

Abstract
In genetically predisposed individuals, ingestion of wheat gliadin provokes a T-cell-mediated enteropathy, celiac disease. Gliadin fragments were previously reported to induce phenotypic maturation and Th1 cytokine production by human dendritic cells (DCs) and to boost their capacity to stimulate allogeneic T cells. Here, we monitor the effects of gliadin on migratory capacities of DCs. Using transwell assays, we show that gliadin peptic digest stimulates migration of human DCs and their chemotactic responsiveness to the lymph node-homing chemokines CCL19 and CCL21. The gliadin-induced migration is accompanied by extensive alterations of the cytoskeletal organization, with dissolution of adhesion structures, podosomes, as well as up-regulation of the CC chemokine receptor (CCR) 7 on cell surface and induction of cyclooxygenase (COX)-2 enzyme that mediates prostaglandin E2 (PGE2) production. Blocking experiments confirmed that gliadin-induced migration is independent of the TLR4 signalling. Moreover, we showed that the α-gliadin-derived 31–43 peptide is an active migration-inducing component of the digest. The migration promoted by gliadin fragments or the 31–43 peptide required activation of p38 mitogen-activated protein kinase (MAPK). As revealed using p38 MAPK inhibitor SB203580, this was responsible for DC cytoskeletal transition, CCR7 up-regulation and PGE2 production in particular. Taken together, this study provides a new insight into pathogenic features of gliadin fragments by demonstrating their ability to promote DC migration, which is a prerequisite for efficient priming of naive T cells, contributing to celiac disease pathology.
Keywords: celiac disease, gliadin, dendritic cell, chemotaxis, migration
Introduction
Celiac disease, a chronic inflammatory disorder of the small intestine, is caused by a pathological immune response to ingested wheat gliadin. The disease develops in genetically susceptible individuals who bear human leukocyte antigen (HLA)-DQ2 or HLA-DQ8 molecules, and is characterized by villous atrophy and crypt hyperplasia, which cause malabsorbtion and myriad of other clinical symptoms [1–3]. In the gut mucosa of patients with active celiac disease, proteolytically resistant gliadin fragments cross the small intestinal epithelium, and are presented to DQ2- or DQ8-restricted CD4+ T cells. The immunogenicity of gliadin peptides generally increases on deamidation by tissue transglutaminase. This enhances binding affinity of the peptides for HLA-DQ2 or HLA-DQ8 molecules on antigen-presenting cells (APCs), such as dendritic cells (DCs) or macrophages [4–6]. An example of such immunodominant CD4+ T-cell peptide is the 33mer peptide, encompassing residues 56–88 of α-gliadin [7]. Once activated by peptide-loaded HLA molecules of APCs, gliadin-specific CD4+ T cells expand and produce high levels of INF-γ and other pro-inflammatory cytokines which in turn stimulate other cells, such as natural killer T cells and CD8+ intraepithelial T lymphocytes in the intestinal mucosa, thus amplifying inflammation and damage of the small intestine [8, 9].
Nevertheless, growing evidence indicates that innate immune cells, including epithelial cells [10, 11], monocytes/macrophages [12–14] and DCs [15–17] contribute directly to disease pathogenesis. For example, the non-immunodominant α-gliadin-derived 31–43 peptide, which does not induce T-cell-specific responses in the gut, was reported to have immune-stimulating effects [10, 18]. The activation of innate immune response might, indeed, provide the danger signal that contributes to the breakdown of oral tolerance to wheat gliadin, thus participating in the initiation of disease pathogenesis [19, 20].
DCs are powerful APCs that bridge innate and adaptive immune responses. In an immature stage, they reside in peripheral tissues, where they sample antigens. Upon encounter of molecules that signal ‘danger’, DCs mature to the functional phenotype of APCs, capable of stimulating T cells, and migrate to lymphoid organs, where they display complexes of MHC molecules with antigen-derived peptides. This allows selection of rare circulating naive T cells for their expansion and differentiation into effector T cells [21].
The ability of DCs to migrate into draining lymph nodes (LNs) determines their capacity to activate naive T cells. Importantly, not all stimuli that activate DC maturation do induce sufficient DC migration. The induction of DC migration depends on alterations of the cytoskeletal organization and cell adhesion, as well as on the changes in expression and/or sensitization of chemokine receptors [22, 23]. The prominent features of immature/early mature DCs are low-speed migration and strong interaction with extracellular matrix components via podosomes [24–27], the structures consisting of actin-dense core surrounded by a ring of vinculin, paxillin, talin and other integrin-linked adaptor molecules, respectively [28]. The presence of podosomes appears to be incompatible with high-speed migration observed in mature DCs [24]. In contrast, highly motile mature DCs reduce the number and strength of cell-substratum contacts and possess the characteristic actin-rich protrusions called dendrites, or veils, which maximize the contact area for scanning of the potentially reactive naive T cells [27, 29, 30].
DC migration to LNs is directed by the chemokine receptor CCR7 [31], expression of which is up-regulated during DC maturation. The receptor responds to two ligands, the chemokines CCL19 and CCL21 [32–34], which are produced by peripheral lymphatic endothelial cells and LN stromal cells and are responsible for guiding of maturing DCs [22]. The homing CCR7 receptor, however, can be expressed in a biologically ‘insensitive’ state and requires additional signals, such as cysteinyl leukotrienes and/or prostaglandin E2 (PGE2) for sensitization [35–37]. The mechanism, by which these signals alter CCR7 function, is unclear. Most probably, it involves an alteration of the signalling cascades activated upon CCR7 binding of ligands [23, 38] and/or mediation of the DC transition from the adhesive to the high migratory phenotype [24].
Human DCs that are treated with gliadin peptic fragments enhance expression of maturation markers, Th1 cytokine production and in vitro capacity to stimulate allogeneic T cells [16]. Nevertheless, no data are currently available on the migratory state and chemotactic responsiveness of DCs that are stimulated by gliadin fragments. As migration of DCs to LNs is essential for the selection, expansion and differentiation of circulating naive T cells, the present study was designed to investigate the effect of gliadin peptic fragments on migratory capacity of DCs and on evaluating the signalling mechanisms that underlie these changes.
Materials and methods
Antibodies and reagents
The following anti-human mAbs were used: CD14-APC-ALEXA FLUOR 750, CD11c-APC and CD83-PE (CALTAG Laboratories, Burlingame, CA, USA); CD86-PE (Immunotech, Marseille, France); HLA DR-FITC and CD40-FITC (BD Biosciences Pharmingen, San Diego, CA, USA) and CCR7-PE (R&D System, Minneapolis, MN, USA). Rabbit polyclonal COX-2 Ab was purchased from Cell Signaling Technology (Beverly, MA, USA). The p38 MAPK inhibitor SB203580 (Sigma, St. Louis, MO, USA) was dissolved in DMSO (Sigma). CCL19 and CCL21 were purchased from PeproTech (London, UK).
α-gliadin-derived peptides and gliadin peptic fragments generation
The α-gliadin-derived 33mer peptide [amino acid (AA) 56–88, Leu-Gln-Leu-Gln-Pro-Phe-Pro-Gln-Pro-Gln-Leu-Pro-Tyr-Pro-Gln-Pro-Gln-Leu-Pro-Tyr-Pro-Gln-Pro-Gln-Leu-Pro-Tyr-Pro-Gln-Pro-Gln-Pro-Phe] and 31–43 peptide (AA 31–43, Leu-Gly-Gln-Gln-Gln-Pro-Phe-Pro-Pro-Gln-Gln-Pro-Tyr) were synthesized using the Fmoc/tBu protection strategy on aminoethyl copoly (styrene-1% divinylbenzene) resin with a Knorr linker. After cleavage from the resin, the peptides were purified using high-performance liquid chromatography and characterized by AA analysis and liquid chromatography/mass spectrometry (System Waters 2690 Separation Module and Waters 2487 Dual λ Absorbance Detector, connected to a Micromass Platform L.C., Waters, Milford, MA, USA).
Peptic fragments of gliadin (Sigma) were prepared by incubation of 7 ml of 1% gliadin in 0.1 M HCl, pH 1.8 with 5 ml of pepsin-agarose gel (ICN, Biomedicals, Inc., OH, USA) at 37°C for 45 min. Enzymatic digestion was stopped by removal of the pepsin-agarose gel by a 10-min. centrifugation at 1500 ×g, and the digest was divided into aliquots and frozen at –20°C.
To exclude any contamination by lipopolysaccharide (LPS), the gliadin digest and all other reagents underwent the E-toxate test (Sigma), and only reagents that were below the LPS detection limit were used in the study.
Generation, handling and treatment of DCs
DCs were generated, handled and treated as described [16]. In brief, human PBMCs were isolated from buffy coats of healthy donors (provided by the Department of Blood Transfusion at Thomayer’s Hospital, Prague, Czech Republic) by Ficoll-Paque plus gradient centrifugation (GE Healthcare, Uppsala, Sweden). Cells were incubated at a concentration of 3 × 106 cells/ml in 75-cm2 plastic culture flasks (Nunc, Roskilde, Denmark). After 2 hrs, the non-adherent fraction of cells was thoroughly washed away, and the isolated adherent monocytes were cultured in the presence of human granulocyte macrophage-colony stimulating factor (500 U/ml; Molgramostin, Gentaur, Belgium) and recombinant human interleukin (IL)-4 (20 ng/ml; PeproTech) in RPMI 1640 (BioWhittaker, Lonza, Belgium), supplemented with L-glutamine (2 mM, Sigma), penicillin/streptomycin (100 U penicillin/ml, 100 μg streptomycin/ml) and 10% fetal bovine serum (FBS) (BioWhittaker) at 37°C in a 5% CO2 atmosphere.
After 5–6 days, the generated DCs were harvested and seeded at 1 × 106 cells/ml in 24-well plates for treatment with gliadin digest (at 100 or 200 μg/ml), α-gliadin-derived synthetic 33mer or 31–43 peptide (100 μg/ml), or LPS (1 μg/ml; E. coli, Sigma) for the indicated time intervals. DC generation and maturation were assessed by flow cytometry. The expression of MHC class II and costimulatory molecules and of DC-specific markers was consistent with previous findings [16].
To block the TLR4-signalling pathway, DCs were pre-incubated with blocking anti-TLR4 mouse mAb (20 μg/ml, eBioscience, San Diego, CA, USA) for 1 hr at 37°C and the cells were further stimulated by gliadin fragments (200 μg/ml) or LPS (1 μg/ml) in the presence of antibody for 24 hrs.
To block p38 MAPK signalling, DCs were pre-incubated with 10 μM or 20 μM SB203580 for 30 min. at 37°C in a 5% CO2 atmosphere. The cells were further stimulated with the indicated agent in the presence of inhibitor for 12, 24 or 48 hrs, respectively, as indicated in the text.
DC migration in transwell assays
DC migration was measured in 24-well transwell cell culture chambers with 5-μm pore size polycarbonate filters (Corning Costar, Cambridge, MA, USA). The lower chambers of the transwell plates were filled with 600 μl of serum-free RPMI 1640 medium (BioWhittaker), with or without CCL19 or CCL21 (200 ng/ml). In total, 2 × 105 DCs, diluted in 100 μl serum-free medium were deposited in the upper chamber of the transwell inserts. After 2 hrs of incubation at 37°C in a 5% CO2 atmosphere, the transwell inserts were removed, and cells in the lower chamber were stained with Hoechst 33342 (Invitrogen, Camarillo, CA, USA) and counted with the CompuCyte iCys Research imaging cytometer. The migration index, defined as the fold increase of number of migrated DCs over the number of untreated, migrated DCs in the absence of chemokines, was used to quantify cell migration.
Fluorescence microscopy of DC cytoskeleton
For fluorescence microscopy of the DC cytoskeleton, cover slips were coated with fibronectin (20 μg/ml, Roche, Mannheim, Germany) in PBS for 1 hr at room temperature. Then, 2 × 105 DCs were seeded on fibronectin-coated cover slips and treated with the indicated agents for 24 hrs at 37°C in a 5% CO2 atmosphere before being fixed with 4% paraformaldehyde in PBS (20 min., room temperature). Cells were permeabilized with 0.1% Triton X-100 in PBS (5 min.) and blocked with 10% AB serum in PBS for 30 min. The cells were incubated with anti-vinculin Ab (1:1000, Sigma) in 2% bovine serum albumin (BSA)-PBS for 45 min., followed by incubation with Alexa Fluor 488-labeled secondary Ab (1:1000, Molecular Probes, Eugene, OR, USA) and tetramethylrhodamine isothiocyanate (TRITC)-conjugated phalloidin (0.5 μg/ml, Sigma) in 2% BSA-PBS for 45 min. Images were taken using an Olympus CellR fluorescence microscope (Olympus, Hamburg, Germany).
RT-PCR
The CCR7, COX-2 and GAPDH mRNA was detected by RT-PCR after total RNA isolation with the NucleoSpinRNA II kit (Macherey-Nagel, Bethlehem, PA, USA). The RNA was reverse-transcribed to cDNA with random nonamers (Sigma-Aldrich, St. Louis, MO, USA), and amplification of cDNA was performed with primers for the human CCR7 and COX-2 (SA Biosciences, Frederick, MD, USA); and GAPDH (Sigma Genosys, St. Louis, MO, USA) genes on a thermocycler T Gradient (Biometra, Goettingen, Germany).
FACS analysis of CCR7 surface expression
CCR7 surface expression was analysed after DC incubation with CCR7-PE Ab (R&D System) on ice for 30 min. Cells were washed and resuspended in ice-cold PBS with 0.1% NaN3 and subjected to flow cytometry on a BD LSR II cytometer (BD Biosciences, San Jose, CA, USA). Staining with Hoechst 33258 (Invitrogen) was performed to assess cell viability. Mean fluorescence intensity (MFI) values of the samples were determined using flow cytometry analysis software (FlowJo Version 8.3.3, Tree Star, Inc., Ashland, OR, USA)
COX-2 protein expression and PGE2 production
DCs were seeded at 1 × 106 cells/ml in 24-well plate, and left untreated for 24 hrs or exposed to gliadin digest (200 μg/ml) or LPS (1 μg/ml) added at 0, 12 or 18 hrs after seeding of immature DCs (iDC) into plates. This resulted in treatment of DCs for 24, 12 and 6 hrs, respectively. After 24 hrs of cell culture, all DCs were taken for COX-2 and/or PGE2 production analysis; the supernatants were subjected to enzyme immunoassay (EIA) to determine PGE2 production, and cell pellets were used to analyse COX-2 protein expression.
DC pellets were washed with PBS and lysed in 70 μl of ice-cold lysis buffer containing 1% Nonidet P-40, 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 10 mM ethylenediaminetetraacetic acid, 10 mM Na4P2O7, 1 mM Na3VO4, 50 mM NaF, 10 nM CalyculinA and Complete Mini protease inhibitors per sample. Samples were then incubated for 15 min. on ice, briefly vortexed, clarified by centrifugation (3 min., 10,000 ×g) and mixed with Laemmli buffer. After being heated for 5 min. at 95°C, the proteins were separated by 10% SDS-PAGE, transferred onto a nitrocellulose membrane, and probed overnight with anti-Cox2 Ab (1:2000, Cell Signaling Technology). Membranes were revealed by HRP-conjugated secondary Ab (1:4000, GE Healthcare) using the West Femto Maximum Sensitivity Substrate (Pierce, Rockford, IL, USA). Western blot signals were detected using the LAS-1000 (Luminiscence Analyzing System, Fuji, Tokyo, Japan) and processed with AIDA 1000/1D Image Analyzer software, version 3.28 (Raytest Isotopenmessgeraete GmbH, Straubenhardt, Germany). After stripping, the membranes were reprobed with anti-actin Ab (1:5000, Abcam, Cambridge, MA, USA).
The supernatants of DCs were used to determine PGE2 levels by EIA (Cayman Chemicals, Ann Arbor, MI, USA), according to the manufacturer’s instructions.
Statistical analysis
Significance of differences in values was assessed by parametric Student’s t-test. A value of P < 0.05 was considered significant.
Results
Gliadin fragments promote DC migration and chemotactic responsiveness to CCL19 and CCL21
To examine whether gliadin fragments induce DC migration and chemotactic responsiveness to CCL19 or CCL21, migration of human monocyte derived DCs after 24 or 48 hrs exposure to gliadin fragments was measured in an in vitro chemotaxis assay. Unstimulated iDC, or DCs after stimulation with gliadin fragments or LPS, were loaded into the upper chamber of transwell plate, while serum-free medium with or without chemokines was placed into the lower chamber. Already in the absence of chemokines, DCs treated with gliadin or LPS exhibited higher spontaneous migration through the filter, as compared to control iDC (Fig. 1A). The effect of gliadin on spontaneous migration was dose dependent (100 versus 200 μg/ml of gliadin fragments) and was more pronounced after 48 hrs of stimulation. Moreover, the addition of CCL19 or CCL21 chemokine to the lower transwell chamber led to a further increase of the numbers of migrating DCs, following exposure to gliadin or LPS. In contrast, migration of control untreated iDC increased only slightly (Fig. 1A). These results suggest that gliadin fragments, like LPS, not only promoted spontaneous migration of DCs but also enhanced DC chemotactic response to the LN-homing chemokines CCL19 and CCL21.
Fig 1.
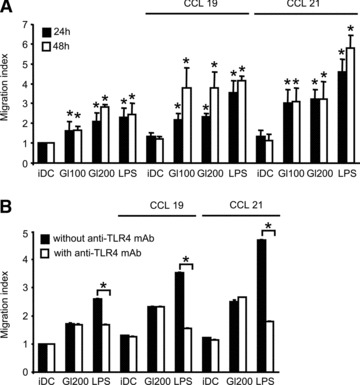
Gliadin fragments promote spontaneous as well as chemotactic DC migration. (A) Spontaneous DC migration (migration of DCs towards the chamber without chemokines) and chemotactic DC migration (migration towards the chamber supplemented with 200 ng/ml of CCL19 or CCL21) were measured after 24 or 48 hrs of cell exposure to gliadin fragments (Gl100, 100 μg/ml; Gl200, 200 μg/ml), or LPS (1 μg/ml). Data are expressed as a migration index and represent the means ± S.D. of at least six independent experiments. *P < 0.05 versus iDC (unstimulated DCs). (B) To confirm that migration was not due to any potential LPS contamination of the gliadin digest, DCs were pretreated with blocking TLR4 mAb at 20 μg/ml for 1 hr and then stimulated for 24 hrs with gliadin fragments (Gl200 μg/ml) or LPS (1 μg/ml). The DC migration was assessed as above. Data represent the means ± S.D. for at least two independent experiments. With versus without anti-TLR4 mAb; *P < 0.05.
In order to exclude that the stimulatory activity of gliadin digest might have been biased by a potential LPS contamination, we employed the anti-TLR4 blocking antibody, thereby disrupting the LPS-dependent signal transduction. When DCs were treated by LPS in the presence of this antibody, LPS-induced spontaneous, as well as chemotactic, migration was effectively inhibited. In turn, no effect of anti-TLR4 on gliadin-induced DC migratory behaviour was observed (Fig. 1B), showing that gliadin-induced migratory behaviour of DCs was not due to LPS contamination.
Gliadin fragments induce cytoskeletal remodelling
Results obtained from the transwell assays revealed that gliadin fragments enhanced DC migration even in the absence of chemokines. We, therefore, supposed that gliadin fragments promote DC cytoskeletal changes and thereby mediate the transition of DCs from an adhesive to a highly migratory phenotype [24]. To verify the association between the induction of DC migration and the reorganization of the cytoskeleton, morphology of cells treated with gliadin fragments or LPS was analysed by staining for F-actin and for the cytoskeletal protein vinculin. Unstimulated control iDC exhibited the classical podosome structures, characterized by the actin-dense core surrounded by a ring of vinculin (Fig. 2A). After treatment of DCs with gliadin fragments (200 μg/ml) or LPS (1 μg/ml) for 24 hrs, an induction of actin-rich protrusions, as well as substantial podosome dissolution, were observed (Fig. 2A). The extent of the podosome loss induced by gliadin fragments was dose-dependent (100 versus 200 μg/ml), as quantified in Figure 2B. These data, thus, demonstrate that gliadin fragments induce cytoskeletal remodelling and mediate the morphological transition from iDC to mature DC.
Fig 2.
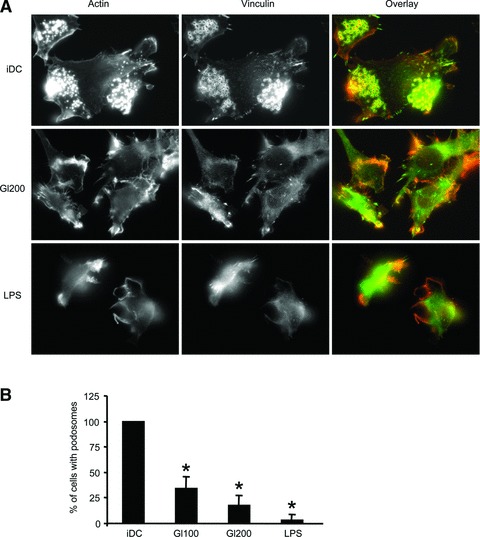
Gliadin fragments induce cytoskeletal remodelling in DCs. (A) DCs plated on fibronectin-coated cover slips were stimulated with gliadin fragments (Gl200, 200 μg/ml) or LPS (1 μg/ml) for 24 hrs. Following fixation with 4% paraformaldehyde, the cells were stained with TRITC-conjugated phalloidin to detect F-actin (red) and anti-vinculin Ab (green). The micrographs are representative of at least six independent experiments. (B) The percentage of cells with podosomes was determined after cell treatment with gliadin fragments (Gl100, 100 μg/ml; Gl200, 200 μg/ml), or LPS (1 μg/ml) for 24 hrs. Results represent the means ± S.D. of at least three independent experiments, in which at least 100 cells per condition per experiment were counted (the percentage of cells containing podosomes of untreated control iDC was arbitrarily set to 100%). *P < 0.05 versus iDC.
Gliadin fragments up-regulate CCR7 expression in DCs
Since gliadin fragments also promoted chemotactic responsiveness of DCs to CCL19 and CCL21 chemokines, we next examined whether gliadin fragments induce the expression of the chemokine receptor CCR7. As assessed by RT-PCR, gliadin digest up-regulated CCR7 mRNA expression after 24 hrs of cell stimulation (Fig. 3A). These data were confirmed by flow cytometry, which revealed enhanced surface expression of CCR7 after 24 hrs of cell stimulation with gliadin fragments (Fig. 3B). Moreover, after prolonged stimulation for 48 hrs, surface expression of CCR7 induced by gliadin fragments was even more pronounced, as observed for both of the gliadin digest doses (100 and 200 μg/ml), (Fig. 3B). This demonstrates that gliadin fragments promote chemotactic responsiveness of DCs through up-regulation of CCR7 expression.
Fig 3.
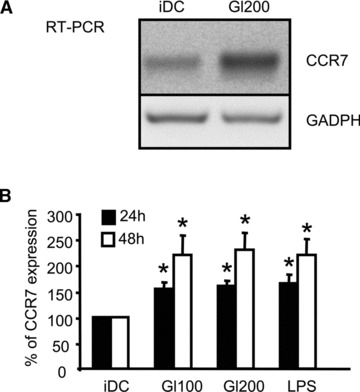
Gliadin fragments up-regulate CCR7 in DCs. (A) Total RNA was isolated from unstimulated control iDC or DCs stimulated with gliadin fragments (Gl200, 200 μg/ml) for 24 hrs. The levels of CCR7 mRNA and total GAPDH mRNA (internal control) were determined by RT-PCR. The shown data are representative of four independent experiments that yield similar results. (B) After cell stimulation for 24 or 48 hrs with gliadin fragments (Gl100, 100 μg/ml; Gl200, 200 μg/ml), or LPS (1 μg/ml), the surface expression of CCR7 was determined by flow cytometry. Data represent the means ± S.D. of at least eight independent experiments, in which the MFI of untreated control iDC was arbitrarily set to 100%. *P < 0.05 versus iDC.
Gliadin fragments up-regulate COX-2 expression, leading to PGE2 production
Enhancement of surface expression of CCR7 is necessary but not sufficient for effective chemotactic migration of DCs toward CCL19 or CCL21 [36, 38, 39]. Thus, we examined the expression of the inducible and rate-limiting enzyme in prostaglandin synthesis, COX-2 [40, 41] and prostaglandin PGE2 production by gliadin-treated cells.
In cells treated with gliadin fragments or LPS, COX-2 mRNA level increased after 6 hrs, after which it slowly decreased throughout 24 hrs (Fig. 4A). The same kinetics was also observed for the COX-2 protein level, as determined by Western blots of cell lysates (Fig. 4B).
Fig 4.
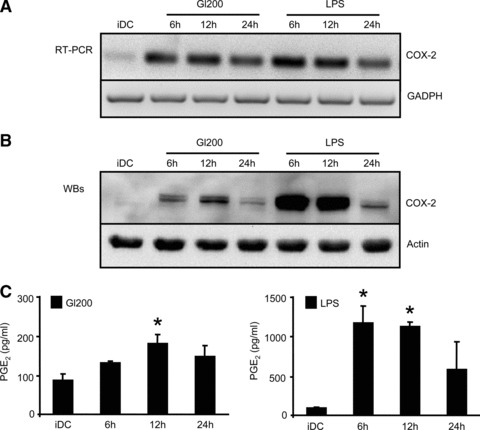
Gliadin fragments up-regulate COX-2 expression and PGE2 production. (A) After DC stimulation with gliadin fragments (Gl200, 200 μg/ml) or LPS (1 μg/ml), COX-2 mRNA levels were determined by RT-PCR. GAPDH mRNA was used as an internal control. The shown data are representative of four independent experiments that gave similar results. (B) COX-2 protein levels and total actin (loading control) in whole cell lysates were detected by Western blot. Results are representative of at least six independent experiments. (C) Levels of PGE2 in culture supernatants were measured by EIA. The shown data are representative experiment of two independent experiments. *P < 0.05 versus iDC.
To verify the functionality of the induced COX-2 enzyme, we further measured PGE2 production by DCs exposed to gliadin fragments or LPS, using EIA. Indeed, PGE2 synthesis was induced by gliadin fragments, albeit at markedly lower levels (reaching 200 pg/ml in 12 hrs), as compared to LPS treatment (reaching 1200 pg/ml in 12 hrs), (Fig. 4C). The kinetics of PGE2 production, as well as the amount of secreted PGE2, correlated well with COX-2 protein levels detected by Western blot (Fig. 4B versus C). Hence, gliadin fragments promote expression of functional COX-2 enzyme leading to an increase in PGE2 production.
α-gliadin-derived 31–43 peptide is an active migration-inducing component of the digest
We sought to identify the active component of the mixture of gliadin fragments responsible for induction of DC migration. Therefore, we tested the capacity of the previously characterized α-gliadin-derived 31–43 and 33mer peptides [7, 18] to induce DC migration. As shown in Figure 5, stimulation of cells with 100 μg/ml of the synthetic 31–43 peptide for 24 hrs promoted spontaneous as well as chemotactic DC migration. On the other hand, 24 hrs treatment with 100 μg/ml of the synthetic 33mer peptide did not activate DC chemotactic responsiveness to CCL19 and CCL21. Moreover, CCL21-induced migration of iDC into the lower chamber of transwell plate was dampened by the DC exposition for 24 hrs to the 33mer peptide. Furthermore, when DCs were treated simultaneously with both peptides (mixed at a ratio 1:1), the stimulatory effect of the 31–43 peptide on DC chemotactic migration was inhibited in the presence of the 33mer peptide (Fig. 5). Hence, the immune stimulating 31–43 peptide, rather than the 33mer peptide, known to be immunodominant for CD4+ T cells, was the active component of the DC migration-inducing gliadin digest.
Fig 5.
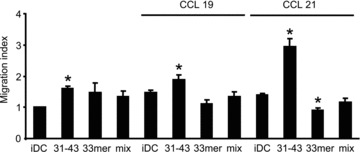
The α-gliadin-derived 31–43 peptide is an active migration-inducing component of the digest. DC migration was measured in transwell assays at 24 hrs of cell treatment with the synthetic 31–43 or 33mer peptides (100 μg/ml) or a mixture of both peptides (100:100 μg/ml, ratio 1:1) for 24 hrs. Data are expressed as the means ± S.D. of at least five independent experiments. *P < 0.05 versus iDC.
Gliadin signalling through the p38 MAPK pathway promotes DC migration, cytoskeletal remodelling, CCR7 expression and PGE2 production
To corroborate on the mechanism of induction of DC migration and chemotactic responsiveness by gliadin fragments, we sought to characterize the role of p38 MAPK in this process.
As shown in Figure 6, when the p38 MAPK inhibitor SB203580 was present during DC stimulation by gliadin fragments or LPS, induction of spontaneous (Fig. 6A), as well as CCL19- and CCL21-stimulated chemotactic migration was substantially impaired (Fig. 6B and C). Diminished numbers of DCs migrating towards the lower transwell chamber with the chemokines were observed upon 24 hrs of cell stimulation with gliadin fragments or LPS in the presence of SB203580, as compared to the numbers of cells migrating after stimulation in the absence of the inhibitor. After 48 hrs both spontaneous, as well as chemotactic DC migration stimulated by gliadin fragments or LPS, were markedly reduced. Moreover, a similar requirement for the p38 MAPK activity was also revealed for DC migration and chemotactic responsiveness induced by the 31–43 peptide (Fig. 6A–C). Importantly, viability of DCs after treatment with SB203580 (20 μM) was verified by flow cytometry upon Hoechst 33258 staining. The viability ranged between 84–88% in 24 hrs and 82–84% in 48 hrs of DC incubation for all samples. No statistically significant difference in cell viability was revealed by Student’s t-test (P < 0.05) using data from three independent flow cytometry experiments.
Fig 6.
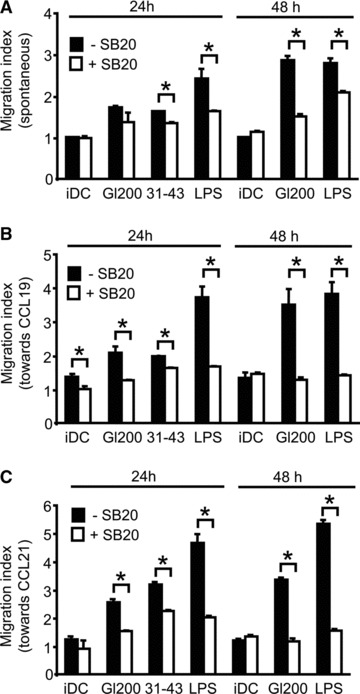
p38 MAPK activation is crucial for gliadin-induced DC migration. Following 30 min. of pre-incubation with 20 μM SB203580 (+SB20) or without treatment with SB203580 (–SB20), DCs were exposed to gliadin fragments (Gl200, 200 μg/ml) or LPS (1 μg/ml) for 24 or 48 hrs, or to the synthetic 31–43 peptide (100 μg/ml) for 24 hrs, washed, and evaluated for spontaneous (A) and chemotactic migration (B, C) in transwell assays, respectively. Data represent the means ± S.D. for at least three independent experiments. With versus without 20 μM SB203580 (SB20); *P < 0.05.
To corroborate these results, therefore, we dissected the role of p38 MAPK signalling pathway in gliadin-induced DC migration. First, we assessed the blocking effect of SB203580 on the dissolution of the adhesive structures, podosomes. Activation of p38 MAPK was required for podosome loss induced by gliadin fragments or LPS (Fig. 7A and B). Moreover, Figure 7C documents a similar requirement for p38 activity in the surface up-regulation of CCR7 triggered by gliadin fragments or LPS. The p38 MAPK inhibitor prevented up-regulation of CCR7 induced by gliadin fragments or LPS, respectively. Finally, we demonstrated the requirement for the p38 activation in COX-2 induction and PGE2 synthesis (Fig. 7D and E).
Fig 7.
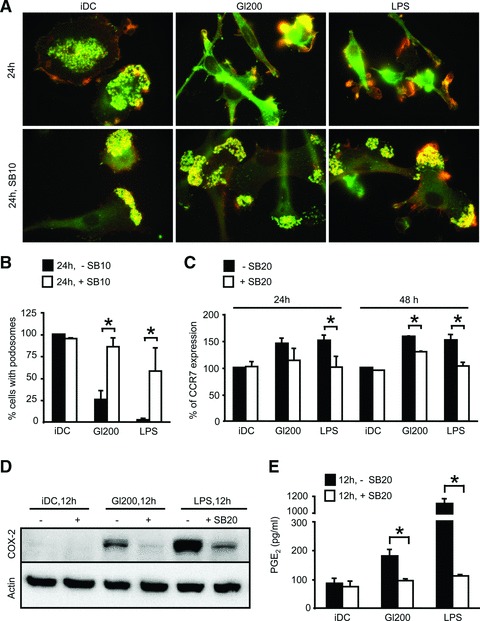
Gliadin fragments signalling through the p38 MAPK pathway promotes cytoskeletal remodelling, CCR7 up-regulation and PGE2 production. (A, B) DCs plated on fibronectin-coated cover slips were pretreated with 10 μM SB203580 (+SB10) for 30 min. or left without SB203580 treatment (–SB10), exposed to gliadin fragments (Gl200, 200 μg/ml) or LPS (1 μg/ml) for 24 hrs, and stained for F-actin (red) and vinculin (green). The percentage of cells with podosomes was counted from at least 100 cells per condition per experiment (the percentage of cells containing podosomes of untreated control iDC was arbitrarily set to 100%) and the shown data represent the means ± S.D. for three independent experiments. With versus without 10 μM SB203580 (SB10); *P < 0.05; (C) Following 30 min. of pretreatment with 20 μM SB203580 (+SB20) or without pretreatment (–SB20), cells were exposed to gliadin fragments (Gl200, 200 μg/ml) or LPS (1 μg/ml) for 24 or 48 hrs, and the surface expression of CCR7 was determined by flow cytometry. Data represent the means ± S.D. of at least four independent experiments, in which the MFI of untreated control iDC was arbitrarily set to 100%. With versus without 20 μM SB203580 (SB20); *P < 0.05; (D, E) Cells were treated with 20 μM SB203580 (+SB20) or not (–SB20) for 30 min. prior to exposure to gliadin fragments (Gl200, 200 μg/ml) or LPS (1 μg/ml). After 12 hrs, COX-2 protein levels and total actin (loading control) in whole cell lysates were detected by Western blot (D), and the level of PGE2 in the culture supernatants was measured by EIA (E). The Western blot is representative of four independent experiments that gave similar results. The PGE2 levels are from representative experiment of two independent experiments, performed in duplicate. With versus without 20 μM SB203580 (SB20); *P < 0.05.
In summary, these results document the critical role of the p38 MAPK signalling in gliadin-induced DC migration via control of cytoskeletal remodelling, surface CCR7 up-regulation and induction of COX-2 and PGE2 synthesis.
Discussion
DCs have a crucial role in maintaining the balance between immunity and tolerance via continuous sampling, transport and presentation of both pathogenic and harmless antigens. Under normal, steady-state conditions, DCs continually migrate from the periphery to LNs, which might contribute to the tolerance of T cells against self and non-dangerous antigens [42, 43]. DCs can, however, be mobilized by molecules signalling ‘danger’ to mature and to increase their migration from the periphery to the LNs, which consequently leads to the generation of adaptive immune responses [22, 43]. We found that fragments resulting from peptic digestion of the food component gliadin promote migration of DCs in vitro. The gliadin peptic fragments significantly enhanced the migration of DCs in the presence, as well as in the absence, of the LN-homing chemokines CCL19 and CCL21. We further demonstrated that the non-immunodominant α-gliadin-derived 31–43 peptide is the active migration-inducing component of the digest.
It has, indeed, been recently suggested that stimulation of intestinal DCs by inflammatory tissue signals and their trafficking into the mesenteric LNs following gliadin ingestion, may result in the induction of anti-gliadin immune response. Importantly, we show here that migratory phenotype of DCs results directly from DC exposure to gliadin fragments, without the need for involvement of pro-inflammatory mediators in the intestinal environment [19]. The question how the inflammation is controlled in healthy individuals remains to be answered. Gliadin fragments activate innate immune cells of all individuals, irrespective of HLA-DQ2 or HLA-DQ8 genotype, or activity of the disease [14, 17, 44]. Nevertheless, the in vitro production of tumour necrosis factor-α and IL-8 by monocytes and the stimulation of autologous T cells by gliadin-activated DCs is higher in active celiac patients, compared to other groups, suggesting that immune cells of healthy controls may dampen more effectively the gliadin-induced responses [14, 17]. In genome-wide association studies of celiac disease, indeed, the risk regions which harbour genes controlling immune responses were identified [45–48]. In addition, the myosin IXB (MYO9B) gene, proposed to regulate actin remodelling in epithelial enterocytes and thus the intestinal barrier permeability and flux of gliadin fragments into the deeper mucosal layer, has been reported to be genetically associated in a Dutch [49], but not in a British cohort [50]. Other mucosal factors and environmental co-factors, comprising drugs, viruses and/or intestinal bacteria may, however, also contribute to dysregulation of intestinal homeostasis, including functionality of T regulatory cells and production of cytokines, and to be involved in disease development [2, 51–53]. Taken together, the activation of immune system and gliadin flux has to be controlled in the intestine of healthy donors, and the identification of the exact mechanisms will be of great importance.
It was reported previously that gliadin fragments induce activation of p38 MAPK signalling pathway in human DCs, which was shown to be crucial for gliadin-triggered up-regulation of maturation markers and cytokine production [16]. As summarized in the model shown in Figure 8, we report that the molecular mechanism by which gliadin promotes DC migration might also rely on the activation of the p38 MAPK signalling pathway, leading to a cytoskeletal remodelling that involves podosome dissolution, as well as to the surface up-regulation of CCR7 and increased synthesis of PGE2.
Fig 8.
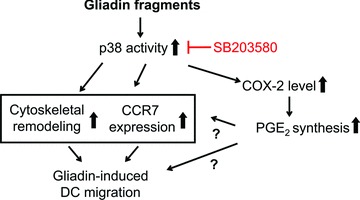
Model of gliadin-induced DC migratory behaviour and chemotactic responsiveness. The migration of DCs promoted by gliadin fragments is under control of the p38 MAPK pathway, and would result from cytoskeletal remodelling, involving dissolution of adhesion structures podosomes, as well as surface CCR7 up-regulation, and induction of COX-2 enzyme that mediates PGE production, respectively.
Indeed podosomes, specialized adhesion structures, were reported to impede high-speed migration of mature DCs [24], and CCR7 is the dominant regulator of DC mobilization from the periphery to LNs [31]. Nevertheless, the surface expression of CCR7 is not predictive of DC chemotactic responsiveness. For example, Bouchon et al. reported that LPS-stimulated DCs exhibited a stronger chemotactic response compared with triggering receptor expressed on myeloid cells 2 (TREM-2) stimulated DCs, although LPS-induced surface up-regulation of CCR7 was weaker than that induced by TREM-2 activation [54]. The function of CCR7 was, indeed, reported to be positively regulated by many extracellular signals, such as PGE2 and/or cysteinyl leukotrienes [35, 36].
We demonstrate that gliadin digest up-regulates the expression of COX-2 enzyme that leads to PGE2 production. Nevertheless, the contribution of the induced PGE2 production to gliadin-stimulated migration is questionable, as the gliadin digest induced PGE2 production was markedly lower than that induced by LPS. Indeed gliadin activation of other, not yet identified signalling pathways yielding CCR7 sensitization, may also contribute to gliadin-induced DC migration. Importantly, gliadin digest enhanced DC migration equally well in serum free medium (CellGro DC, Cell Genix and/or Stemline Dendritic Cell Maturation Medium, Sigma; data not shown), which excludes the possibility of a contribution of serum components (cytokines or growth factor) to gliadin-induced migration.
We also found that the non-immunodominant α-gliadin-derived 31–43 peptide is an active migration component of the digest. Interestingly, addition of the immunodominant 33mer peptide during cell stimulation effectively dampened the migration induced by the 31–43 peptide. We assume, hence, that the 33mer peptide inhibits signals by the 31–43 peptide, either by the direct physical interaction with the peptide or its receptor, or by the activation of an inhibitory receptor signalling. In the same manner, indeed, inhibition of K562(S) cell agglutinating activity of wheat gliadin peptides by a 10 amino acid residue peptide, isolated from durum wheat gliadin, was previously reported [55, 56].
The molecular details of the interaction of gliadin fragments with DCs and/or monocytes/macrophages have not yet been clarified [57]. The complexity of gliadin fragments in the digest might, indeed, enable individual gliadin peptides to target various membrane receptors, although the non-receptor mediated transport of small gliadin peptides (self-penetration of the peptides) into cell cytosol and subsequent activation of intracellular receptors should also be considered [58]. Recently, binding of gliadin to the CXCR3 in the intestinal mucosa was reported [59]. The gliadin-induced effects were further shown to depend on the MyD88 adapter molecule, while being independent of the TLR4- and TLR2- signalling [13]. Consistent with this, we also report here that the gliadin-induced DC migration is independent of the TLR4 signalling.
In summary, this study unravels for the first time the in vitro ability of gliadin fragments to promote DC migration via p38 MAPK activation, leading to cytoskeletal remodelling, surface up-regulation of CCR7 and induction of PGE2 synthesis. The results indicate that gliadin fragments might trigger DC trafficking to LNs in vivo, which would result in generation and expansion of differentiated gliadin-specific T cells, thereby supporting the induction of harmful local and systemic immune responses and development of celiac disease.
Acknowledgments
The authors thank Ondrej Hovorka and Zdenek Cimburek for their assistance with Compucyte imagining. Zdenek Cimburek is further acknowledged for assistance with flow cytometry. This work was supported by grants GA310/07/0414, GD310/08/H077 and GA310/08/0447 from the Czech Science Foundation; grants IAA500200801 and IAA500200914 from the Grant Agency of AS CR; Institutional Research Concept Grant AV0Z50200510 and grant 2B06155 from the Czech Ministry of Education, Youth and Sports.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Sollid LM, Markussen G, Ek J, et al. Evidence for a primary association of celiac disease to a particular HLA-DQ alpha/beta heterodimer. J Exp Med. 1989;169:345–50. doi: 10.1084/jem.169.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Sabatino A, Corazza GR. Coeliac disease. Lancet. 2009;373:1480–93. doi: 10.1016/S0140-6736(09)60254-3. [DOI] [PubMed] [Google Scholar]
- 3.Spurkland A, Sollid LM, Polanco I, et al. HLA-DR and -DQ genotypes of celiac disease patients serologically typed to be non-DR3 or non-DR5/7. Hum Immunol. 1992;35:188–92. doi: 10.1016/0198-8859(92)90104-u. [DOI] [PubMed] [Google Scholar]
- 4.Molberg O, McAdam SN, Korner R, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998;4:713–7. doi: 10.1038/nm0698-713. [DOI] [PubMed] [Google Scholar]
- 5.Schuppan D. Current concepts of celiac disease pathogenesis. Gastroenterology. 2000;119:234–42. doi: 10.1053/gast.2000.8521. [DOI] [PubMed] [Google Scholar]
- 6.Villanacci V, Not T, Sblattero D, et al. Mucosal tissue transglutaminase expression in celiac disease. J Cell Mol Med. 2009;13:334–40. doi: 10.1111/j.1582-4934.2008.00325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shan L, Molberg O, Parrot I, et al. Structural basis for gluten intolerance in celiac sprue. Science. 2002;297:2275–9. doi: 10.1126/science.1074129. [DOI] [PubMed] [Google Scholar]
- 8.Nilsen EM, Jahnsen FL, Lundin KE, et al. Gluten induces an intestinal cytokine response strongly dominated by interferon gamma in patients with celiac disease. Gastroenterology. 1998;115:551–63. doi: 10.1016/s0016-5085(98)70134-9. [DOI] [PubMed] [Google Scholar]
- 9.Sollid LM. Intraepithelial lymphocytes in celiac disease: license to kill revealed. Immunity. 2004;21:303–4. doi: 10.1016/j.immuni.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 10.Hue S, Mention JJ, Monteiro RC, et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity. 2004;21:367–77. doi: 10.1016/j.immuni.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 11.Meresse B, Chen Z, Ciszewski C, et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity. 2004;21:357–66. doi: 10.1016/j.immuni.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 12.Tuckova L, Novotna J, Novak P, et al. Activation of macrophages by gliadin fragments: isolation and characterization of active peptide. J Leukoc Biol. 2002;71:625–31. [PubMed] [Google Scholar]
- 13.Thomas KE, Sapone A, Fasano A, et al. Gliadin stimulation of murine macrophage inflammatory gene expression and intestinal permeability are MyD88-dependent: role of the innate immune response in Celiac disease. J Immunol. 2006;176:2512–21. doi: 10.4049/jimmunol.176.4.2512. [DOI] [PubMed] [Google Scholar]
- 14.Cinova J, Palova-Jelinkova L, Smythies LE, et al. Gliadin peptides activate blood monocytes from patients with celiac disease. J Clin Immunol. 2007;27:201–9. doi: 10.1007/s10875-006-9061-z. [DOI] [PubMed] [Google Scholar]
- 15.Nikulina M, Habich C, Flohe SB, et al. Wheat gluten causes dendritic cell maturation and chemokine secretion. J Immunol. 2004;173:1925–33. doi: 10.4049/jimmunol.173.3.1925. [DOI] [PubMed] [Google Scholar]
- 16.Palova-Jelinkova L, Rozkova D, Pecharova B, et al. Gliadin fragments induce phenotypic and functional maturation of human dendritic cells. J Immunol. 2005;175:7038–45. doi: 10.4049/jimmunol.175.10.7038. [DOI] [PubMed] [Google Scholar]
- 17.Rakhimova M, Esslinger B, Schulze-Krebs A, et al. In vitro differentiation of human monocytes into dendritic cells by peptic-tryptic digest of gliadin is independent of genetic predisposition and the presence of celiac disease. J Clin Immunol. 2009;29:29–37. doi: 10.1007/s10875-008-9228-x. [DOI] [PubMed] [Google Scholar]
- 18.Maiuri L, Ciacci C, Ricciardelli I, et al. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet. 2003;362:30–7. doi: 10.1016/S0140-6736(03)13803-2. [DOI] [PubMed] [Google Scholar]
- 19.Jabri B, Sollid LM. Tissue-mediated control of immunopathology in coeliac disease. Nat Rev Immunol. 2009;9:858–70. doi: 10.1038/nri2670. [DOI] [PubMed] [Google Scholar]
- 20.Brandtzaeg P. The changing immunological paradigm in coeliac disease. Immunol Lett. 2006;105:127–39. doi: 10.1016/j.imlet.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 21.Banchereau J, Briere F, Caux C, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 22.Alvarez D, Vollmann EH, von Andrian UH. Mechanisms and consequences of dendritic cell migration. Immunity. 2008;29:325–42. doi: 10.1016/j.immuni.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol. 2005;5:617–28. doi: 10.1038/nri1670. [DOI] [PubMed] [Google Scholar]
- 24.van Helden SF, Krooshoop DJ, Broers KC, et al. A critical role for prostaglandin E2 in podosome dissolution and induction of high-speed migration during dendritic cell maturation. J Immunol. 2006;177:1567–74. doi: 10.4049/jimmunol.177.3.1567. [DOI] [PubMed] [Google Scholar]
- 25.De Vries IJ, Krooshoop DJ, Scharenborg NM, et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003;63:12–7. [PubMed] [Google Scholar]
- 26.Burns S, Thrasher AJ, Blundell MP, et al. Configuration of human dendritic cell cytoskeleton by Rho GTPases, the WAS protein, and differentiation. Blood. 2001;98:1142–9. doi: 10.1182/blood.v98.4.1142. [DOI] [PubMed] [Google Scholar]
- 27.Burns S, Hardy SJ, Buddle J, et al. Maturation of DC is associated with changes in motile characteristics and adherence. Cell Motil Cytoskeleton. 2004;57:118–32. doi: 10.1002/cm.10163. [DOI] [PubMed] [Google Scholar]
- 28.Linder S, Aepfelbacher M. Podosomes: adhesion hot-spots of invasive cells. Trends Cell Biol. 2003;13:376–85. doi: 10.1016/s0962-8924(03)00128-4. [DOI] [PubMed] [Google Scholar]
- 29.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 30.Benvenuti F, Hugues S, Walmsley M, et al. Requirement of Rac1 and Rac2 expression by mature dendritic cells for T cell priming. Science. 2004;305:1150–3. doi: 10.1126/science.1099159. [DOI] [PubMed] [Google Scholar]
- 31.Martin-Fontecha A, Sebastiani S, Hopken UE, et al. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med. 2003;198:615–21. doi: 10.1084/jem.20030448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dieu MC, Vanbervliet B, Vicari A, et al. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J Exp Med. 1998;188:373–86. doi: 10.1084/jem.188.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sallusto F, Schaerli P, Loetscher P, et al. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur J Immunol. 1998;28:2760–9. doi: 10.1002/(SICI)1521-4141(199809)28:09<2760::AID-IMMU2760>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 34.Sozzani S, Allavena P, D’Amico G, et al. Differential regulation of chemokine receptors during dendritic cell maturation: a model for their trafficking properties. J Immunol. 1998;161:1083–6. [PubMed] [Google Scholar]
- 35.Robbiani DF, Finch RA, Jager D, et al. The leukotriene C(4) transporter MRP1 regulates CCL19 (MIP-3beta, ELC)-dependent mobilization of dendritic cells to lymph nodes. Cell. 2000;103:757–68. doi: 10.1016/s0092-8674(00)00179-3. [DOI] [PubMed] [Google Scholar]
- 36.Scandella E, Men Y, Gillessen S, et al. Prostaglandin E2 is a key factor for CCR7 surface expression and migration of monocyte-derived dendritic cells. Blood. 2002;100:1354–61. doi: 10.1182/blood-2001-11-0017. [DOI] [PubMed] [Google Scholar]
- 37.Kabashima K, Sakata D, Nagamachi M, et al. Prostaglandin E2-EP4 signaling initiates skin immune responses by promoting migration and maturation of Langerhans cells. Nat Med. 2003;9:744–9. doi: 10.1038/nm872. [DOI] [PubMed] [Google Scholar]
- 38.Scandella E, Men Y, Legler DF, et al. CCL19/CCL21-triggered signal transduction and migration of dendritic cells requires prostaglandin E2. Blood. 2004;103:1595–601. doi: 10.1182/blood-2003-05-1643. [DOI] [PubMed] [Google Scholar]
- 39.Legler DF, Krause P, Scandella E, et al. Prostaglandin E2 is generally required for human dendritic cell migration and exerts its effect via EP2 and EP4 receptors. J Immunol. 2006;176:966–73. doi: 10.4049/jimmunol.176.2.966. [DOI] [PubMed] [Google Scholar]
- 40.Teloni R, Giannoni F, Rossi P, et al. Interleukin-4 inhibits cyclo-oxygenase-2 expression and prostaglandin E production by human mature dendritic cells. Immunology. 2007;120:83–9. doi: 10.1111/j.1365-2567.2006.02482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fogel-Petrovic M, Long JA, Knight DA, et al. Activated human dendritic cells express inducible cyclo-oxygenase and synthesize prostaglandin E2 but not prostaglandin D2. Immunol Cell Biol. 2004;82:47–54. doi: 10.1111/j.1440-1711.2004.01213.x. [DOI] [PubMed] [Google Scholar]
- 42.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 43.Cerovic V, McDonald V, Nassar MA, et al. New insights into the roles of dendritic cells in intestinal immunity and tolerance. Int Rev Cell Mol Biol. 2009;272:33–105. doi: 10.1016/S1937-6448(08)01602-X. [DOI] [PubMed] [Google Scholar]
- 44.Harris KM, Fasano A, Mann DL. Cutting edge: IL-1 controls the IL-23 response induced by gliadin, the etiologic agent in celiac disease. J Immunol. 2008;181:4457–60. doi: 10.4049/jimmunol.181.7.4457. [DOI] [PubMed] [Google Scholar]
- 45.Dubois PCA, Trynka G, Franke L, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010 doi: 10.1038/ng.543. http://dx.doi.org/10.1038/ng.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hunt KA, Zhernakova A, Turner G, et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet. 2008;40:395–402. doi: 10.1038/ng.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Heel DA, Franke L, Hunt KA, et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat Genet. 2007;39:827–9. doi: 10.1038/ng2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhernakova A, van Diemen CC, Wijmenga C. Detecting shared pathogenesis from the shared genetics of immune-related diseases. Nat Rev Genet. 2009;10:43–55. doi: 10.1038/nrg2489. [DOI] [PubMed] [Google Scholar]
- 49.Monsuur AJ, de Bakker PI, Alizadeh BZ, et al. Myosin IXB variant increases the risk of celiac disease and points toward a primary intestinal barrier defect. Nat Genet. 2005;37:1341–4. doi: 10.1038/ng1680. [DOI] [PubMed] [Google Scholar]
- 50.Hunt KA, Monsuur AJ, McArdle WL, et al. Lack of association of MYO9B genetic variants with coeliac disease in a British cohort. Gut. 2006;55:969–72. doi: 10.1136/gut.2005.086769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Palma G, Cinova J, Stepankova R, et al. Pivotal advance: bifidobacteria and gram-negative bacteria differentially influence immune responses in the proinflammatory milieu of celiac disease. J Leukoc Biol. 2010;87:765–78. doi: 10.1189/jlb.0709471. [DOI] [PubMed] [Google Scholar]
- 52.Gianfrani C, Levings MK, Sartirana C, et al. Gliadin-specific type 1 regulatory T cells from the intestinal mucosa of treated celiac patients inhibit pathogenic T cells. J Immunol. 2006;177:4178–86. doi: 10.4049/jimmunol.177.6.4178. [DOI] [PubMed] [Google Scholar]
- 53.Granzotto M, dal Bo S, Quaglia S, et al. Regulatory T-cell function is impaired in celiac disease. Dig Dis Sci. 2009;54:1513–9. doi: 10.1007/s10620-008-0501-x. [DOI] [PubMed] [Google Scholar]
- 54.Bouchon A, Hernandez-Munain C, Cella M, et al. A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J Exp Med. 2001;194:1111–22. doi: 10.1084/jem.194.8.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Vincenzi M, Dessi MR, Giovannini C, et al. Agglutinating activity of wheat gliadin peptide fractions in coeliac disease. Toxicology. 1995;96:29–35. doi: 10.1016/0300-483x(94)02912-e. [DOI] [PubMed] [Google Scholar]
- 56.De Vincenzi M, Stammati A, Luchetti R, et al. Structural specificities and significance for coeliac disease of wheat gliadin peptides able to agglutinate or to prevent agglutination of K562(S) cells. Toxicology. 1998;127:97–106. doi: 10.1016/s0300-483x(98)00034-1. [DOI] [PubMed] [Google Scholar]
- 57.Barone MV, Gimigliano A, Castoria G, et al. Growth factor-like activity of gliadin, an alimentary protein: implications for coeliac disease. Gut. 2007;56:480–8. doi: 10.1136/gut.2005.086637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vilasi S, Sirangelo I, Irace G, et al. Interaction of ‘toxic’ and ‘immunogenic’ A-gliadin peptides with a membrane-mimetic environment. J Mol Recognit. 2010;23:322–8. doi: 10.1002/jmr.987. [DOI] [PubMed] [Google Scholar]
- 59.Lammers KM, Lu R, Brownley J, et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology. 2008;135:194–204. doi: 10.1053/j.gastro.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]