in the nuclear envelop distinctive from IGF-1 action.
Materials and methods
Cell culture and treatment
Human embryonic kidney (HEK) 293T cells having no detectable endogenous cav-1 and cav-2 [36, 37] and rat 1 fibroblast and COS-7 (African green monkey kidney) cells endogenously expressing cav-1 and cav-2 [13, 38] were grown in DMEM (Gibco/BRL, Grand Island, NY, USA) containing 5 mM D-glucose supplemented with 10% (v/v) foetal bovine serum (FBS) (Cambrex Bio Science, Verviers, Belgium) and 0.5% penicillin/streptomycin (Sigma-Aldrich, St Louis, MO, USA) in a 5% CO2 incubator at 37°C. Human insulin receptor-expressed rat 1 fibroblast (Hirc-B) cells having cav-2 alone [13–15] were grown in DMEM containing 5 mM D-glucose supplemented with 10% (v/v) FBS, 100 nM methotrexate (Sigma-Aldrich) and 0.5% gentamycin (Gibco/BRL) in a 5% CO2 incubator at 37°C as previously described [13–15, 39]. The HEK293T, rat-1, COS-7 and Hirc-B cells express both IR and IGF-1R and are shown to be sensitive to both insulin and IGF-1 [13, 24, 40–44]. The cells were grown to 80% confluence before serum starvation for 18 hrs and then stimulated with various concentrations of insulin (human insulin; Novo Nordisk A/S, Bagsvaerd, Denmark) and IGF-1 (Sigma-Aldrich) for 10 min. as described in figure legends.
Plasmid constructs, mutagenesis and transfection
A full-length Rattus norvegicus cav-2 cDNA (GenBank Accession No. NM_131914) was subcloned into pDonor207 entry vector (Invitrogen, Carlsbad, CA, USA) by a PCR-based gateway cloning method as described previously [15, 45]. Additional entry vectors cav-2-related encoding deletion mutants (47–162, 71–162 and 1–119 a. as) were generated by using the entry vector full-length cDNAs (1–162 a. as) as template via EZchange site-directed mutagenesis kit (Enzynomics, Daejeon, Korea). The resulting entry vectors of wild-type (WT) and mutants were converted into self-constructed FLAG tagging destination expression vector (pDS_FLAG-XB vector). All expression vectors were verified by sequencing.
Point mutations were performed by the methods described previously [14, 15] and confirmed by sequencing. The various cav-2 combination mutants including Y19A, AAA, AVA, AVS, SVA, SAS and ASVS were constructed by inserting the appropriate residues (Y19A, S154A, V155A, S156A and S153A) in the pcDNA3-cav-2 (Rattus norvegicus). A full-length Homo sapiens cav-2 cDNA (GenBank accession no. AAB88492.1) was subcloned into pcDNA3 vector using XbaI-HindIII (NEW ENGLAND BioLabs, Beverly, MA, USA).
For transient expression of the cDNA constructs and mutants, HEK293T, Hirc-B and COS-7 cells were transfected using a modified calcium phosphate precipitation protocol. In brief, the cDNA constructs were introduced into the cells in the DMEM, which was replaced with 30 μl of 2.5 M CaCl2 and 0.5 ml of 1× Hepes-buffered saline and incubated for 24 hrs at 37°C. After washing with phosphate-buffered saline (PBS), fresh medium was added to the cells and incubated for another 24 hrs. Recombinant expression was confirmed by SDS-PAGE / immunoblot analysis.
Immunostaining and image acquisition
Cells were seeded into individual wells of a 24-well plate containing 12-mm glass cover slips and grown until 50% confluent before the transfection of cav-2 variant mutants. Transfected cells were serum starved for 18 hrs. Cells were then incubated with or without insulin (100 nM) or IGF-1 (10 nM) for 10 min., washed with PBS, fixed with 3.7% paraformaldehyde in PBS for 20 min. at room temperature (RT) and permeabilized with 0.1 or 0.5% Triton X-100 in PBS for 30 or 5 min., respectively, at RT. Non-specific protein binding sites were saturated with 1% bovine serum albumin (BSA) in PBS for 30 min. at RT. Cells were washed with PBS and incubated with anti-cav-2, anti-lamin A/C, anti-FLAG and anti-phospho-ERK antibodies in 1% BSA in PBS for 24 hrs at 4°C. After washing three times with PBS, the primary antibodies were detected with either tetramethyl- rhodamine isothiocyanate (TRITC)- or fluorescein isothiocyanate (FITC)-conjugated antimouse or anti-rabbit secondary antibodies (Calbiochem, San Diego, CA, USA) in 1% BSA in PBS for 2 hrs at RT. Nuclei were fluorescently labelled with 4′, 6-diamidino-2-phenylindole (DAPI, D8417; Sigma-Aldrich). The cover slips were washed and mounted on glass slides. Fluorescent images were obtained using an Olympus Fluoview 1000 confocal microscope attached to an Olympus BX61 vertical microscope equipped with a fluorescein. FV10-ASW software (Olympus, Tokyo, Japan) was used to merge the images from differential interference contrast (DIC), FITC and TRITC.
FLAG and DAPI were visualized by an Olympus BX51 fluorescence microscope and imaged with Olympus DP-71 digital camera with an image processing system equipped with Image-ProPlus 6.1 (MediaCybernetics, Silver Spring, MD, USA). Neither labelling in the absence of the primary antibody nor cross-reactivity between secondary and primary antibodies was observed.
Immunofluorescence and co-localization intensity measurements
The distribution of various cav-2 variant fluorescence intensity (FI) values was measured to determine whether insulin promotes translocation of the cav-2 variants to the nuclear envelope as compared to control without insulin stimulation in the cav-2 variant-transfected HEK293T cells. Measurements of the cav-2 variant brightness involved six steps. (1) As a control, the cells were stained for the cav-2 immunoreactivity with TRITC-conjugated secondary antibody without cav-2 primary antibody. (2) Camera gain was calibrated on the cells stained with the secondary antibody, which by definition had no specific fluorescence. (3) Digital images were obtained from cells stained for the cav-2 immunoreactivity with cav-2 primary antibody followed by TRITC-conjugated secondary antibody with camera gain set as for the calibration. (4) Red-green-blue (RGB) images were converted to 8-bit grey-scale images using Image J (National Institutes of Health, Bethesda, MD, USA). (5) FI was determined for each pixel using Image J. (6) The cav-2 FI of these pixels was measured using the Surface Plotter Plugin of Image J on each of the digital images described for cells with or without insulin treatment (five separate images from five individual cells per condition, n= 3).
Measurement of co-localization of cav-2 with lamin A/C was performed with the Colocalization Finder Plugin of Image J [14, 15]. Digital images of the red (cav-2-labelled with TRITC-conjugated secondary antibody) and green (lamin A/C-labelled with FITC-conjugated secondary antibody) channels of the same field were scanned on the confocal microscope (×60 objective). The amount of co-localization of cav-2 with lamin A/C, expressed as the percentage of pixels with threshold lamin A/C immunoreactivity that co-localized with threshold cav-2 staining, was calculated as the number of co-localized pixels divided by the number of threshold pixels in the lamin A/C image. Averages and standard errors were computed over five images per condition for a minimum of 20 cells per condition (n= 3).
Nuclear translocation was determined with binary image masks, which were created of FLAG and DAPI positive staining to define region of interest (ROI) for analysis. The DAPI mask was subtracted from the FLAG mask defining the cytoplasmic ROI using the image calculator from Image J [14, 15]. Nuclear and cytoplasmic staining intensities were compared to give the nuclear : cytoplasmic ratio.
Quantitation of cav-2 FI and co-localization of cav-2 with lamin A/C or phospho-ERK were performed with the Line Profile Tool of Image-ProPlus 6.1 (MediaCybernetics).
Nuclear fractionation
Nuclear fractions were prepared as described [46]. Briefly, HEK293T cells transfected with cav-2 variants were scraped with hypotonic lysis buffer [10 mM Tris-HCl, pH 7.5, 10 mM NaCl, 3 mM MgCl2, 1 mM sodium ortho-vanadate, 5 μg/ml aprotinin, 3 μg/ml pepstatin, 5 μg/ml leupeptin, 1 mM ethylenediaminetetraacetic acid (EDTA) and 1 mM dithiothreitol (DTT)] and incubated on ice for 15 min. Cells were then homogenized using 10 strokes of a Dounce homogenizer and centrifuged at 1000 rpm for 3 min. at 4°C. The crude nuclear pellet was resuspended in nuclear isolation buffer [0.5% Nonidet P-40 (Igepal CA-630, octylphenoxylpolyethoxylethanol, 198596; ICN Biomedicals, Illkirch, France), 10 mM Tris-HCl, pH 7.5, 10 mM NaCl, 3 mM MgCl2, 1 mM sodium ortho-vanadate, 5 μg/ml aprotinin, 3 μg/ml pepstatin, 5 μg/ml leupeptin, 1 mM EDTA and 1 mM DTT] and incubated on ice for 5 min. to solubilize the membrane contaminations, and the nuclei were pelleted by centrifugation at 1000 rpm for 3 min. The initial supernatant was centrifuged at 12,500 ×g for 15 min. to yield the crude cytoplasmic fraction (supernatant) and the membrane pellet comprising mitochondria, endoplasmic reticulum and plasma membrane (pellet). The crude cytoplasmic fraction was centrifuged for 60 min. at 100,000 ×g using a TLA 100.3 rotor (Beckman) to yield the pure cytoplasmic fraction.
Immunoprecipitation
Serum-starved cells were incubated with or without insulin (1 or 100 nM) or IGF-1 (10 nM) for 10 min., washed with ice-cold PBS and lysed in buffer A containing 1% Triton X-100, 150 mM NaCl, 10 mM Tris-HCl (pH 7.4), 1 mM EDTA, 1 mM ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) (pH 8.0), 0.2 mM sodium ortho-vanadate, 0.2 mM phenylmethylsulfonyl fluoride (PMSF), 0.5% Nonidet P-40 and 60 mM n-octylglucoside (OG, 494460, Calbiochem) [14, 15]. The cell lysates were centrifuged at 12,000 rpm for 20 min. at 4°C and the supernatants were subjected to immunoprecipitation with either anti-cav-2, anti-lamin A/C, anti-FLAG, anti-IR or anti-ERK antibodies. The lysates were rotated overnight at 4°C, then 30 μl of protein G or A plus Agarose (Calbiochem) was added and the mixture was rotated for 4 hrs at 4°C. The immunocomplexes were collected by centrifugation at 12,000 rpm for 10 min. 4°C and washed three times with ice-cold buffer B [1% Triton X-100, 150 mM NaCl, 10 mM Tris-HCl (pH 7.4), 1 mM EDTA, 1 mM EGTA (pH 8.0), 0.2 mM sodium ortho-vanadate, 0.2 mM PMSF and 0.5% Nonidet P-40]. After the final wash the pellet was resuspended in 30 μl of 2× SDS-PAGE sample buffer. The immunoprecipitates were then resolved, separated by SDS-PAGE, and subjected to immunoblot analysis.
SDS-PAGE and immunoblot analysis
For the protein extraction, cells were washed twice with ice-cold PBS and lysed with RIPA buffer (50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 150 mM NaCl, 100 mM Tris-HCl [pH 8.0], 0.25% deoxycholic acid, 0.1% SDS, 5 mM EDTA, 10 mM NaF, 5 mM DTT, 1 mM PMSF, 1 mM sodium ortho-vanadate, 20 μM leupeptin and 100 μM aprotinin). The cell lysates were put on ice for 30 min. and microcentrifuged at 12,000 rpm for 20 min. at 4°C. Aliquots from the clear supernatant were taken for protein quantification as determined by the Bradford assay (Bio-Rad, Hercules, CA, USA). Equal amounts of samples (50 μg) were separated on 15% (w/v) SDS-PAGE and transferred to polyvinylidene difluoride membrane (Millipore, Bedford, MA, USA). Transfers were blocked overnight at 4°C with 5% (v/v) non-fat dry milk in Tris-buffered saline (TBS), 0.1% (v/v) Tween 20 and then incubated for 2 hrs at RT in the primary antibody. The primary antibodies used were as follows: cav-2 (BD 610685), cav-1 (BD 610406), phosphotyrosine-PY20 (BD 610000), ERK1 (BD 610031) and flotillin-1 (BD 610820) antibodies from BD Transduction Laboratories (San Diego, CA, USA); F-actin (sc-1616), α-tubulin (sc-5286), lamin A/C (sc-20681) and IR-β (sc-711) antibodies from Santa Cruz Biotechnology (Santa Cruz, CA, USA); phospho-ERK (Thr202 and Tyr204) (#9101) and p44/42 MAPK (#9102) antibodies from Cell Signaling (Invitrogen, Carlsbad, CA, USA); pY19-cav-2 (ab3417) antibody from Abcam (Cambridge, MA, USA); FLAG® M2 (F 1804) antibody from Sigma-Aldrich. The membranes were washed with TBS, 0.1% (v/v) Tween 20 and incubated for 1 hr at RT in horseradish peroxidase-conjugated anti-rabbit (#W4011) or antimouse (#W4021) secondary antibodies (Promega, Madison, WI, USA) in 5% (v/v) non-fat dry milk in TBS, 0.1% (v/v) Tween 20. The immunoblots were developed using the ECL detection reagent (RPN2106; Amersham Biosciences, Buckinghamshire, UK).
Densitometry and statistical analysis
Chemiluminescent images of immunoblots were analysed by scanning densitometry using a Kodak Gel Logic 100 Imaging System (Eastman Kodak Co., New Haven, CT, USA). Multiple exposure of each bolt was use to obtain grey-scale images of each chemiluminescent band. Bands were visualized on a UV transilluminator and photographed. Data are expressed as mean ± S.E. An unpaired Student’s t-test was used to compare treatment groups with significance established at a level of P < 0.05.
Results
Cav-2 translocation to the nuclear envelope and association with Lamin A/C in the inner nuclear membrane in response to insulin
We have previously demonstrated that cav-2 is translocated with phospho-ERK to the nucleus in response to insulin in Hirc-B cells [13–15]. When immunostaining of the nuclear localized cav-2 was examined using confocal microscopy in cav-2-transfected HEK293T cells, the cav-2 displayed a strong signal in the region of nuclear envelope in response to insulin as compared to control (Fig. 1A). Analysis of changes in height of peaks in surface plots of cav-2 FI showed that insulin induced translocation of cav-2 to the nuclear envelope. Quantitation of cav-2 FI using line profile analysis re-confirmed the nuclear envelope targeted cav-2 in response to insulin (Fig. 1A). As nuclear fraction of cav-2-transfected HEK293T cells was subjected to immunoprecipitation analysis, cav-2 interaction not only with lamin A/C but also with phospho-ERK was increased by insulin in the nucleus (Fig. 1B). The insulin-induced increase in cav-2 interaction with lamin A/C was observed in Hirc-B and Rat-1 cells. Insulin up-regulated association between lamin A/C and phospho-ERK as well (Fig. 1C). Confocal microscopic image analysis showed that cav-2 was distributed throughout intracellular compartments in control cells as observed in Figure 1A. Insulin, however, induced a profound change in localization of cav-2 to nuclear envelope and increased co-localization of cav-2 with lamin A/C in the inner nuclear membrane in Hirc-B and COS-7 cells (Fig. 1D). The quantitative correlation analysis showed the expected increase in the inner nuclear membrane co-localization (%) from merge intensity of cav-2 with lamin A/C staining (54.8% and 57.2% increases in Hirc-B and COS-7 cells, respectively). Quantitative line profile analysis confirmed that cav-2 was co-localized with lamin A/C in the inner nuclear membrane in response to insulin in Hirc-B (Fig. 1D, panels a and b) and COS-7 (Fig. 1D, panels c and d) cells. These results verify that insulin translocates cav-2 to the nuclear envelope and the cav-2 is subsequently localized to the inner nuclear membrane where cav-2, phospho-ERK and lamin A/C interact together.
Fig 1.
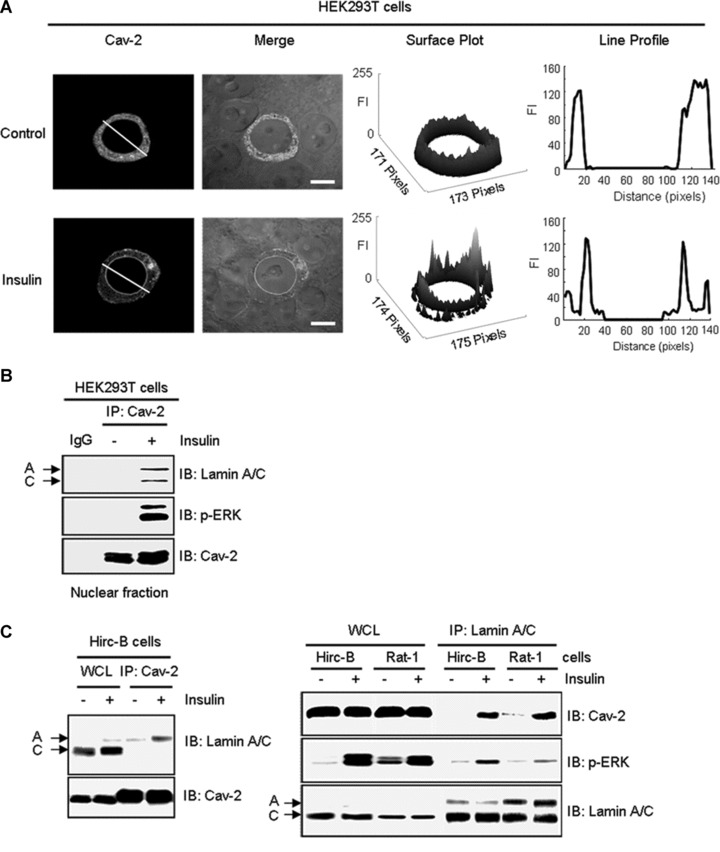
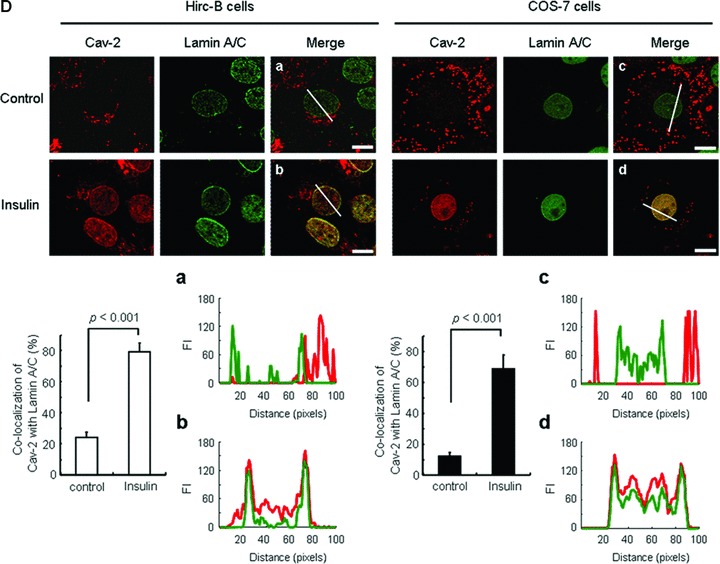
Insulin-induced interaction between cav-2 and lamin A/C in the inner nuclear membrane. (A) HEK293T cells were transfected with pcDNA-cav-2. Thirty-six hours after transfection, cells were treated with or without insulin (100 nM) for 10 min. Cells were stained with anti-cav-2 antibody followed by TRIRC-conjugated antibody. The cells were visualized by confocal microscopy. Results are representative images of cells from three independent experiments. Grey: cav-2; Merge: cav-2 + DIC. Scale bars: 10 μm. Height of peaks representing cav-2 FI in surface plots illustrates localization of cav-2. Cav-2 FI was measured using the Surface Plotter Plugin of Image J as described under ‘Materials and methods’. The white lines in cav-2 panel were converted to line profiles using the Line Profile Tool of Image-ProPlus 6.1. (B) The cav-2 transfected cells were subjected to nuclear fractionation as described under ‘Materials and methods’. Equal amounts of protein from the nuclear fractions were immunoprecipitated with anti-cav-2 antibody and subjected to SDS-PAGE and immunoblotting with antibodies against lamin A/C, phospho-ERK and cav-2. Shown is a representative experiment that was repeated three times. (C) Hirc-B and Rat-1 cells were treated with or without insulin (100 nM) for 10 min. Whole cell lysates (WCL) were immunoprecipitated with anti-cav-2 or anti-lamin A/C antibodies and subjected to immunoblotting using antibodies specific for cav-2, phospho-ERK and lamin A/C. Shown is a representative experiment that was repeated three times. (D) Hirc-B and COS-7 cells were treated with or without insulin (100 nM) for 10 min. Cells were immunostained with anti-cav-2 and anti-lamin A/C antibodies followed by TRITC- and FITC-conjugated antibodies, respectively, and visualized by confocal microscopy. Results are representative images of cells from three independent experiments. Red: cav-2; Green: lamin A/C; Merge: lamin A/C + cav-2. Scale Bars: 10 μm. Co-localization of cav-2 with lamin A/C in the nuclear envelope was quantified by the Colocalization Finder Plugin of Image J as described under ‘Materials and methods’. The results represent the mean ± S.E. of three independent experiments. (a–d) The white lines in merge panels were converted to line profiles using the Line Profile Tool of Image-ProPlus 6.1. Red line: cav-2; Green line: lamin A/C.
Prevention of nuclear localization of cav-2 and phospho-ERK by deletion of amino acids 120–162 at the C-terminus
To identify molecular domains in cav-2 required for targeting to nucleus, we constructed domain deletion mutants of cav-2 and generated a series of fusion proteins, which comprised the N-terminal regions of cav-2 fused to FLAG as shown schematically in Figure 2A. HEK293T cells were transfected with the mutants and subjected to nuclear fractionation to examine their effects on insulin-induced nuclear translocation of the cav-2 and phospho-ERK. FLAG-Cav-21–162 and phospho-ERK were significantly increased in nuclear fraction of insulin-stimulated cells (Fig. 2B, lanes 1 versus 2). N-terminal FLAG-cav-247–162 and 71–162 and C-terminal FLAG-cav-21–119 mutants, however, were not detected in the nuclear fraction in response to insulin. Insulin-induced nuclear translocation of phospho-ERK was completely abolished by transfection of the mutants (Fig. 2B, lanes 2 versus 4, 6 and 8). These results indicate that both N-terminal (residues 1–46) and C-terminal (residues 120–162) domains are required for nuclear localization of cav-2 and phospho-ERK in response to insulin.
Fig 2.
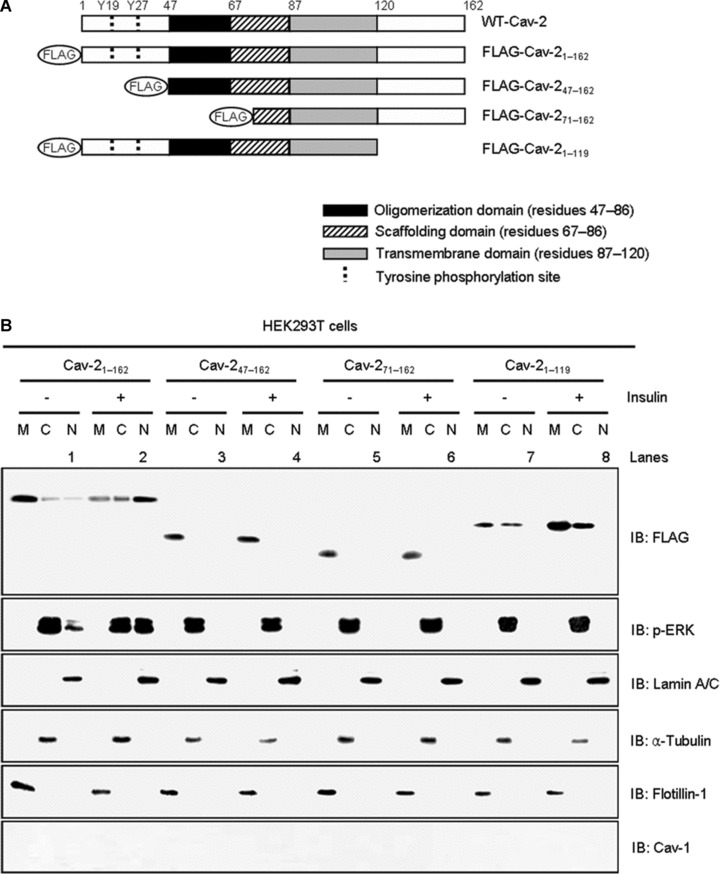
Effects of cav-2 domain deletion mutants on insulin-induced nuclear translocation of cav-2 and phospho-ERK. (A) A diagram of cav-2 deletion mutants. (B) HEK293T cells expressing FLAG-cav-21–162 or FLAG-cav-247–162, FLAG-cav-271–162, or FLAG-cav-21–119 mutants were treated with or without insulin (100 nM) for 10 min. and subjected to nuclear fractionation as described under ‘Materials and methods’. The fractions were analysed by SDS-PAGE and immunoblotted using antibodies against FLAG, phospho-ERK and cav-1, and detected with antibodies specific for lamin A/C, α-tubulin and flotillin-1 as markers for nuclear (N), cytoplasmic (C) and membrane (M) fractions, respectively. Shown is a representative experiment that was repeated three times.
Requirement of amino acids 120–162 at the C-terminus of cav-2 for insulin-induced phosphorylation of pY19-cav-2 and cav-2 interaction with phospho-ERK
Tyrosine 19 in the N-terminus of cav-2 is essential for insulin-induced nuclear localization of cav-2 with phospho-ERK [14, 15]. The retardation observed by N-terminal FLAG-cav-247–162 and 71–162 mutants (Fig. 2B) is in agreement with our previous findings. Accordingly, we tested effect of C-terminal truncation mutation (FLAG-cav-21–119) on insulin-induced phosphorylation of pY19-cav-2 and ERK. As demonstrated in Figure 3A, tyrosine 19 on FLAG-cav-21–119 mutant was not phosphorylated and the mutant attenuated insulin-stimulated ERK activation by a 1.5-fold as compared to FLAG-cav-21–162-transfected cells. Insulin-induced interaction of FLAG-cav-21–119 with phospho-ERK was decreased by a 6.1-fold as compared to FLAG-cav-21–162-transfected cells (Fig. 3B). When immunofluorescence localization was examined, FLAG-cav-21–119 mutant was not detected in the nucleus whereas FLAG-cav-21–162 was translocated to the nucleus in response to insulin (Fig. 3C). Consistent with the results, quantitative data showed a significant decrease in nuclear : cytoplasmic ratio of FLAG-cav-21–119 mutant as compared to FLAG-cav-21–162 staining in response to insulin. Thus, these data in accordance with the results from Figure 2B suggest that C-terminal domain (residues 120–162) is important for phosphorylation of pY19-cav-2 and ERK and their interaction, and is required for nuclear targeting of cav-2 and phospho-ERK in response to insulin.
Fig 3.
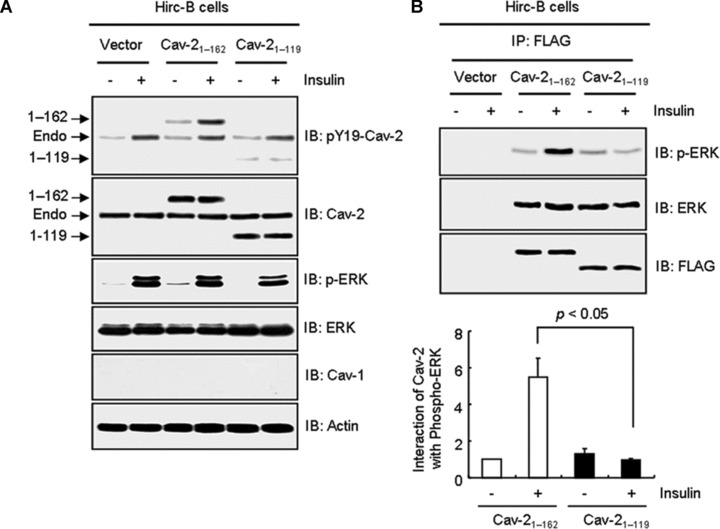
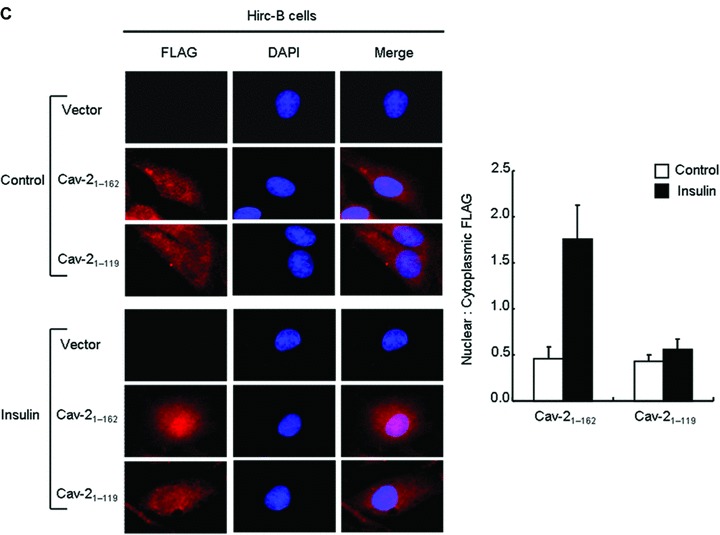
Retardation of insulin-induced phosphorylation of pY19-cav-2 and ERK, interaction of cav-2 with phospho-ERK, and nuclear translocation of cav-2 by deletion of C-terminal domain (residues 120–162). Hirc-B cells expressing pDS_FLAG-XB vector, FLAG-cav-21–162, or FLAG-cav-21–119 mutant were treated with or without insulin (100 nM) for 10 min. (A) WCL were subjected to immunoblotting using antibodies specific for pY19-cav-2, cav-2, phospho-ERK, ERK, cav-1 and actin. Shown is a representative experiment that was repeated three times. (B) WCL were immunoprecipitated with anti-FLAG antibody and subjected to immunoblot analysis with anti-phospho-ERK, anti-ERK and anti-FLAG antibodies as indicated. Interaction of cav-2 with phospho-ERK was quantified by densitometry. The results represent the mean ± S.E. of three independent experiments. (C) Cells were fixed, immunostained with anti-FLAG antibody, and visualized by TRITC-conjugated antibody. DNA was stained using DAPI to visualize the nucleus. Results are representative images of cells from three independent experiments. Red: FLAG; Blue: DAPI; Merge: FLAG + DAPI. Nuclear translocation of FLAG-cav-21–162 and FLAG-cav-21–119 mutant was quantified by the Image Calculator of Image J as described under ‘Materials and methods’. Nuclear : cytoplasmic ratios of FLAG staining are shown.
Effects of Ser154–Val155–Ser156 domain mutants on insulin-induced phosphorylation of pY19-cav-2
Upon a detailed mutagenesis analysis of the C-terminal domain, a three amino acid sequence composed of residues Ser154, Val155 and Ser156 (SVS) (Fig. 4A) was identified to regulate insulin-induced phosphorylation and nuclear translocation of cav-2. To characterize function of this domain, we first replaced the residues to Ala and generated AAA; S154A, V155A and S156A and AVA; S154A and S156A mutants. Insulin-induced phosphorylation of pY19-cav-2 was completely inhibited by the AAA and AVA mutants in Hirc-B and COS-7 cells (Fig. 4B and C). When single point mutants (AVS; S154A and SVA; S156A mutants) in the Ser154–Val155–Ser156 domain were examined, AVS mutant abolished phosphorylation of pY19-cav-2 in response to insulin as compared to WT-cav-2-transfected cells (Fig. 4D and E). Of interest, phosphorylation of pY19-cav-2 was up-regulated by SVA mutant even without insulin stimulation and the phosphorylation status was unchanged by insulin treatment in Hirc-B and COS-7 cells (Fig. 4D and E).
Fig 4.
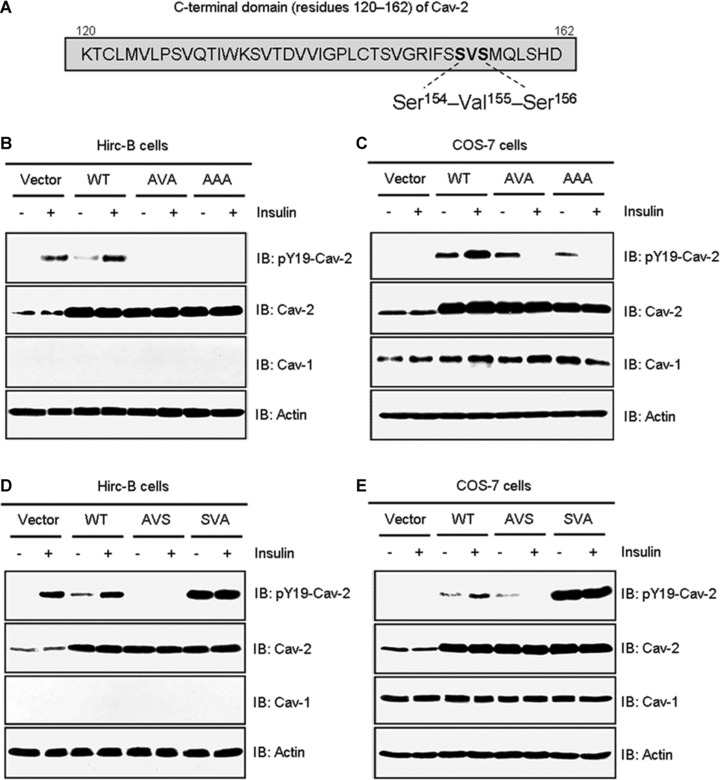
Effects of mutations in the Ser154–Val155–Ser156 domain on phosphorylation of pY19-cav-2 in response to insulin. (A) The Ser154–Val155–Ser156 domain in C-terminal residues 120–162 of cav-2. Hirc-B (B) and COS-7 (C) cells expressing pcDNA3 vector, WT-cav-2, AVA (S154A and S156A), or AAA (S154A, V155A and S156A) mutants were treated with or without insulin (100 nM) for 10 min. WCL were subjected to immunoblotting using antibodies specific for pY19-cav-2, cav-2, cav-1 and actin. Shown is a representative experiment that was repeated three times. Hirc-B (D) and COS-7 (E) cells expressing pcDNA3 vector, WT-cav-2, AVS (S154A) or SVA (S156A) mutants were treated with or without insulin (100 nM) for 10 min. WCL were subjected to immunoblotting using antibodies specific for pY19-cav-2, cav-2, cav-1 and actin as indicated. Shown is a representative experiment that was repeated three times.
The Ser154–Val155–Ser156 domain as a nuclear trafficking signal in cav-2
To verify requirement of the Ser154–Val155–Ser156 domain for insulin-induced translocation of cav-2 to the nucleus, WT-cav-2 and various Ser154–Val155–Ser156 domain mutants were expressed in HEK293T cells and analysed for their immunofluorescence localization by confocal microscopy (Fig. 5A). As shown in Figure 1A, WT-cav-2 was re-localized to the nuclear envelope in response to insulin (Fig. 5A, panel 1). AAA, AVA and AVS mutants were not translocated to the nuclear envelope (Fig. 5A, panels 2–4) whereas SVA mutant was (Fig. 5A, panel 5) in response to insulin. When we checked Val155 with the Val155 single mutant (SAS: V155A mutant) and Ser153 before the Ser154 with the Ser153 single mutant (ASVS; S153A mutant), SAS mutant was not but ASVS mutant translocated to the nuclear envelope in response to insulin (Fig. 5A, panels 6 and 7). Analysis of changes in height of peaks in surface plots of the cav-2 FI confirmed the retardation of nuclear envelope translocation of the AAA, AVA, AVS and SAS mutants, and translocation of WT-cav-2 and SVA and ASVS mutants to the nuclear envelope in response to insulin (Fig. 5A). When effects of the various mutants were further examined for insulin-induced nuclear localization of cav-2 and phospho-ERK by nuclear fractionation, WT-cav-2 and phospho-ERK were predominantly detected in membrane and cytoplasmic fractions, respectively, in untreated cells and their localization to nucleus was significantly increased by insulin treatment (Fig. 5B, lanes 1 and 2). AAA, AVA, AVS and SAS mutants were not detected in nuclear fraction in response to insulin. Insulin-stimulated nuclear localization of phospho-ERK was not observed in the mutant-transfected cells (Fig. 5B, lanes 4, 6, 8 and 10). Consistent with the results in Figure 5A, nuclear localization of SVA and ASVS mutants with phospho-ERK was initiated by insulin treatment (Fig. 5B, lanes 12 and 14). Together with the results demonstrated in Figures 2–4, these data point out that Ser154–Val155–Ser156 domain in C-terminal (residues 120–162) of cav-2 is required for phosphorylation of pY19-cav-2 and ERK and its interaction with phospho-ERK, and regulates translocation of cav-2 with phospho-ERK to the nuclear envelope in response to insulin.
Fig 5.
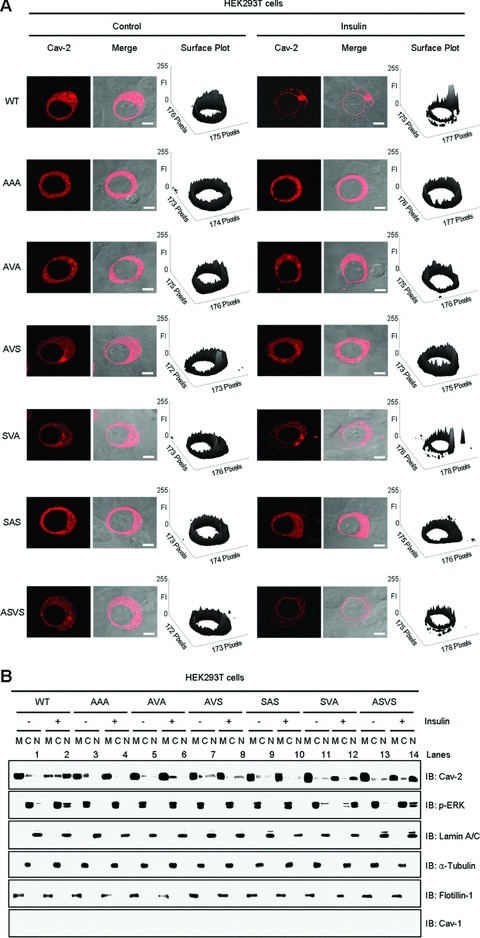
Effects of exogenous transfection of Ser154–Val155–Ser156 domain mutants on insulin-induced nuclear transport of cav-2 and phospho-ERK. HEK293T cells expressing WT-cav-2, AAA (S154A, V155A and S156A), AVA (S154A and S156A), AVS (S154A), SAS (V155A), SVA (S156A) or ASVS (S153A) mutants were treated with or without insulin (100 nM) for 10 min. (A) Cells were immunostained with anti-cav-2 antibody followed by TRITC-conjugated antibody and visualized by confocal microscopy. Results are representative images of cells from three independent experiments. Red: cav-2; Merge: cav-2 + DIC. Scale Bars: 10 μm. Height of peaks representing cav-2 FI in surface plots illustrates localization of cav-2. FI of the cav-2 variants were measured using the Surface Plotter Plugin of Image J as described under ‘Materials and methods’. (B) WCL were subjected to nuclear fractionation as described under ‘Materials and methods’. The fractions were analysed by SDS-PAGE and immunoblotted using antibodies against cav-2, phospho-ERK and cav-1, and detected with antibodies specific for lamin A/C, α-tubulin and flotillin-1 as markers for nuclear (N), cytoplasmic (C) and membrane (M) fractions, respectively. Shown is a representative experiment that was repeated four times.
Insulin-specific interaction of phospho-ERK with cav-2
To verify association of phospho-ERK with cav-2 is an insulin-specific action in comparison to IGF-1, we examined phosphorylation of ERK and the interaction by insulin versus IGF-1 in Hirc-B cells having endogenous cav-2 alone [13–15]. In response to insulin, phosphorylation of ERK was increased 3.0-, 5.1- and 8.1-fold by 1, 10 and 100 nM insulin, respectively (Fig. 6A). IGF-1 elicited a response at 10 nM concentration, consistent with effects mediated by low affinity binding to IR [24, 40, 47]. ERK was associated with cav-2 independently of their phosphorylation [14] (Fig. 6B). In comparison, that interaction of phospho-ERK with cav-2 was increased by 1 and 100 nM insulin, the interaction was not affected by 10 nM IGF-1 (Fig. 6B), although phosphorylation of ERK was induced by 10 nM IGF-1 (Fig. 6A). When the insulin-specific phospho-ERK interaction with cav-2 was examined in COS-7 cells having endogenous cav-1 and cav-2, the interaction was induced by insulin, but not IGF-1 (Fig. 6C). These results demonstrate that ERK is phosphorylated by both insulin and IGF-1, but phosphorylation of ERK in the ERK-cav-2 complex is an insulin-specific signalling event.
Fig 6.
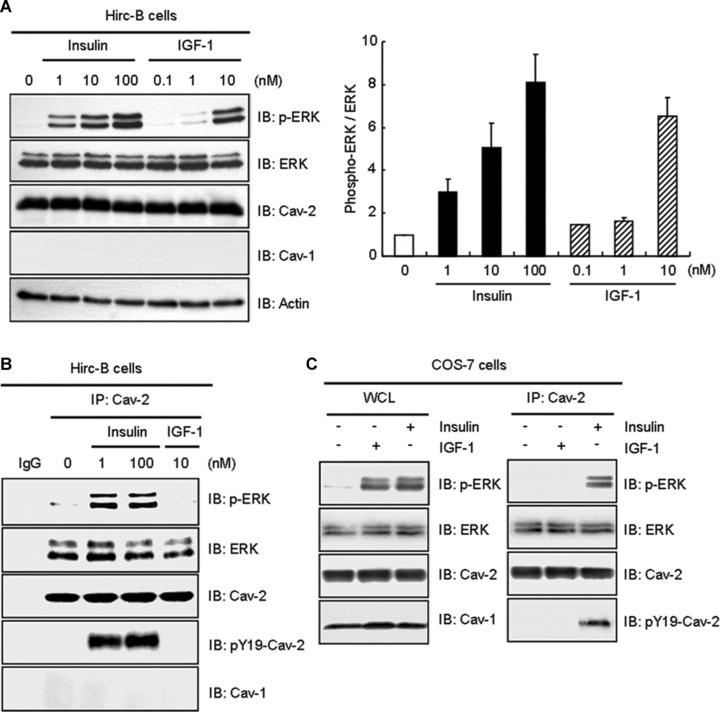
Insulin-induced interaction of phospho-ERK with cav-2 distinctively from IGF-1. Hirc-B cells were treated with the indicated nanomolar concentration of insulin or IGF-1 for 10 min. (A) WCL were subjected to immunoblot analysis using anti-phospho-ERK, anti-ERK, anti-cav-2, anti-cav-1 and anti-actin antibodies. The densitometry ratios of phospho-ERK to total ERK were illustrated. The results represent the mean ± S.E. of three independent experiments. (B) WCL were immunoprecipitated with anti-cav-2 antibody and subjected to immunoblot analysis with anti-phospho-ERK, anti-ERK, anti-cav-2, anti-pY19-cav-2 and anti-cav-1 antibodies as indicated. Shown is a representative experiment that was repeated three times. (C) COS-7 cells were treated with or without IGF-1 (10 nM) or insulin (100 nM) for 10 min. Equal amount of WCL were immunoprecipitated with anti-cav-2 antibody and subjected to immunoblot analysis with anti-phospho-ERK, anti-ERK, anti-cav-2, anti-pY19-cav-2 and anti-cav-1 antibodies as indicated. Shown is a representative experiment that was repeated three times.
Insulin-specific cav-2-dependent activation of ERK and IR
To characterize further the differential effect of insulin and IGF-1 on activation of ERK, HEK293T cells having no detectable endogenous cav-1 and cav-2 [36, 37] were incubated with various concentrations of insulin and IGF-1. Phosphorylation of ERK was unaffected by insulin treatment at various concentrations [24, 48]. But 10 nM IGF-1 induced phosphorylation of ERK (Fig. 7A) as previously shown in HEK293T cells [24]. We next examined the IGF-1- and insulin-induced ERK activation in WT-cav-2-transfected HEK293T cells. IGF-1 treatment showed an increase in ERK activation as observed in Figure 7A by a 3.4-fold in vector control (Fig. 7B). WT-cav-2 transfection itself up-regulated phosphorylation of ERK by a 3.5-fold as compared to unstimulated vector control, and ERK activation was increased only by a 1.3-fold upon IGF-1 treatment (Fig. 7B). In contrast, although ERK activation was 1.2-fold increased by insulin treatment in vector control, the activation became increased by a 2.5-fold in WT-cav-2-transfected cells. We have isolated and sequenced the cDNA encoding Rattus norvegicus (rat) cav-2. Analysis of deduced amino acid sequence indicates that the rat cav-2 shares 88.9% identity with Homo sapiens (human) cav-2 sequence (Fig. S1). To test whether there is any different effect on insulin-stimulated phosphorylation of ERK by rat versus human cav-2, we expressed both full-length constructs in HEK293T cells and examined phosphorylation of ERK by insulin treatment. As shown in Figure 7C, almost the same level of insulin-induced phosphorylation was observed. Cav-2 is required for prolonged tyrosine kinase activation of IR [49]. When we investigated insulin-specific cav-2-dependent expression and activation of IR in WT-cav-2-transfected HEK293T cells, expression of IR was not changed (Fig. 7D). Insulin triggered tyrosine kinase activation of IR by a 5.0-fold in vector control. WT-cav-2 promoted phosphorylation of IR by a 7.6-fold in response to insulin. By contrast, IGF-1-induced IR activation was unchanged by WT-cav-2 transfection (Fig. 7D). Thus, these data reveal that rescue of cav-2 loading restores insulin-specific activation of ERK by promoting tyrosine kinase activation of IR.
Fig 7.
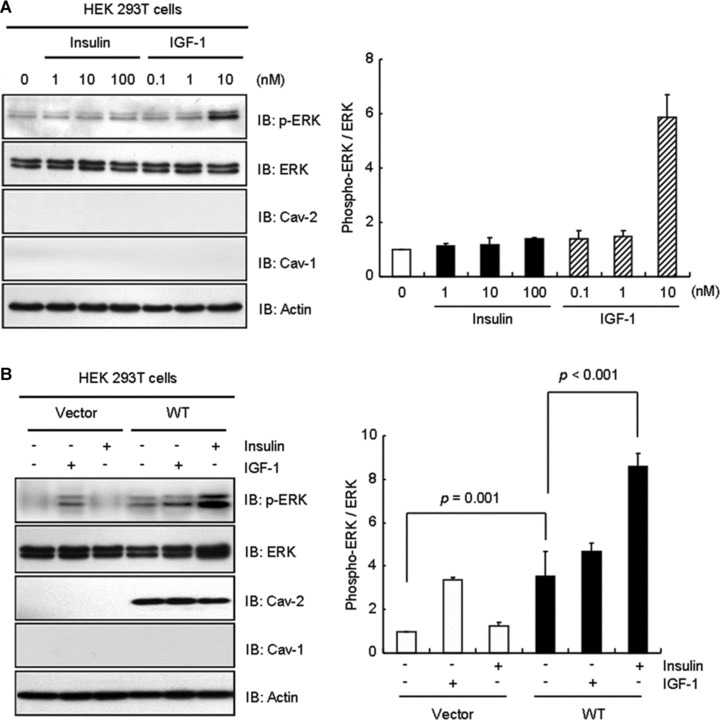
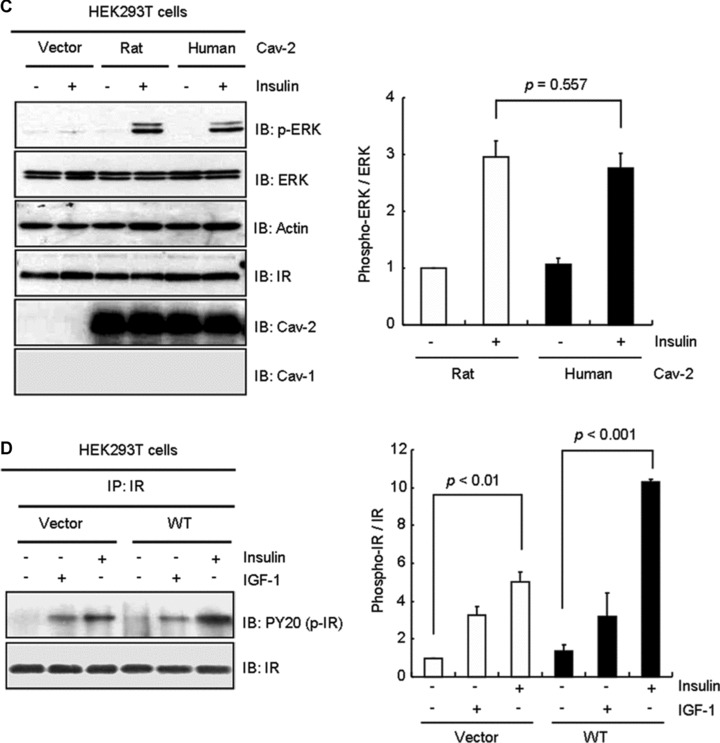
Effect of cav-2 transfection on activation of ERK and IR in response to insulin and IGF-1. (A) HEK 293T cells were treated with the indicated nanomolar concentration of insulin or IGF-1 for 10 min. WCL were subjected to immunoblot analysis using anti-phospho-ERK, anti-ERK, anti-cav-2, anti-cav-1 and anti-actin antibodies. The densitometry ratios of phospho-ERK to total ERK were illustrated. The results represent the mean ± S.E. of three independent experiments. (B) Cells were transfected with pcDNA3 vector or WT-cav-2. Thirty-six hours after transfection, the cells were treated with or without IGF-1 (10 nM) or insulin (100 nM) for 10 min. WCL were subjected to immunoblotting using antibodies specific for phospho-ERK, ERK, cav-2, cav-1 and actin. The densitometry ratios of phospho-ERK to total ERK were illustrated. The results represent the mean ± S.E. of four independent experiments. (C) Cells were transfected with pcDNA3 vector, rat or human cav-2. Thirty-six hours after transfection, the cells were treated with or without insulin (100 nM) for 10 min. WCL were subjected to immunoblotting using antibodies specific for phospho-ERK, ERK, actin, IR, cav-2 and cav-1. The densitometry ratios of phospho-ERK to total ERK were illustrated. The results represent the mean ± S.E. of three independent experiments. (D) Cells expressing pcDNA3 vector or WT-cav-2 were treated with or without IGF-1 (10 nM) or insulin (100 nM) for 10 min. WCL were immunoprecipitated with anti-IR antibody and subjected to immunoblot analysis with anti-phosphotyrosine PY20 and anti-IR antibodies. The densitometry ratios of phospho-IR to total IR were illustrated. The results represent the mean ± S.E. of three independent experiments.
Insulin-specific pY19-cav-2-dependent phosphorylation of ERK
pY19-Cav-2 was phosphorylated by insulin, but not IGF-1 and insulin increased association between cav-2 and phospho-ERK in Hirc-B and COS-7 cells as shown in Figure 6B and C. When HEK293T cells were transfected with WT-cav-2, insulin induced interaction between ERK and pY19-cav-2 (Fig. 8A). To verify if the insulin-specific ERK activation requires insulin-induced phosphorylation of cav-2 at tyrosine 19, cells were transfected with Y19A mutant. As shown in Figure 8B, WT-cav-2 enhanced insulin-induced activation of ERK by 2.4-fold as compared to control. The ERK activation, however, was inhibited by Y19A mutant. Thus, these data demonstrate that ERK activation is dependent on phosphorylation of cav-2 at tyrosine 19 in response to insulin.
Fig 8.
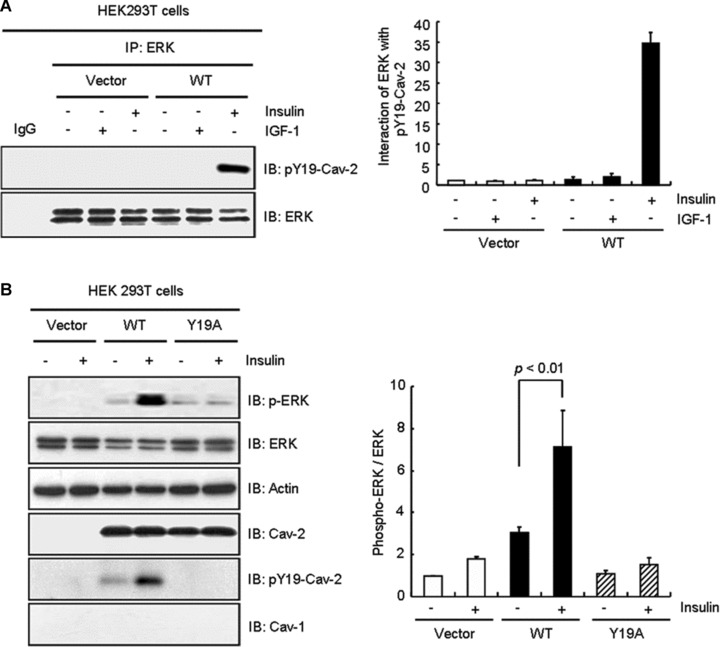
Attenuation of insulin-induced phosphorylation of ERK by Y19A mutant. (A) HEK293T cells expressing pcDNA3 vector or WT-cav-2 were treated with or without IGF-1 (10 nM) or insulin (100 nM) for 10 min. WCL were immunoprecipitated with anti-ERK antibody and subjected to immunoblot analysis with anti-pY19-cav-2 and anti-ERK antibodies as indicated. Interaction of ERK with pY19-cav-2 was quantified by densitometry. The results represent the mean ± S.E. of three independent experiments. (B) Cells were transfected with pcDNA3 vector, WT-cav-2 or Y19A mutant. Thirty-six hours after transfection, the cells were treated with or without insulin (100 nM) for 10 min. WCL were subjected to immunoblotting using antibodies specific for phospho-ERK, ERK, actin, cav-2, pY19-cav-2 and cav-1. The densitometry ratios of phospho-ERK to total ERK were illustrated. The results represent the mean ± S.E. of four independent experiments.
Insulin-specific cav-2-dependent nuclear envelope localization of phospho-ERK
Epidermal growth factor, but not insulin, enhanced phosphorylation and nuclear translocation of ERK in HEK293T cells [24, 50]. We examined whether insulin-induced nuclear translocation of phospho-ERK can be constituted in the cells by introducing exogenous WT-cav-2. When the effect of cav-2 transfection on immunofluorescence localization of phospho-ERK was investigated using confocal microscopy (Fig. 9A), phospho-ERK was not detected in the nucleus in vector control cells with or without insulin stimulation (Fig. 9A, panels 1 and 2). When cells were transfected with WT-cav-2, phospho-ERK was observed in the cytoplasm without insulin and IGF-1 stimulation (Fig. 9A, panel 4). Upon insulin treatment, translocation of phospho-ERK to the nucleus was detected in the WT-cav-2-transfected cells. And the phospho-ERK was co-localized with the cav-2 in the nuclear envelope (Fig. 9A, panel 5). In contrast, IGF-1 induced nuclear translocation of phospho-ERK in both vector- and WT-cav-2-transfected cells (Fig. 9A, panels 3 and 6) but failed to re-localize the cav-2 to the nuclear envelope (Fig. 9A, panel 6). Quantitative line profile analyses of cav-2 and phospho-ERK FI showed the co-localization of phospho-ERK with cav-2 in the nuclear envelope in response to insulin, but not IGF-1 (Fig. 9A, panels a–c). These results suggest that insulin-specific nuclear envelope localization of phospho-ERK is mediated by cav-2. As we further investigated the cav-2 effect on insulin-induced nuclear translocation of phospho-ERK by nuclear fractionation of WT-cav-2-transfected HEK293T cells, insulin induced a 6.0-fold increase of phospho-ERK localization in nuclear fraction (Fig. 9B). In parallel, cav-2 was localized 90.6% in membrane and 2.1% in nuclear fractions in untreated cells. However, in insulin-stimulated cells, cav-2 was found 28.7% in membrane and 60.5% in nuclear fractions (Fig. 9B). These data again demonstrate that cav-2 regulates insulin-specific nuclear targeting of phospho-ERK.
Fig 9.
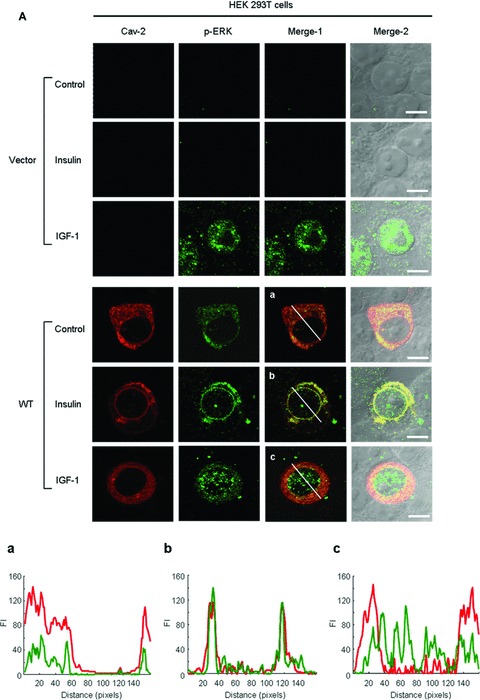
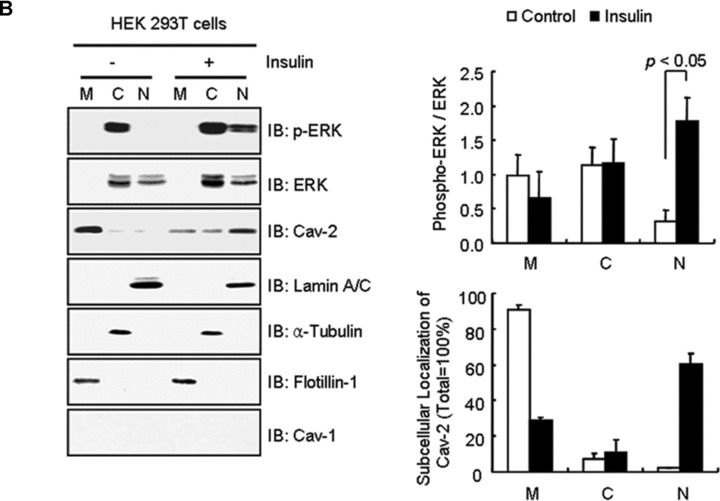
Restoration of insulin-induced nuclear envelope targeting of phospho-ERK and cav-2 by exogenous cav-2 transfection. (A) HEK293T cells were transfected with pcDNA3 vector and WT-cav-2. Thirty-six hours after transfection, cells were treated with or without insulin (100 nM) or IGF-1 (10 nM) for 10 min. The cells were then stained with anti-cav-2 and anti-phospho-ERK antibodies followed by TRITC- or FITC-conjugated antibodies, respectively, and visualized by confocal microscopy. Results are representative images of cells from three independent experiments. Red: cav-2; Green: phospho-ERK; Merge-1: cav-2 + phospho-ERK; Merge-2: cav-2 + phospho-ERK + DIC. Scale Bars: 10 μm. (a–c) The white lines in merge-1 panel were converted to line profiles using the Line Profile Tool of Image-ProPlus 6.1. Red line: cav-2; Green line: phospho-ERK. (B) WT-cav-2-transfected cells were treated with or without insulin (100 nM) for 10 min. and subjected to nuclear fractionation as described under ‘Materials and methods’. The fractions were analysed by SDS-PAGE and immunoblotted using antibodies against cav-2, phospho-ERK and cav-1 and detected with antibodies specific for lamin A/C, α-tubulin and flotillin-1 as markers for nuclear (N), cytoplasmic (C) and membrane (M) fractions, respectively. The densitometry ratios of phospho-ERK to total ERK and percentage of localization of cav-2 in each fraction were illustrated. The results represent the mean ± S.E. of five independent experiments.
Discussion
Previously, we have reported that cav-2 regulates ERK-mediated mitogenesis of insulin signalling in Hirc-B cells [13–15]. Hirc-B cells were shown to be sensitive to both insulin and IGF-1 [43, 44]. IGF-1 binds to IR with low affinity (less than 1%) but high concentration of IGF-1 promotes activation of IR [47]. As we examined activation and interaction of ERK with cav-2 in response to various concentrations of IGF-1 and insulin in the cells, IGF-1-activated ERK was not associated with cav-2 but insulin-activated ERK interacted with cav-2 (Fig. 6A and B). To better understand the regulatory role of cav-2 in insulin versus IGF-1 action, we explored the activation of ERK and association and nuclear targeting of phospho-ERK in HEK293T cells that express endogenous cav-2 at extremely low levels [36, 37]. We have demonstrated that exogenous addition of cav-2 constitutes insulin-induced activation of ERK and co-localization of phospho-ERK and cav-2 with nuclear lamin in the inner nuclear membrane (Figs 1A and B, 7B and 9A). Unlike insulin, although ERK was phosphorylated and translocated to the nucleus by IGF-1, cav-2 remained in the cytoplasm (Fig. 9A). Our investigation of cav-2-dependent activation of IR by insulin versus IGF-1 in cav-2-transfected HEK293T cells showed that cav-2 did not affect the protein level of IR (Fig. 7D); in contrast that cav-1 has been shown to increase IR expression in cav-1-transfected HEK293T cells [51]. By cav-2 transfection, however, tyrosine kinase activation of IR was increased in response to insulin, but not IGF-1 (Fig. 7D). In agreement with this, we have recently demonstrated that cav-2 prolongs IR activation by preventing IR interaction with suppressor of cytokine signaling-3 (SOCS-3) [49]. Taken together, our data suggest that cav-2 elevates insulin sensitivity to IR, thereby enhancing phosphorylation of cav-2 and ERK, subsequently their interaction, and, in turn, cav-2-dependent nuclear targeting of phospho-ERK.
We investigated insulin-specific ERK signalling in the various cell lines: (i) Hirc-B cells having cav-2 alone with no cav-1 gene expression (Fig. 6A) [13–15]; (ii) COS-7 cells having endogenous cav-1 and cav-2 (Fig. 6C) and (iii) HEK293T cells expressing extremely low endogenous cav-1 and cav-2 (Fig. 7A) [36, 37]. Our investigation showed that insulin-induced activation and nuclear translocation of ERK are dependent on cav-2 irrespective of cav-1 expression (Figs 6–9). Cav-2 knockout mice showed pulmonary defects without change in cav-1 protein expression [11] and displayed hyperproliferative phenotype in the lung endothelial cells [52]. Thus, these and our present studies point out the specific physiological function of cav-2 independent of cav-1.
Our recent works have shown that tyrosines 19 and 27 in cav-2 are essential for nuclear targeting of phospho-ERK with cav-2, that are demonstrated by using siRNA, overexpression, Y19A, Y27A and Y19A/Y27A tyrosine mutations, and immunofluorescence localization of cav-2 [14, 15]. In the present study, the findings are confirmed and extended to identify the nuclear trafficking signal domain in the C-terminal by the analyses of cav-2 domain deletion mutants. First, the N-terminal domain (residues 1–46) containing tyrosines 19 and 27 was required for nuclear localization of cav-2 and phospho-ERK and its deletion abolished the localization (Fig. 2B). Second, the C-terminal domain (residues 120–162) was necessary for the phosphorylation of tyrosine 19, cav-2 interaction with phospho-ERK and nuclear targeting of phospho-ERK with cav-2. And its truncation caused inhibition of the phosphorylation, interaction and translocation (Figs 2B and 3). These results suggest that the N- and C-terminal domains are needed for the exit of cav-2 with phospho-ERK from intracellular compartments or conversely that the truncation causes an increased retention in the compartments in response to insulin.
What information does the domain of cav-2 itself contain regarding the protein’s nuclear traffic? We have demonstrated that the C-terminal domain (residues 120–162) is necessary for cav-2 to target the nucleus in response to insulin (Figs 2B and 3C). The C-terminal domain does not contain the canonical NLS, but there are three serine residues (Ser153–Ser154–Val155–Ser156), which are conserved and detected in nuclear proteins such as reverse transcriptase, nucleotide-binding protein 1, adenosine monophosphate (AMP)-dependent ligase, RNA polymerase β-subunit and acetyltransferase, identified from sequence analysis using Basic Local Alignment Search Tool (http://www.ncbi.nlm.nih.gov/BLAST/) [53]. Here, we have taken a mutational approach to determine the role of the serine residues (Ser153–Ser154–Val155–Ser156). Two serines among 12 residues conserved in the caveolin family of primary sequences [54], Ser80 in cav-1 was reported to regulate cav-1 entry into secretory pathway but mutation of Ser168 to Ala did not alter the subcellular localization of cav-1 [55]. We found that mutation of Ser153, which aligns with Ser168 in cav-1, of cav-2 C-terminal to Ala (ASVS mutant) did not affect the insulin-induced nuclear translocation of cav-2 (Fig. 5). Our detailed mutagenesis studies on the Ser153–Ser154–Val155–Ser156 sequence reveal that Ser154–Val155–Ser156 domain is important for the nuclear translocation of cav-2 and identify the domain as a non-conventional NLS.
It is interesting to notice that Ser154–Val155–Ser156 in the Ser153–Ser154–Val155–Ser156 sequence is conserved only in cav-2 among caveolin family and the Ser154–Val155–Ser156 domain regulates nuclear trafficking of cav-2 (Fig. 5). Insulin-induced phosphorylation of pY19-cav-2 was markedly decreased by AVS (S154A) mutant. SVA (S156A) mutant, however, exhibited significant increase in pY19-cav-2 phosphorylation even without insulin stimulation. The increased phosphorylation status was no longer changed by insulin treatment (Fig. 4D and E). Although the phosphorylation of pY19-cav-2 was up-regulated independently of insulin in SVA mutant-transfected cells, cav-2 was unable to translocate to the nucleus without insulin stimulation. Indeed, the cav-2 translocation to the nucleus required insulin action (Fig. 5). These data indicate that Ser154 and Ser156 might function as a positive and negative regulator, respectively, in insulin-induced phosphorylation of pY19-cav-2 and cav-2 localization to the nucleus. Insulin-mediated phosphorylation and/or dephosphorylation of the serine residues of Ser154–Val155–Ser156 domain and their possible regulation of the cav-2 nuclear translocation, however, remain to be answered.
Active ERK interacts with lamin A/C and the nuclear envelope-bound ERK functions as a molecular switch for rapid mitogen-dependent transcriptional activation [17]. The present data demonstrate that insulin-specific nuclear translocation of phospho-ERK requires cav-2 and that the nuclear localized phospho-ERK with cav-2 associates with lamin A/C in the inner nuclear membrane (Figs 1 and 9). In the context of the evidence, present findings suggest that nuclear targeted cav-2 regulates interaction between phospho-ERK and lamin A/C in the inner nuclear membrane, thereby promoting activation of transcriptional factors such as c-Jun and STAT3, cell cycle regulators and DNA synthesis for cell growth and proliferation in ERK-mediated mitogenesis of insulin signalling as we previously demonstrated [13–15].
How does cav-2 move into the nucleus? It has been well documented that movements of caveolin-containing vesicles are controlled by microtubules [56, 57]. Our investigation of the nuclear trafficking of cav-2 has shown that cav-2 is retro-transported to the nucleus through microtubules driven by kinesin molecular motor in response to insulin (Jeong K., Kwon H., Pak Y., manuscript in preparation).
Acknowledgments
This study was supported in part by grants from the National R&D Program for Cancer Control, Ministry for Health and Welfare, Republic of Korea (0920110) and the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (2010-0007897) to Y.P. H.K. is a recipient of the Science and Technology Scholarship from Korea Student Aid Foundation (2009–2010). K.J. is a recipient of the Graduate Research Fellowship from Gyeongsang National University (2009–2010).
Conflict of interest
The authors confirm that there are no conflicts of interest.
Supporting Information
Amino acid sequence alignment of rat cav-2 with human cav-2 using the LALIGN program. * indicate identical amino acid sequences.
References
- 1.Fra AM, Williamson E, Simons K, et al. De novo formation of caveolae in lymphocytes by expression of VIP21-caveolin. Proc Natl Acad Sci USA. 1995;92:8655–9. doi: 10.1073/pnas.92.19.8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu P, Rudick M, Anderson RG. Multiple functions of caveolin-1. J Biol Chem. 2002;277:41295–98. doi: 10.1074/jbc.R200020200. [DOI] [PubMed] [Google Scholar]
- 3.Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev. 2002;54:431–67. doi: 10.1124/pr.54.3.431. [DOI] [PubMed] [Google Scholar]
- 4.Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol. 2007;8:185–94. doi: 10.1038/nrm2122. [DOI] [PubMed] [Google Scholar]
- 5.Head BP, Insel PA. Do caveolins regulate cells by actions outside of caveolae. Trends Cell Biol. 2007;17:51–7. doi: 10.1016/j.tcb.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 6.Goetz JG, Lajoie P, Wiseman SM, et al. Caveolin-1 in tumor progression: the good, the bad and the ugly. Cancer Metastasis Rev. 2008;27:715–35. doi: 10.1007/s10555-008-9160-9. [DOI] [PubMed] [Google Scholar]
- 7.Lajoie P, Goetz JG, Dennis JW, et al. Lattices, rafts, and scaffolds: domain regulation of receptor signaling at the plasma membrane. J Cell Biol. 2009;185:381–5. doi: 10.1083/jcb.200811059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng Y, Venema VJ, Venema RC, et al. VEGF induces nuclear translocation of Flk-1/KDR, endothelial nitric oxide synthase, and caveolin-1 in vascular endothelial cells. Biochem Biophys Res Commun. 1999;256:192–7. doi: 10.1006/bbrc.1998.9790. [DOI] [PubMed] [Google Scholar]
- 9.Gaus K, Le Lay S, Balasubramanian N, et al. Integrin-mediated adhesion regulates membrane order. J Cell Biol. 2006;174:725–34. doi: 10.1083/jcb.200603034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanna E, Miotti S, Mazzi M, et al. Binding of nuclear caveolin-1 to promoter elements of growth-associated genes in ovarian carcinoma cells. Exp Cell Res. 2007;313:1307–17. doi: 10.1016/j.yexcr.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Razani B, Wang XB, Engelman JA, et al. Caveolin-2-deficient mice show evidence of severe pulmonary dysfunction without disruption of caveolae. Mol Cell Biol. 2002;22:2329–44. doi: 10.1128/MCB.22.7.2329-2344.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lahtinen U, Honsho M, Parton RG, et al. Involvement of caveolin-2 in caveolar biogenesis in MDCK cells. FEBS Lett. 2003;538:85–8. doi: 10.1016/s0014-5793(03)00135-2. [DOI] [PubMed] [Google Scholar]
- 13.Kim S, Pak Y. Caveolin-2 regulation of the cell cycle in response to insulin in Hirc-B fibroblast cells. Biochem Biophys Res Commun. 2005;330:88–96. doi: 10.1016/j.bbrc.2005.02.130. [DOI] [PubMed] [Google Scholar]
- 14.Kwon H, Jeong K, Pak Y. Identification of pY19-caveolin-2 as a positive regulator of insulin-stimulated actin cytoskeleton-dependent mitogenesis. J Cell Mol Med. 2009;13:1549–64. doi: 10.1111/j.1582-4934.2008.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwon H, Jeong K, Hwang EM, et al. Caveolin-2 regulation of STAT3 transcriptional activation in response to insulin. Biochim Biophys Acta. 2009;1793:1325–33. doi: 10.1016/j.bbamcr.2009.04.015. [DOI] [PubMed] [Google Scholar]
- 16.Zehorai E, Yao Z, Plotnikov A, et al. The subcellular localization of MEK and ERK–A novel nuclear translocation signal (NTS) paves a way to the nucleus. Mol Cell Endocrinol. 2010;314:213–20. doi: 10.1016/j.mce.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 17.González JM, Navarro-Puche A, Casar B, et al. Fast regulation of AP-1 activity through interaction of lamin A/C, ERK1/2, and c-Fos at the nuclear envelope. J Cell Biol. 2008;183:653–66. doi: 10.1083/jcb.200805049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taniura H, Glass C, Gerace L. A chromatin binding site in the tail domain of nuclear lamins that interacts with core histones. J Cell Biol. 1995;131:33–44. doi: 10.1083/jcb.131.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mariappan I, Parnaik VK. Sequestration of pRb by cyclin D3 causes intranuclear reorganization of lamin A/C during muscle cell differentiation. Mol Biol Cell. 2005;16:1948–60. doi: 10.1091/mbc.E04-02-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu JP, Baker J, Perkins AS, et al. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- 21.Jhun BH, Meinkoth JL, Leitner JW, et al. Insulin and insulin-like growth factor-I signal transduction requires p21ras. J Biol Chem. 1994;269:5699–704. [PubMed] [Google Scholar]
- 22.LeRoith D, Werner H, Beitner-Johnson D, et al. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev. 1995;16:143–63. doi: 10.1210/edrv-16-2-143. [DOI] [PubMed] [Google Scholar]
- 23.Di Cola G, Cool MH, Accili D. Hypoglycemic effect of insulin-like growth factor-1 in mice lacking insulin receptors. J Clin Invest. 1997;99:2538–44. doi: 10.1172/JCI119438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.El-Shewy HM, Lee MH, Obeid LM, et al. The insulin-like growth factor type 1 and insulin-like growth factor type 2/mannose-6-phosphate receptors independently regulate ERK1/2 activity in HEK293 cells. J Biol Chem. 2007;282:26150–7. doi: 10.1074/jbc.M703276200. [DOI] [PubMed] [Google Scholar]
- 25.Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–12. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 26.Baserga R, Hongo A, Rubini M, et al. The IGF-I receptor in cell growth, transformation and apoptosis. Biochim Biophys Acta. 1997;1332:F105–26. doi: 10.1016/s0304-419x(97)00007-3. [DOI] [PubMed] [Google Scholar]
- 27.Dupont J, Fernandez AM, Glackin CA, et al. Insulin-like growth factor 1 (IGF-1)-induced twist expression is involved in the anti-apoptotic effects of the IGF-1 receptor. J Biol Chem. 2001;276:26699–707. doi: 10.1074/jbc.M102664200. [DOI] [PubMed] [Google Scholar]
- 28.Dupont J, Khan J, Qu BH, et al. Insulin and IGF-1 induce different patterns of gene expression in mouse fibroblast NIH-3T3 cells: identification by cDNA microarray analysis. Endocrinology. 2001;142:4969–75. doi: 10.1210/endo.142.11.8476. [DOI] [PubMed] [Google Scholar]
- 29.Myers MG, Jr, Sun XJ, Cheatham B, et al. IRS-1 is a common element in insulin and insulin-like growth factor-I signaling to the phosphatidylinositol 3′-kinase. Endocrinology. 1993;132:1421–30. doi: 10.1210/endo.132.4.8384986. [DOI] [PubMed] [Google Scholar]
- 30.Sun XJ, Wang LM, Zhang Y, et al. Role of IRS-2 in insulin and cytokine signalling. Nature. 1995;377:173–7. doi: 10.1038/377173a0. [DOI] [PubMed] [Google Scholar]
- 31.Lehr S, Kotzka J, Herkner A, et al. Identification of major tyrosine phosphorylation sites in the human insulin receptor substrate Gab-1 by insulin receptor kinase in vitro. Biochemistry. 2000;39:10898–907. doi: 10.1021/bi000982k. [DOI] [PubMed] [Google Scholar]
- 32.Pronk GJ, McGlade J, Pelicci G, et al. Insulin-induced phosphorylation of the 46- and 52-kDa Shc proteins. J Biol Chem. 1993;268:5748–53. [PubMed] [Google Scholar]
- 33.Pandini G, Frasca F, Mineo R, et al. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J Biol Chem. 2002;277:39684–95. doi: 10.1074/jbc.M202766200. [DOI] [PubMed] [Google Scholar]
- 34.Al-Khalili L, Kotova O, Tsuchida H, et al. ERK1/2 mediates insulin stimulation of Na(+),K(+)-ATPase by phosphorylation of the alpha-subunit in human skeletal muscle cells. J Biol Chem. 2004;279:25211–8. doi: 10.1074/jbc.M402152200. [DOI] [PubMed] [Google Scholar]
- 35.Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. Biochem J. 2000;346:561–76. [PMC free article] [PubMed] [Google Scholar]
- 36.Vial C, Evans RJ. Disruption of lipid rafts inhibits P2×1 receptor-mediated currents and arterial vasoconstriction. J Biol Chem. 2005;280:30705–11. doi: 10.1074/jbc.M504256200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Torres VA, Tapia JC, RodrÌguez DA, et al. Caveolin-1 controls cell proliferation and cell death by suppressing expression of the inhibitor of apoptosis protein survivin. J Cell Sci. 2006;119:1812–23. doi: 10.1242/jcs.02894. [DOI] [PubMed] [Google Scholar]
- 38.Labrecque L, Nyalendo C, Langlois S, et al. Src-mediated tyrosine phosphorylation of caveolin-1 induces its association with membrane type 1 matrix metalloproteinase. J Biol Chem. 2004;279:52132–40. doi: 10.1074/jbc.M409617200. [DOI] [PubMed] [Google Scholar]
- 39.Pak Y, Paule CR, Bao YD, et al. Insulin stimulates the biosynthesis of chiro-inositol-containing phospholipids in a rat fibroblast line expressing the human insulin receptor. Proc Natl Acad Sci USA. 1993;90:7759–63. doi: 10.1073/pnas.90.16.7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stumpo DJ, Blackshear PJ. Cellular expression of mutant insulin receptors interferes with the rapid transcriptional response to both insulin and insulin-like growth factor I. J Biol Chem. 1991;266:455–60. [PubMed] [Google Scholar]
- 41.Girard M, McPherson PS. RME-8 regulates trafficking of the epidermal growth factor receptor. FEBS Lett. 2008;582:961–6. doi: 10.1016/j.febslet.2008.02.042. [DOI] [PubMed] [Google Scholar]
- 42.Roudabush FL, Pierce KL, Maudsley S, et al. Transactivation of the EGF receptor mediates IGF-1-stimulated shc phosphorylation and ERK1/2 activation in COS-7 cells. J Biol Chem. 2000;275:22583–9. doi: 10.1074/jbc.M002915200. [DOI] [PubMed] [Google Scholar]
- 43.Sasaoka T, Rose DW, Jhun BH, et al. Evidence for a functional role of Shc proteins in mitogenic signaling induced by insulin, insulin-like growth factor-1, and epidermal growth factor. J Biol Chem. 1994;269:13689–94. [PubMed] [Google Scholar]
- 44.Dalle S, Ricketts W, Imamura T, et al. Insulin and insulin-like growth factor I receptors utilize different G protein signaling components. J Biol Chem. 2001;276:15688–95. doi: 10.1074/jbc.M010884200. [DOI] [PubMed] [Google Scholar]
- 45.Park JY, Hwang EM, Yarishkin O, et al. TRPM4b channel suppresses store-operated Ca2+ entry by a novel protein-protein interaction with the TRPC3 channel. Biochem Biophys Res Commun. 2008;368:677–83. doi: 10.1016/j.bbrc.2008.01.153. [DOI] [PubMed] [Google Scholar]
- 46.Behrmann I, Smyczek T, Heinrich PC, et al. Janus kinase (Jak) subcellular localization revisited: the exclusive membrane localization of endogenous Janus kinase 1 by cytokine receptor interaction uncovers the Jak.receptor complex to be equivalent to a receptor tyrosine kinase. J Biol Chem. 2004;279:35486–93. doi: 10.1074/jbc.M404202200. [DOI] [PubMed] [Google Scholar]
- 47.Sciacca L, Costantino A, Pandini G, et al. Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene. 1999;18:2471–9. doi: 10.1038/sj.onc.1202600. [DOI] [PubMed] [Google Scholar]
- 48.Herbert TP, Kilhams GR, Batty IH, et al. Distinct signalling pathways mediate insulin and phorbol ester-stimulated eukaryotic initiation factor 4F assembly and protein synthesis in HEK 293 cells. J Biol Chem. 2000;275:11249–56. doi: 10.1074/jbc.275.15.11249. [DOI] [PubMed] [Google Scholar]
- 49.Kwon H, Pak Y. Prolonged tyrosine kinase activation of insulin receptor by pY27-caveolin-2. Biochem Biophys Res Commun. 2010;391:49–55. doi: 10.1016/j.bbrc.2009.10.159. [DOI] [PubMed] [Google Scholar]
- 50.Burack WR, Shaw AS. Live cell imaging of ERK and MEK: simple binding equilibrium explains the regulated nucleocytoplasmic distribution of ERK. J Biol Chem. 2005;280:3832–7. doi: 10.1074/jbc.M410031200. [DOI] [PubMed] [Google Scholar]
- 51.Cohen AW, Razani B, Wang XB, et al. Caveolin-1-deficient mice show insulin resistance and defective insulin receptor protein expression in adipose tissue. Am J Physiol Cell Physiol. 2003;285:C222–35. doi: 10.1152/ajpcell.00006.2003. [DOI] [PubMed] [Google Scholar]
- 52.Xie L, Frank PG, Lisanti MP, et al. Endothelial cells isolated from caveolin-2 knockout mice display higher proliferation rate and cell cycle progression relative to their wild type counterparts. Am J Physiol Cell Physiol. 2010;298:C693–701. doi: 10.1152/ajpcell.00401.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McGinnis S, Madden TL. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004;32:W20–5. doi: 10.1093/nar/gkh435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Minetti C, Sotgia F, Bruno C, et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet. 1998;18:365–8. doi: 10.1038/ng0498-365. [DOI] [PubMed] [Google Scholar]
- 55.Schlegel A, Arvan P, Lisanti MP. Caveolin-1 binding to endoplasmic reticulum membranes and entry into the regulated secretory pathway are regulated by serine phosphorylation. Protein sorting at the level of the endoplasmic reticulum. J Biol Chem. 2001;276:4398–408. doi: 10.1074/jbc.M005448200. [DOI] [PubMed] [Google Scholar]
- 56.Mundy DI, Machleidt T, Ying YS, et al. Dual control of caveolar membrane traffic by microtubules and the actin cytoskeleton. J Cell Sci. 2002;115:4327–39. doi: 10.1242/jcs.00117. [DOI] [PubMed] [Google Scholar]
- 57.Pelkmans L, Helenius A. Endocytosis via caveolae. Traffic. 2002;3:311–20. doi: 10.1034/j.1600-0854.2002.30501.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Amino acid sequence alignment of rat cav-2 with human cav-2 using the LALIGN program. * indicate identical amino acid sequences.