

Abstract
Interleukin-1β (IL-1β) is a prototypical multifunctional cytokine that plays an important role in intestinal inflammation of Crohn’s disease and other inflammatory conditions of the gut. Previous studies have shown that IL-1β causes an increase in intestinal epithelial tight junction (TJ) permeability both in in vivo animal and in vitro cell culture model systems. The IL-1β-induced increase in intestinal epithelial TJ permeability has been postulated to be an important pathogenic mechanism contributing to intestinal inflammation. However, the signalling pathways and the molecular processes that mediate the IL-1β modulation of intestinal epithelial TJ barrier remain unclear. Here, we show that the IL-1β-induced increase in Caco-2 monolayer TJ permeability was mediated by activation of extracellular signal-regulated kinases 1/2 (ERK1/2) signalling pathway and that inhibition of ERK1/2 activity inhibits the IL-1β-induced increase in Caco-2 TJ permeability. The activation of ERK1/2 pathway caused a downstream activation of nuclear transcription factor Elk-1. The activated Elk-1 translocated to the nucleus and binds to the cis-binding motif on myosin light chain kinase (MLCK) promoter region, triggering MLCK gene activation, MLCK mRNA transcription and MLCK protein synthesis and MLCK catalysed opening of the intestinal epithelial TJ barrier. These studies provide novel insight into the cellular and molecular processes that mediate the IL-1β-induced increase in intestinal epithelial TJ permeability.
Keywords: intestinal tight junction permeability, IL-1β, ERK1/2, Elk-1, myosin light chain kinase
Introduction
Interleukin-1β (IL-1β) is a prototypical multifunctional cytokine that plays an important role in intestinal inflammation of Crohn’s disease (CD) and other inflammatory conditions of the gut [1–3]. CD is an immune mediated disorder characterized by chronic, relapsing inflammation of the gastrointestinal tract [4]. IL-1β is markedly elevated in intestinal tissue and serum of CD patients and a correlation between increasing levels of IL-1β and increasing severity of intestinal inflammation has been demonstrated in patients with CD [5]. An imbalance in IL-1β levels and its naturally occurring antagonist IL-1 receptor antagonist (IL-1ra) occurs in CD patients such that there is an excess of pro-inflammatory and deficiency of anti-inflammatory forms of IL-1 [6–8]. IL-1β gene polymorphism also exists in CD patients that determine the severity of intestinal inflammation in the affected patients [7, 9, 10]. Thus, IL-1β has been identified as an important pro-inflammatory mediator of CD and therapeutic strategies to inhibit IL-1β are being explored [3, 8].
An integral function of epithelial cells lining the intestinal tract is to act as a physical and functional barrier against noxious substances present in the intestinal lumen [11]. The bi-lipid apical membrane of the enterocytes is an effective barrier against transcellular permeation of passively absorbed hydrophilic substances present in the lumen [11, 12]. The intercellular tight junctions (TJs) that encircle the enterocytes at the apico-lateral membrane border act as a barrier against permeation of hydrophilic substances in-between cells (referred to as paracellular barrier function) [11, 12]. It is well established that patients with CD have a defective intestinal TJ barrier characterized by an increase in intestinal permeability to paracellular markers [13–15]. The defective TJ barrier allows paracellular permeation of luminal antigens that stimulate inflammatory response [11, 13]. Clinical studies have shown that therapeutic re-tightening of the intestinal TJ barrier is associated with rapid clinical improvement and prolonged clinical remission [16–18], and persistent elevation in intestinal permeability following medical therapy have been shown to be predictive of refractory disease and early recurrence of the disease [16, 18, 19].
It is well established that IL-1β causes an increase in intestinal epithelial TJ permeability in vivo and in vitro[20–22]. Previous studies from our laboratory suggested that the IL-1β-induced increase in Caco-2 monolayer TJ permeability was mediated by an increase in myosin light chain kinase (MLCK) gene and protein expression and that inhibition of MLCK synthesis or MLCK activity prevented the IL-1β-induced increase in TJ permeability [20]. However, the intracellular signalling pathways and the molecular processes responsible for the increase in MLCK gene and protein expression and the increase in intestinal TJ permeability remain unknown. The major purpose of this study was to delineate the signalling pathway and the molecular mechanisms that mediate the IL-1β-induced increase in intestinal TJ permeability using a commonly used in vitro intestinal epithelial model system consisting of filter-grown Caco-2 intestinal epithelial monolayers [20, 23].
Previous studies have shown that many of the pro-inflammatory actions of IL-1β are mediated by mitogen-activated protein kinases (MAPK) [24, 25]. Extracellular regulated protein kinase 1/2 (ERK1/2) is a key member of MAPK family of proteins and has been found to play an important role in mediating some of the biological actions of IL-1β[26, 27]. ERK1/2 pathways play an integral role in signal transduction processes leading to regulation of cell growth [28], differentiation [29] and gene activity that culminate in the production of cytokines, chemokines, adhesion molecules and effector proteins [30]. In this study, we examined the possibility that ERK1/2 signalling pathway plays a key role in the regulation of IL-1β-induced increase in intestinal epithelial TJ permeability. Our data show that IL-1β-induced activation of ERK1/2 pathways was required for the increase in intestinal TJ permeability. Additionally, our data provide new insight into the cellular and molecular processes that mediate the ERK1/2 kinase-induced activation of MLCK gene and increase in Caco-2 monolayer TJ permeability.
Materials and methods
Chemicals
Cell culture media (DMEM), trypsin, foetal bovine serum (FBS) and related reagents were purchased from Life Technologies (Gaithersburg, MD, USA). Glutamine, penicillin, streptomycin and PBS were purchased from GIBCO-BRL (Grand Island, NY, USA). Anti-ERK1/2 (ERK1/2), Elk-1(Ets-like gene 1), phosphor-ERK1/2, MLCK, AP-1 (activator protein-1) and anti-β-actin antibodies were obtained from Sigma (St. Louis, MO, USA). Horseradish peroxidase (HRP)-conjugated secondary antibodies for Western blot analysis were purchased from Invitrogen (San Francisco, CA, USA). SiRNA of ERK1/2, MLCK, Elk-1 and AP-1 and transfection reagents were obtained from Dharmacon (Lafayette, CO, USA). ERK1/2 inhibitor 2′-amino-3′-methoxyflavone (PD-98059) was purchased from Sigma. All other chemicals were purchased from Sigma, VWR (West Chester, PA, USA) or Fisher Scientific (Pittsburgh, PA, USA).
Cell cultures
Caco-2 cells (passage 18) were purchased from the American Type Culture Collection (Rockville, MD, USA) and maintained at 37°C in a culture medium composed of DMEM with 4.5 mg/ml glucose, 50 U/ml penicillin, 50 U/ml streptomycin, 4 mM glutamine, 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and 10% FBS. The cells were kept at 37°C in a 5% CO2 environment. Culture medium was changed every 2 days. Caco-2 cells were subcultured after partial digestion with 0.25% trypsin and 0.9 mM ethylenediaminetetraacetic acid (EDTA) in Ca2+- and Mg2+-free PBS.
Determination of epithelial monolayer resistance and paracellular permeability
An epithelial voltohmeter (World Precision Instruments, Sarasota, FL, USA) was used for measurements of the transepithelial electrical resistance (TER) of the filter-grown Caco-2 intestinal monolayers as previously reported [31]. The effect of IL-1β on Caco-2 paracellular permeability was determined using an established paracellular marker inulin (m.w. = 5000 g/mol) [32]. For determination of mucosal-to-serosal flux rates of inulin, Caco-2-plated filters having epithelial resistance of 400–500 Ω.cm2 were used. Known concentrations of permeability marker (2 μM) and its radioactive tracer were added to the apical solution. Low concentrations of permeability marker were used to ensure that negligible osmotic or concentration gradient was introduced.
Assessment of protein expression by Western blot analysis
Caco-2 monolayers were treated with IL-1β (10 ng/ml) for varying time periods. At the end of the experimental period, Caco-2 monolayers were immediately rinsed with ice-cold PBS, and cells were lysed with lysis buffer (50 mM Tris.HCl, pH 7.5, 150 mM NaCl, 500 μM NaF, 2 mM EDTA, 100 μM vanadate, 100 μM phenylmethylsulfonyl fluoride (PMSF), 1 μg/ml leupeptin, 1 μg/ml pepstatin A, 40 mM paranitrophenyl phosphate, 1 μg/ml aprotinin and 1% Triton X-100) and scraped, and the cell lysates were placed in Microfuge tubes. Cell lysates were centrifuged to yield a clear lysate. Supernatant was collected, and protein measurement was performed with Bio-Rad Protein Assay kit (Bio-Rad Laboratories, Hercules, CA, USA). Laemmli gel loading buffer was added to the lysate containing 10–20 μg of protein and boiled for 7 min., after which proteins were separated on SDS-PAGE gel. Proteins from the gel were transferred to the membrane (Trans-Blot Transfer Medium, Nitrocellulose Membrane; Bio-Rad Laboratories) overnight. The membrane was incubated for 2 hrs in blocking solution [5% dry milk in tris buffered saline (TBS)-Tween 20 buffer]. The membrane was incubated with appropriate primary antibodies in blocking solution. After being washed in TBS-1% Tween buffer, the membrane was incubated in appropriate secondary antibodies and developed using the Santa Cruz Western Blotting Luminol Reagents (Santa Cruz Biotechnology, Santa Cruz, CA, USA) on the Kodak BioMax MS film (Fisher Scientific).
RNA isolation and reverse transcription
Caco-2 cells (5 × 105/filter) were seeded into 6-well transwell permeable inserts and grown to confluency. Filter-grown Caco-2 cells were then treated with appropriate experimental reagents for desired time periods. At the end of the experimental period, cells were washed twice with ice-cold PBS. Total RNA was isolated using Qiagen RNeasy Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. Total RNA concentration was determined by absorbance at 260/280 nm using SpectrraMax 190 (Molecular Devices, Sunnyvale, CA, USA). The reverse transcription (RT) was carried out using the GeneAmp Gold RNA PCR core kit (Applied Biosystems, Foster City, CA, USA). Two micrograms of total RNA from each sample were reverse transcribed into cDNA in a 40-μl reaction containing 1× RT-PCR buffer, 2.5 mM MgCl2, 250 μM of each dNTP, 20 U RNase inhibitor, 10 mM DL-Dithiothreitol (DTT), 1.25 μM random hexamer and 30 U multiscribe RT. The RT reactions were performed in a thermocycler (PTC-100, MJ Research, Waltham, MA, USA) at 25°C for 10 min., 42°C for 30 min. and 95°C for 5 min.
Quantification of gene expression using real-time PCR
The real-time PCRs were carried out using ABI prism 7900 sequence detection system and Taqman universal PCR master mix kit (Applied Biosystems, Branchburg, NJ, USA) as previously described [32, 33]. Each real-time PCR reaction contained 10 μl RT reaction mix, 25 μl 2× Taqman universal PCR master mix, 0.2 μM probe and 0.6 μM primers. Primer and probe design for the real-time PCR was made with Primer Express version 2 from Applied Biosystems. [The primers used in this study are as follows: MLCK specific primer pairs consisted of 5′-AGGAAGGCAGCATTGAGGTTT-3′[forward], 5′-GCTTTCAGCAGGCAGAGGTAA-3′[reverse]; probe specific for MLCK consisted of FAM 5′-TGAAGATGCTGGCTCC-3′ TAMRA; the internal control glyceraldehyde 3-phosphate dehydrogenase (GAPDH)-specific primer pairs consisted of 5′-CCACCCATGGCAAATTCC-3′[forward], 5′-TGGGATTTCCATTGATGACCAG-3′[reverse]; probe specific for GAPDH consisted of JOE 5′-TGGCACCGTCAAGGCTGAGAACG-3′ TAMRA.] All runs were performed according to the default PCR protocol (50°C for 2 min., 95°C for 10 min., 40 cycles of 95°C for 15 sec. and 60°C for 1 min.). For each sample, real-time PCR reactions were performed in triplicate, and the average threshold cycle (Ct) was calculated. A standard curve was generated to convert the Ct to copy numbers. Expression of MLCK mRNA was normalized with GAPDH mRNA expression. The average copy number of MLCK mRNA expression in control samples was set to 1.0. The relative expression of MLCK mRNA in treated samples was determined as a fold increase compared with control samples.
SiRNA of ERK1/2, MLCK, Elk-1 and AP-1
Targeted siRNAs were obtained from Dharmacon, Inc. (Chicago, IL, USA). Caco-2 monolayers were transiently transfected using DharmaFect transfection reagent (Lafayette). Briefly, 5 × 105 cells/filter were seeded into a 12-well transwell plate and grown to confluency. Caco-2 monolayers were then washed with PBS twice and 1.0 ml Opti-Minimum Essential Media (MEM) medium was added to the apical compartment of each filter and 1.5 ml were added to the basolateral compartment of each filter. Five nanograms of the siRNA of interest and 2 μl of DharmaFect reagent were pre-incubated in Opti-MEM. After 5 min. of incubation, two solutions were mixed and incubated for another 20 min., and the mixture was added to the apical compartment of each filter. The IL-1β experiments were carried out 96 hrs after transfection. The efficiency of silencing was confirmed by Western blot analysis.
ELISA-based in vitro ERK1/2 kinase activity
Biotinylated myelin basic protein (MBP) was diluted in PBS and coated on streptavidin 96-well plates at 37°C for 1 hr. The plates were washed three times with PBS, incubated with blocking solution (1 mg/ml bovine serum albumin in PBS) at 37°C for 1 hr, and then washed three times with PBS. The kinase reaction buffer (90 μl) (20 mM Tris/HCl pH7.5, 10 mM MgCl2, 50 mM NaCl, 1 mM DTT, 1 mM NaF, 50 μM ATP) provided by the manufacturer (MBL International, Woburn, MA, USA) and the samples containing immunoprecipitated ERK1/2 (10 μl) were added to each well, and the kinase reaction (phosphorylation of MBP) was carried out at 37°C for 30–60 min. The reaction was stopped by removing the reaction mixtures and washing the plates three times with washing buffer (20 mM Tris-HCl at pH 7.4, 0.5 M NaCl and 0.05% Tween 20). The washed plates were incubated with the anti-phospho-MBP antibody (5 ng/ml) at room temperature for 1 hr. The plates were washed four times with washing buffer, and goat anti-rabbit IgG antibody (diluted at 1:2000 in washing buffer) was added to the wells, and the plates were incubated at 37°C for 1 hr. The plates were then washed four times and incubated with 100 μl substrate solution tetramethylbenzidine at 37°C for 5–15 min. A stop solution containing 0.5 N H2SO4 (100 μl) was added to stop the reaction. The absorbance at 450 nm was determined using the SpectrraMax 190 (Molecular Devices).
Nuclear extracts and ELISA for transcription factor activation
Filter-grown Caco-2 monolayers were treated with 10 ng/ml IL-1β for 30 min. Caco-2 monolayers were washed with ice-cold PBS, scraped, collected and centrifuged at 14,000 rpm for 30 sec. The cell pellets were resuspended in 200 μl of buffer A (in millimoles: 10 HEPES-KOH, 1.5 MgCl2, 10 KCl, 0.5 DTT and 0.2 PMSF [pH 7.9]), and incubated on ice for 15 min. After centrifugation at 14,000 rpm for 30 sec., pelleted nuclei were resuspended in 30 μl of buffer C (in millimoles: 20 HEPES-KOH (25% glycerol), 420 NaCl, 1.5 MgCl2, 0.2 EDTA, 0.5 DTT and 0.2 PMSF [pH 7.9]). After incubation on ice for 20 min., the lysates were centrifuged at 14,000 rpm for 20 min. Protein concentrations were determined using the Bradford method. The Elk-1, AP-1 and Signal Transducers and Activator-1 (STAT-1) DNA-binding assay was performed with Trans-AM ELISA-based kits from Active Motif according to the manufacturer’s protocol. In brief, the binding reactions contained 1 pM biotinylated probe (Integrated DNA Technologies, Coralville, IA, USA) and 5 μg of nuclear extract in complete binding buffer with a total volume of 50 μl. After 30 min. of incubation, the solution was transferred to an individual well on 96-well plate and incubated for 1 hr. Appropriate antibody (2 μg/ml) was added to the well to bind the target protein in nuclear extract. After incubation for 1 hr, the antibody was removed, and 100 μl of HRP-conjugated secondary antibody was added to the well and incubated for 1 hr. Subsequently, 100 μl of developing solution was added for 2–10 min., and 100 μl of stop solution were added. The absorbance at 450 nm was determined using the SpectraMax 190 (Molecular Devices).
Cloning of the full length MLCK promoter region and deletion constructs
The MLCK promoter region was cloned using GenomeWalker system (Clontech, Mountain View, CA, USA). A 2091-bp DNA fragment (–2109 to –18) was amplified by PCR. The amplification condition was 1 cycle at 94°C for 2 min., followed by 43 cycles at 94°C for 1 min., 50°C for 1 min. and 72°C for 2 min. and 1 cycle at 72°C for 5 min. The resultant PCR product was digested with HindIII and KpnI and inserted into pGL3-basic luciferase reporter vector (Promega, Madison, WI, USA). The sequence was confirmed by DNA services at the University of New Mexico. Construction of MLCK promoter reporter plasmids was carried out using the pGL-3 basic luciferase reporter vector. Deletions of MLCK promoter were done by the PCR method. The primers used for cloning two MLCK promoter deletion constructs are listed in Table 1. The PCR conditions were 1 cycle at 94°C for 2 min., followed by 43 cycles at 94°C for 1 min., 50°C for 1 min. and 72°C for 2 min. and 1 cycle at 72°C for 5 min. The resultant PCR products were cloned into pGL-3 basic luciferase reporter vector and the sequences were confirmed.
Table 1.
Sequences of cloning primers
| Primer name | Sequence (5′-3′) |
|---|---|
| FL-MLCK (+) | GCCGGTACCGAGAAGCAGGAGAGTATTAAATG |
| MLCK-929 (+) | CGGGGTACCCTCTGCCCTCTTGACTTAATC |
| MLCK-313 (+) | GCCGGTACCATGGCCTTCCTCCCTCACCCCT |
Transfection of DNA constructs and assessment of promoter activity
DNA constructs of MLCK promoters were transiently transfected into Caco-2 cells using transfection reagent lipofectamine 2000 (Life Technologies). Renilla luciferase vector (pRL-TK, Promega) was cotransfected with each plasmid construct as an internal control. Cells (5 × 105/filter) were seeded into a 6-well transwell plate and grown to confluency. Caco-2 monolayers were then washed with PBS twice and 1.0 ml Opti-MEM medium was added to the apical compartment of each filter and 1.5 ml were added to the basolateral compartment of each filter. One microgram of each plasmid construct and 0.25 μg pRL-TK or 2 μl lipofectamine 2000 was preincubated in 250 μl Opti-MEM, respectively. After 5 min. of incubation, two solutions were mixed and incubated for another 20 min., and the mixture was added to the apical compartment of each filter. After incubation for 3 hrs at 37°C, 500 μl DMEM containing 10% FBS were added to both sides of the filter to reach a 2.5% final concentration of FBS. Subsequently, media were replaced with normal Caco-2 growth media 16 hrs after transfection. Specific experiments were carried out 48 hrs after transfection. At the completion of specific experimental treatments, Caco-2 cells were washed twice with 1 ml ice-cold PBS, followed by the addition of 400 μl 1× passive lysis buffer, incubated at room temperature for 15 min., scraped and transferred into an Eppendorf tube, and centrifuged for 15 sec. at 13,000 rpm in a microcentrifuge. Luciferase activity was determined using the dual luciferase assay kit (Promega). Twenty microlitres of the supernatant were used for each assay. Luciferase values were determined by Lumat LB 9507 (EG&G Berthold, Oak Ridge, TN, USA). The value of reporter luciferase activities were then divided by that of renilla luciferase activities to normalize for differences in transfection efficiencies. The average activity value of the control samples was set to 1.0. The luciferase activity of MLCK promoter in treated samples was determined relative to the control samples.
Site-directed mutagenesis
Mutagenesis of MLCK promoter was performed with the GeneTailor Site-Directed Mutagenesis System (Invitrogen). Briefly, primers (Mutant primer: forward: 5′-CTGCAGGAAGGCAGCTCCCACAATTCCTTCTTTCTACCCCTGCCA-3′; reverse primer: 5′-TGGGAGCTGCCTTCCTGCAGGTGAAAGGCA-3′) were generated that included the mutation site flanked by a wild-type sequence on either side. A PCR reaction produced a new complete copy of the plasmid containing the mutation coded for by the primers. The linear PCR product was subsequently transformed into DH5-T1 Escherichia coli, which circularized the PCR product and digested any remaining parent plasmid. DNA sequence was then verified by DNA services at the University of New Mexico.
Statistical analysis
Results are expressed as means ± S.E. Statistical significance of differences between mean values was assessed with Student’s t-tests for unpaired data and ANOVA analysis whenever was required. All reported significance levels represent two-tailed P-values. A P-value of <0.05 was used to indicate statistical significance. Each experiment was performed in triplicate or quadruplicates (n= 3 or 4) and all experiments were repeated three to six times to ensure reproducibility.
Results
ERK1/2 signalling pathway mediates the IL-1β induced increase in Caco-2 TJ permeability
The signalling pathways that regulate the IL-1β-induced increase in intestinal epithelial TJ permeability remain unknown. In the following studies, we examined the possibility that the IL-1β-induced increase in Caco-2 monolayer TJ permeability was regulated by ERK1/2 signalling pathways. The time-course effect of IL-1β (10 ng/ml) on ERK1/2 activation was determined by measuring ERK1/2 phosphorylation in filter-grown Caco-2 monolayers and by in vitro kinase assay of immunoprecipitated ERK1/2. IL-1β (10 ng/ml) caused a rapid increase in ERK1/2 phosphorylation in Caco-2 cells starting at about 10 min. and continuing up to 60 min. as determined by phospho-ERK1/2 immunoblotting (Fig. 1A). The total ERK1/2 level was not affected by the IL-1β treatment (Fig. 1A). The IL-1β effect on ERK1/2 kinase activity was also determined by an ELISA-based in vitro kinase assay. Following IL-1β treatment of filter-grown Caco-2 monolayers, Caco-2 cells were lysed and ERK1/2 immunoprecipitated using ERK1/2 antibody. The kinase activity of immunoprecipitated ERK1/2 was measured in vitro using MBP as the substrate as described in the Methods section. IL-1β caused a rapid increase in Caco-2 ERK1/2 kinase activity starting at about 5–10 min. (Fig 1B). The IL-1β-induced increase in ERK1/2 kinase activity paralleled the increase in ERK1/2 phosphorylation. These results indicated that IL-1β causes a rapid activation (within minutes) of ERK1/2.
Fig 1.
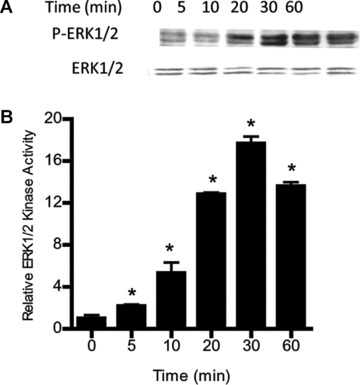
Time-course effect of IL-1β on Caco-2 ERK1/2 activation. (A) Time-course effect of IL-1β (10 ng/ml) on Caco-2 ERK1/2 phosphorylation (total ERK1/2 was used for equal protein loading). (B) Time-course effect of IL-1β on ERK1/2 activity was determined by ELISA-based in vitro kinase activity using MBP as the substrate. IL-1β caused a time-dependent increase in Caco-2 ERK1/2 activity (means ± S.E., n= 4).*P < 0.001 versus control.
To determine the involvement of ERK1/2 pathway in IL-1β-induced increase in Caco-2 TJ permeability, the effect of ERK1/2 inhibition was examined. The IL-1β effect on Caco-2 TJ permeability was determined by measuring TER and transepithelial flux rate of paracellular marker inulin (Fig. 2A and B) [20, 32]. IL-1β caused a progressive drop in Caco-2 TER and an increase in transepithelial flux of inulin starting at about 12 hrs (Fig. 2A and B). The ERK1/2 inhibitor PD98059 (100 μM) inhibited the IL-1β-induced activation of ERK1/2 (Fig. 2C) and prevented the drop in TER (Fig. 2D) and the increase in inulin flux (Fig. 2E). These results showed that IL-1β-induced increase in Caco-2 TJ permeability required ERK1/2 activation. To further validate the requirement of ERK1/2 on IL-1β effect on Caco-2 TJ permeability, ERK1/2 expression was knocked down by Caco-2 transfection of ERK1/2 siRNA. The transfection of ERK1/2 siRNA caused a near-complete depletion of ERK1/2 in filter-grown Caco-2 monolayers (Fig. 3A). The knockdown of ERK1/2 inhibited the IL-1β-induced drop in Caco-2 TER (Fig. 3B) and increase in inulin flux (Fig. 3C), confirming the regulatory role of ERK1/2 in IL-1β-induced increase in Caco-2 TJ permeability.
Fig 2.
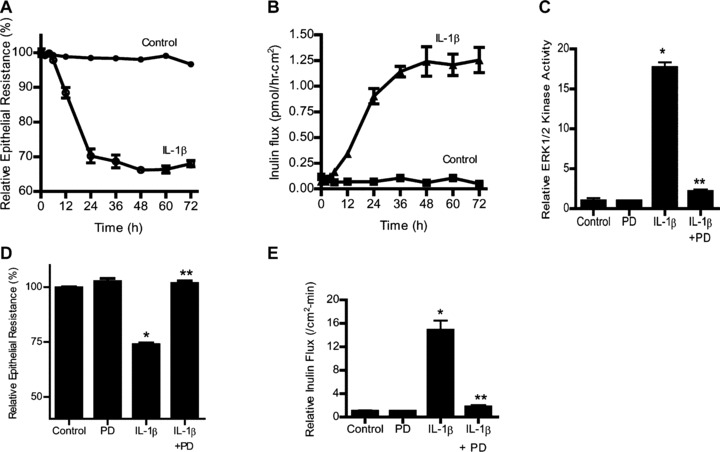
Effect of ERK1/2 inhibition by pharmacologic inhibitor (PD-98059) (100 μM) on Caco-2 ERK1/2 activity, TER, and paracellular permeability. (A) The effect of IL-1β (10 ng/ml) on Caco-2 TER and (B) mucosal-to-serosal flux of paracellular marker inulin (2 μM) were measured sequentially over the 72-hr experimental period as described in the section ‘Materials and methods’. IL-1β caused a time-dependent drop in Caco-2 TER and increase in paracellular permeability to inulin (C) PD-98059 (100 μM) pre-treatment inhibited the IL-1β-induced increase in ERK1/2 in vitro kinase activity. *P < 0.001 versus control; **P < 0.001 versus IL-1β. (D) PD-98059 prevented the IL-1β-induced drop in Caco-2 TER. *P < 0.001 versus control; **P < 0.001 versus IL-1β. (E) pre-treatment with PD-98059 prevented the IL-1β increase in mucosal-to-serosal inulin flux (means ± S.E., n= 5). *P < 0.001 versus control; **P < 0.001 versus IL-1β treatment.
Fig 3.
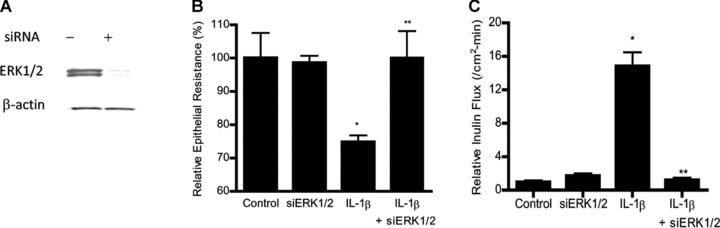
Effect of siRNA-induced ERK1/2 knockdown on IL-1β-induced increase in Caco-2 TJ permeability. (A) ERK1/2 siRNA transfection resulted in a near complete depletion in ERK1/2 protein expression as determined by Western blot analysis. (B) ERK1/2 siRNA transfection prevented the IL-1β-induced drop in Caco-2 TER. (C) ERK1/2 siRNA transfection prevented the IL-1β-induced increase in inulin flux (means ± S.E., n= 5). *P < 0.001 versus control; **P < 0.001 versus IL-1β treatment.
ERK1/2 signalling pathway is involved in the regulation of MLCK gene and protein expression
Previous studies from our laboratory indicated that the IL-1β-induced increase in Caco-2 TJ permeability was mediated by an increase in MLCK protein expression and activity [20]. However, the intracellular pathways and the molecular processes involved in the IL-1β regulation of MLCK gene and protein expression remain unclear. In the following studies, we examined the possibility that ERK1/2 pathways also mediate the IL-1β-induced up-regulation of MLCK gene and protein expression. As shown in Figure 4A and B, IL-1β caused an increase in Caco-2 MLCK protein and mRNA expression. The siRNA-induced knockdown of MLCK inhibited the IL-1β-induced drop in Caco-2 TER [20] and increase in inulin flux (Fig. 4C). The inhibition of ERK1/2 activity with pharmacologic inhibitor PD 98059 (100 μM) inhibited the IL-1β-induced increase in MLCK mRNA and protein expression (Fig. 4B and D). The siRNA-induced knockdown of Caco-2 ERK1/2 also inhibited the IL-1β-induced increase in MLCK mRNA and protein level (Fig. 4E and F). These findings indicated that ERK1/2 signalling pathway was involved in IL-1β regulation of MLCK mRNA and protein expression.
Fig 4.
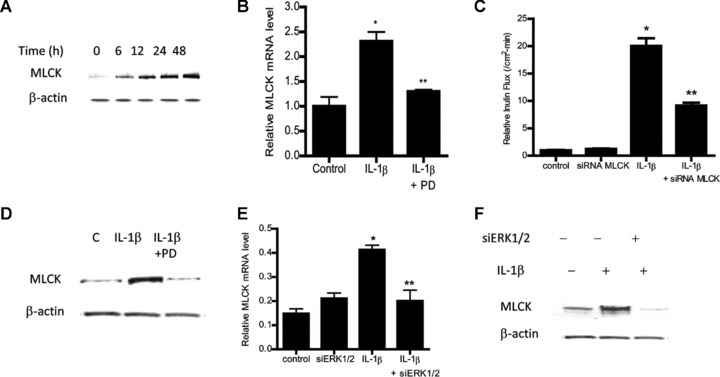
ERK1/2 inhibition prevented the IL-1β-induced up-regulation of Caco-2 MLCK. (A) Time-course effect of IL-1β on Caco-2 MLCK protein expression (β-actin was used as an internal control for protein loading). (B) IL-1β treatment caused an increase in Caco-2 MLCK mRNA. ERK1/2 inhibitor PD-98059 prevented the increase in MLCK mRNA levels. MLCK mRNA level was expressed relative to the control level which was assigned a value of 1. The average copy number of MLCK mRNA in controls was 4.63 × 1011 (means ± S.E., n= 5). *P < 0.001 versus control; **P < 0.001 versus IL-1β treatment. (C) MLCK siRNA transfection prevented the IL-1β-induced drop in Caco-2 TER. (D) MLCK siRNA-induced knockdown prevented the IL-1β-induced increase in inulin flux (means ± S.E., n= 5). *P < 0.001 versus control; **P < 0.001 versus IL-1β treatment. (E) ERK1/2 inhibitor PD-98059 prevented the IL-1β-induced up-regulation of MLCK protein expression. Effect of siRNA ERK1/2 transfection on IL-1β-induced increase in MLCK mRNA level (F) and increase in MLCK protein expression (G). siRNA-induced knockdown of ERK1/2 prevented the IL-1β-induced increase in MLCK mRNA and MLCK protein levels (means ± S.E., n= 4). *P < 0.001 versus control; **P < 0.001 versus IL-1β treatment.
IL-1β induces MLCK promoter activation
To delineate the molecular processes involved in IL-1β modulation of MLCK mRNA and protein expression, the IL-1β effect on MLCK promoter activity was determined. The IL-1β effect on MLCK promoter activity was determined by measuring luciferase activity (reporter gene) following transfection of filter-grown Caco-2 monolayers with plasmid vector containing the MLCK promoter region [34]. The IL-1β (10 ng/ml) treatment of MLCK promoter transfected Caco-2 cells resulted in an increase in MLCK promoter activity (Fig. 5), indicating that IL-1β has a stimulatory effect on MLCK gene activity.
Fig 5.
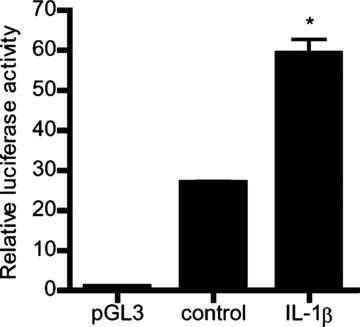
IL-1β effect on MLCK promoter activity. IL-1β effect on the activity of the full-length MLCK promoter region (2091bp) in Caco-2 monolayers. pGL-3 basic vector containing the MLCK promoter region was transfected into the filter-grown Caco-2 cells. Caco-2 cells were treated with IL-1β (10 ng/ml) for 6 hrs. The MLCK promoter activity was determined by the luciferase assay and expressed as relative luciferase activity (means ± S.E., n= 8). *P < 0.0001 versus control.
Elk-1 regulates the ERK1/2 signalling cascade induced increase in MLCK gene expression
In the following series of studies, the nuclear transcription factors involved in IL-1β/ERK1/2 signalling pathway induced increase in Caco-2 MLCK mRNA and protein was determined. Previous studies in different cell types suggested that transcription factors AP-1, Elk-1 and STAT-1 may be involved in IL-1β and ERK1/2 regulation of various biological activities [25, 35]. Using Genomatix/Promoter Inspector software, transcription factor binding sequences for AP-1, Elk-1 and STAT-1 were identified on the MLCK promoter region, suggesting the possibility that these transcription factors may have a regulatory role on MLCK gene activity. The IL-1β effect on transcription factor activation was determined by binding of the activated transcription factors to their respective DNA binding site using ELISA-based DNA binding assay [32, 36]. IL-1β treatment of Caco-2 cells resulted in an activation of Elk-1 and AP-1 but not STAT-1 (Fig. 6A–C), suggesting that AP-1 or Elk-1 (but not STAT-1) may be involved in IL-1β modulation of MLCK gene expression. Next, the involvement of AP-1 or Elk-1 in IL-1β-induced increase in MLCK mRNA and protein was determined by selective knockdown of AP-1 or Elk-1 via siRNA transfection. The siRNA-induced knockdown of AP-1 (Fig. 7A) did not affect the IL-1β-induced increase in MLCK gene or protein level (Fig. 7B and C). In contrast, siRNA-induced knockdown of Elk-1 (Fig. 8A) almost completely inhibited the IL-1β-induced increase in Caco-2 MLCK mRNA and protein levels (Fig. 8B and C). The siRNA-induced knockdown of Elk-1 also inhibited the IL-1β-induced drop in Caco-2 TER (Fig. 8D) and increase in inulin flux (Fig. 8E). Together, these results indicated that Elk-1 was required for the IL-1β-induced increase in MLCK gene and protein expression and increase in Caco-2 monolayer TJ permeability. Next, the involvement of ERK1/2 signalling pathway on Elk-1 activation was determined. The inhibition of IL-1β-induced activation of ERK1/2 by PD-98059 (100 μM) inhibited the activation of Elk-1 (Fig. 9).
Fig 6.
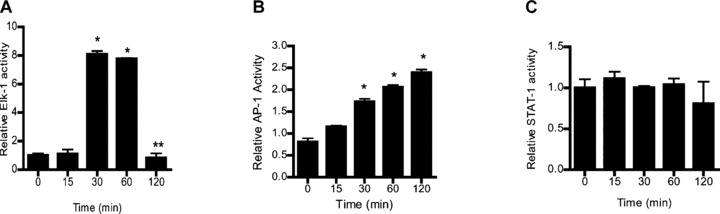
ELISA-based DNA binding assay of transcription factors Elk-1, AP-1 and STAT-1. (A and B) IL-1β treatment caused a significant increase in Caco-2 Elk-1 and AP-1 binding to the DNA probe. (C) IL-1β did not affect STAT-1 binding to its DNA binding site in Caco-2 monolayers (means ± S.E., n= 4). *P < 0.0001 versus control. Relative activity of transcription factors was determined by comparing the relative binding of the activated transcription factor of interest to its DNA binding site following IL-1β treated cells versus control cells.
Fig 7.
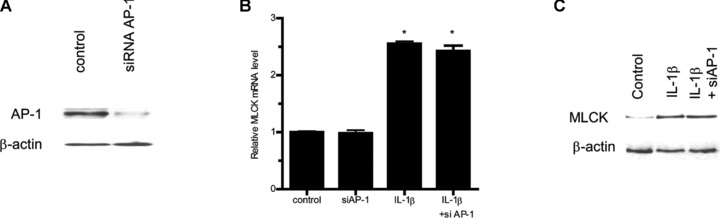
siRNA-induced AP-1 knockdown did not prevent IL-1β-induced increase in Caco-2 MLCK protein expression. (A) AP-1 siRNA transfection resulted in a near complete depletion in AP-1 protein expression as determined by Western blot analysis. (B) AP-1 siRNA transfection did not prevent the IL-1β-induced increase in Caco-2 MLCK mRNA levels (means ± S.E., n= 5). *P < 0.001 versus control. (C) AP-1 siRNA transfection did not inhibit the IL-1β-induced up-regulation of Caco-2 MLCK protein expression.
Fig 8.
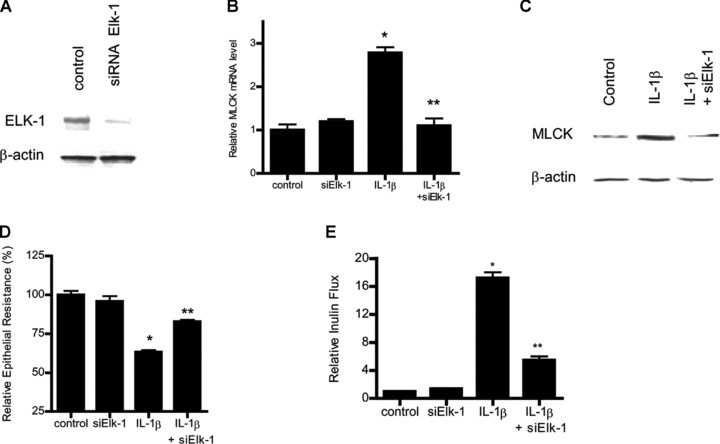
siRNA-induced Elk-1 knockdown prevented the IL-1β-induced increase in Caco-2 MLCK mRNA and protein expression. (A) Elk-1 siRNA transfection resulted in a near complete depletion in Elk-1 protein expression. (B) Elk-1 siRNA transfection significantly prevented the IL-1β-induced increase in Caco-2 MLCK mRNA levels (means ± S.E., n= 5). *P < 0.001 versus control; **P < 0.001 versus IL-1β treatment. (C) Elk-1 silencing by siRNA transfection abolished the IL-1β up-regulation of Caco-2 MLCK protein expression. (D) Elk-1 siRNA transfection prevented the IL-1β-induced drop in Caco-2 TER (n= 4). *P < 0.001 versus control; **P < 0.001 versus IL-1β treatment. (E) Elk-1 silencing inhibited the IL-1β increase in mucosal-to-serosal inulin flux (n= 5). *P < 0.0001 versus control; **P < 0.0001 versus IL-1β treatment.
Fig 9.
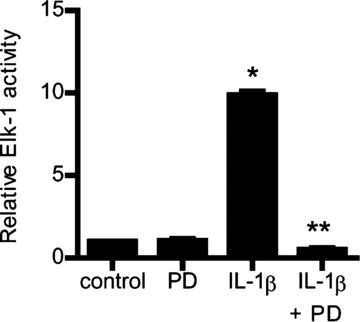
Effect of ERK1/2 inhibitor PD-98059 on Elk-1 activity. ERK1/2 inhibitor PD-98059 (100 μM) prevented the IL-1β-induced activation of Elk-1 as assessed by ELISA-binding assay (means ± S.E., n= 5). *P < 0.0001 versus control; **P < 0.001 versus IL-1β treatment.
Elk-1 regulates the MLCK promoter activity
To ascertain the molecular processes involved in IL-1β regulation of MLCK gene transcription, the involvement of Elk-1 in the regulation of MLCK promoter activity was determined. The siRNA knockdown of Elk-1 resulted in an inhibition of IL-1β-induced increase in MLCK promoter activity (Fig. 10), suggesting that Elk-1 was required for the activation of MLCK promoter. Next, to delineate the molecular determinants involved in IL-1β modulation of MLCK promoter activity, Elk-1 binding sequences were identified using Genomatix/Promoter Inspector software. Two Elk-1 binding motifs were identified on MLCK promoter region at –1118 to –1102 (TGGCCTTCCTCCCTC) designated as ‘site A’ and at –310 to –296 (GAAAATGGAAGTCCAAG) designated as ‘site B’ (Fig. 11A). Three MLCK promoter deletion constructs were generated that contained both Elk-1 binding sites (A and B) or those containing only the downstream binding site B (located within the minimal promoter region) (Fig. 11A). (The minimal MLCK promoter region had been previously localized between –313 and +118 [20].) Following IL-1β treatment, deletion constructs encoding only binding site B (MLCK-929 or MLCK –313) had a similar proportional increase in promoter activity as the full length (FL) promoter region containing both sites A and B (Fig. 11B and C), suggesting that binding site B (located within the MLCK minimal promoter region) was sufficient to cause an increase in MLCK promoter activity. To further validate the involvement of site B in the regulation of MLCK promoter activity, the binding site B was mutated via site-directed mutagenesis. The mutation of site B inhibited the increase in MLCK –313 promoter activity (Fig. 11D), confirming the requirement of site B for the up-regulation of promoter activity. Lastly, the effect of siRNA-induced silencing of Elk-1 on IL-1β-induced increase in MLCK promoter activity in MLCK –313 was determined. The siRNA-induced knockdown of Elk-1 inhibited the IL-1β-induced increase in promoter activity in MLCK –313 (Fig. 11E). In combination, these data indicated that Elk-1 binding site B located within the minimal promoter region was the regulatory site responsible for mediating the Elk-1 activation of MLCK promoter activity.
Fig 10.
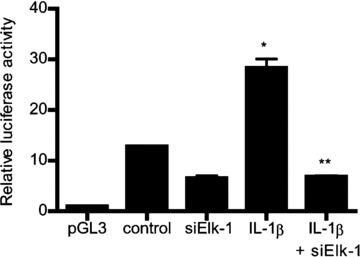
Elk-1 knockdown by siRNA transfection prevented the IL-1β-induced increase in MLCK promoter activity as assessed by luciferase activity. Caco-2 monolayers were co-transfected with siRNA Elk-1 for 96 hrs before IL-1β treatment (means ± S.E., n= 8). *P < 0.0001 versus control; **P < 0.0001 versus IL-1β treatment.
Fig 11.
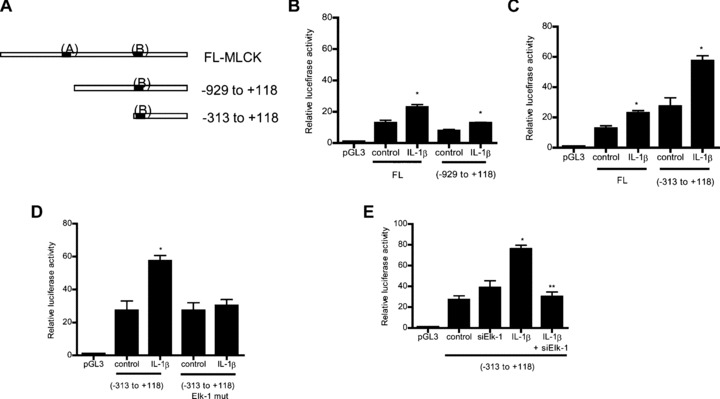
(A) Schematic diagram of the DNA constructs of MLCK promoter (–2109 to –18) containing different combinations of Elk-1 binding sites. The Elk-1 binding sites (A) (GAAAATGGAAGTCCAAG) and (B) (TGGCCTTCCTCCCTC) are represented in black. (B) Effect of IL-1β on the deletion construct of MLCK promoter region (–929 to +118) lacking the downstream Elk-1 binding site (A). IL-1β caused a similar proportional increase in MLCK luciferase activity as compared to the FL construct (means ± S.E., n= 8). *P < 0.001 versus control. (C) IL-1β treatment caused a significant increase in MLCK promoter activity of the deletion construct (–313 to +118) containing only the cis-binding site (B) in Caco-2 monolayers (means ± S.E., n= 8). *P < 0.0001 versus control. (D) Site directed mutagenesis of Elk-1 binding site (B) (TGGCCTTCCTCCCTC) on MLCK –313 prevented the IL-1β-induced increase in MLCK –313 promoter activity (means ± S.E., n= 5). *P < 0.001 versus control. (E) Effect of Elk-1 siRNA co-transfection on the IL-1β-induced increase in MLCK –313 promoter activity. Elk-1 silencing completely prevented the IL-1β-induced increase in MLCK luciferase activity (means ± S.E., n= 8). *P < 0.0001 versus control; **P < 0.0001 versus IL-1β treatment.
Discussion
The cellular and molecular mechanisms that mediate IL-1β regulation of intestinal epithelial TJ barrier remain unclear. Previous studies from our laboratory indicated that the IL-1β-induced increase in Caco-2 monolayer TJ permeability was mediated by an increase in MLCK protein level and enzymatic activity [20]. In these studies, the inhibition of MLCK protein expression or activity prevented the IL-1β-induced increase in Caco-2 TJ permeability [20]. The major aim of the present study was to delineate the signalling pathway and the molecular mechanisms involved in IL-1β modulation of intestinal epithelial TJ barrier using filter-grown Caco-2 monolayers as the intestinal epithelial model system. Our data indicated that the IL-1β-induced increase in Caco-2 TJ permeability was mediated by activation of ERK1/2 signalling cascade. Additionally, our data showed that nuclear transcription factor Elk-1 played a key intermediary role in the regulation of Caco-2 TJ barrier function by targeting the MLCK promoter activity.
The pro-inflammatory cytokines including IFN-γ, TNF-α and IL-1β cause a disturbance in intestinal TJ barrier as demonstrated by an increase in intestinal TJ permeability [32, 37, 38]. In contrast, anti-inflammatory cytokine IL-10 causes an enhancement in the epithelial TJ barrier function [39, 40]. The cytokine-induced alteration in intestinal TJ barrier has been shown to be an important pathogenic factor in the development of intestinal inflammation in murine models of intestinal inflammation [33]. It has been previously shown that the development of enterocolitis in IL-10−/− mice (an immune model of inflammatory bowel disease) is preceded by an initial increase in intestinal permeability [39], suggesting the possibility that an early defect in intestinal TJ barrier may allow an influx of noxious luminal antigens that lead to inflammatory response. In studying the causal relationship between intestinal TJ barrier defect and development of intestinal inflammation in IL-10−/− mouse, Madsen and co-workers recently showed that pharmacologic enhancement of intestinal TJ barrier in IL-10−/− mouse by oral administration of zonulin peptide inhibitor AT-1001 prevented the development of enterocolitis in IL-10−/− mice and they concluded that ‘in the IL-10−/− mouse, abnormal small intestinal permeability not only precedes the development of colitis but is etiologically important’[41]. Similarly, in other murine models of intestinal inflammation, the maintenance of intestinal TJ barrier function also prevented the development of intestinal inflammation or inflammation associated clinical sequelae [42–44]. Thus, elucidating the intracellular signalling pathways and the molecular processes involved in cytokine modulation of intestinal TJ barrier has great significance in understanding the basic physiology of TJ barrier regulation and in developing potential therapeutic targets to enhance or preserve the intestinal TJ barrier function during inflammatory conditions of the gut.
Our results indicated that the IL-1β-induced increase in Caco-2 intestinal epithelial TJ permeability was mediated by ERK1/2 signalling pathway. Our data showed that ERK1/2 activation preceded the IL-1β-induced increase in Caco-2 TJ permeability and inhibition of ERK1/2 activity prevented the increase in TJ permeability. This is the first study to show that ERK1/2 signalling cascade plays a regulatory role in the modulation of intestinal epithelial TJ barrier. In other studies in intestinal epithelial cell systems, ERK1/2 inhibition did not have any effect on intestinal TJ barrier regulation [45, 46]. Reardon et al. reported that the inhibition of ERK1/2 pathway did not affect IFN-γ-induced increase in T84 TJ permeability [45]. Similarly, Savkovic et al. reported that inhibition of ERK1/2 activation did not prevent the enteropathogenic E. coli induced increase in T84 TJ permeability [46]. In contrast, our present data show that ERK1/2 signalling cascade plays an essential role in IL-1β regulation of Caco-2 TJ barrier function. (In separate studies, we also found that IL-1β treatment (10 ng/ml) causes a drop in T84 TER, and that the decrease in T84 TER required activation of ERK1/2 signalling pathway [data not shown].)
Previous studies from our laboratory indicated that the IL-1β-induced increase in Caco-2 TJ permeability was regulated by an increase in MLCK gene and protein expression [20]. These studies showed that the inhibition of IL-1β-induced increase in MLCK transcription or MLCK protein synthesis inhibited the increase in Caco-2 TJ permeability, leading to the conclusion that the IL-1β effect on intestinal epithelial TJ permeability was due to an increase in expression of downstream effector protein MLCK [20]. Our present data expand on those findings to show that IL-1β increase in MLCK gene and protein expression was mediated in part by the activation of ERK1/2 signalling pathway. Our data showed that the inhibition of ERK1/2 activity prevented the IL-1β increase in MLCK gene and protein level and Caco-2 TJ permeability. Thus, our results suggested that the IL-1β-induced increase in Caco-2 TJ permeability was mediated by ERK1/2 signalling pathway regulation of MLCK gene and protein expression. As for the intracellular mechanisms that mediate the MLCK modulation of intestinal TJ barrier, previous studies have suggested that peri-junctional acto-myosin filaments are involved in the TJ barrier opening [47–49]. In brief, these studies showed that MLCK catalyses the phosphorylation of peri-junctional myosin light chain, which in turn, causes a sequential activation of myosin-Mg++ ATPase and ATP-dependent contraction of peri-junctional actin/myosin filaments [47]. The contraction of peri-junctional actomyosin filaments cause mechanical tension near the cellular junctions, leading to a tension-induced retraction of apical membrane and opening of the TJs [47–49].
Our studies also identified Elk-1 as the transcription factor that mediated the IL-1β/ERK1/2 modulation of Caco-2 TJ permeability. Elk-1 belongs to the ETS domain transcription factor family and the ternary complex factor subfamily [50]. Elk-1 forms a ternary complex with the serum response factor protein and binds the serum response element, inducing an activation of target gene activity. Elk-1 contains two domains that serve as docking region for MAP kinases that cause phosphorylation at the C-terminal domain [50]. The Elk-1 phosphorylation is critical for the transcriptional regulation. Our results suggested that the IL-1β-induced activation of Elk-1 was also dependent on ERK1/2 signalling cascade. The inhibition of ERK1/2 activation prevented the IL-1β-induced activation of Elk-1. Our data also showed that Elk-1 played an essential role in IL-1β up-regulation of MLCK gene expression. Although potential binding sites for other ERK1/2 activated transcription factors STAT-1 and AP-1 were also present on the MLCK promoter region, only Elk-1 appeared to have a regulatory role in MLCK mRNA expression. Consistent with our previous data showing the requirement of MLCK up-regulation in IL-1β modulation of TJ barrier, our present data also indicated that Elk-1 modulation of Caco-2 TJ barrier was due to an increase in MLCK gene and protein expression. Our data indicated that Elk-1 activation was required for both the increase in MLCK expression and increase in Caco-2 TJ permeability and that knockdown of MLCK prevented the increase in TJ permeability. As far as we are aware, this is the first study to show that Elk-1 plays a regulatory role in TJ barrier modulation. Interestingly, in previous studies, we also showed that NF-κB activation was required for the IL-1β-induced increase in MLCK gene and protein expression [20]. This appears that both Elk-1 and NF-κB play a critical role in mediating IL-1β regulation of MLCK gene expression. Although the precise mechanisms involved in Elk-1 and NF-κB modulation of MLCK gene activity remain unknown, we suggest that NF-κB and Elk-1 interact with each other to affect their binding to the MLCK promoter and the subsequent activation of MLCK promoter. Further studies are needed to fully elucidate the molecular interaction between NF-κB and Elk-1 that leads to MLCK gene activation.
In this study, we also examined some of the molecular interactions involved in the IL-1β regulation of MLCK gene activity. First, we showed that the increase in MLCK mRNA expression was due to an increase in MLCK promoter activity. Our studies also indicated that ERK1/2 activation caused a downstream activation of Elk-1. The activated Elk-1 translocated to the nucleus, binding to the cis-binding motif located within the minimal promoter region (−310 to −296). The Elk-1 binding to the cis-binding site caused an activation of MLCK promoter, and sequential increase in MLCK transcription and translation.
In conclusion, our data provide new insight into the intracellular pathway and the molecular processes that mediate the IL-1β-induced increase in intestinal epithelial TJ permeability. Our studies show that the IL-1β-induced increase in Caco-2 TJ permeability was mediated by an activation of ERK1/2 signalling cascade (Fig. 12). The ERK1/2 signalling pathway activation caused a downstream activation of transcription factor Elk-1. The activated Elk-1 binds to the cis-binding motif on the MLCK promoter to cause an activation of MLCK promoter activity, resulting in an increase in MLCK protein synthesis and MLCK catalysed opening of the intestinal epithelial TJ barrier (Fig. 12). Thus, our results show for the first time that ERK1/2 signalling pathways and transcription factor Elk-1 play an essential regulatory role in IL-1β regulation of intestinal epithelial TJ barrier function.
Fig 12.
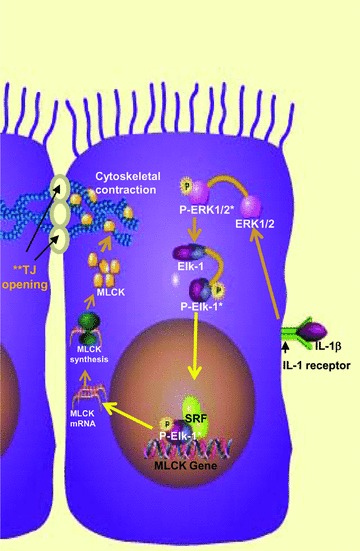
Proposed scheme of the IL-1β-induced increase in intestinal epithelial TJ permeability. IL-1β treatment resulted in activation of ERK1/2 signalling cascade, which then caused an activation of Elk-1- and Elk-1-induced activation of MLCK gene and increase in MLCK protein expression (* indicated activation of ERK1/2 and Elk-1 upon phosphorylation).
Acknowledgments
This research project was supported by a Veterans Affairs Merit Review grant from the Veterans Affairs Research Service and National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-64165 and R01-DK-081429 (to T.Y.M.).
References
- 1.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–147. [PubMed] [Google Scholar]
- 2.Dunne A, O’Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci STKE. 2003;2003:re3. doi: 10.1126/stke.2003.171.re3. [DOI] [PubMed] [Google Scholar]
- 3.O’Neill LA, Dinarello CA. The IL-1 receptor/toll-like receptor superfamily: crucial receptors for inflammation and host defense. Immunol Today. 2000;21:206–9. doi: 10.1016/s0167-5699(00)01611-x. [DOI] [PubMed] [Google Scholar]
- 4.Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–29. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 5.Reinecker HC, Steffen M, Witthoeft T, et al. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn’s disease. Clin Exp Immunol. 1993;94:174–81. doi: 10.1111/j.1365-2249.1993.tb05997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cominelli F, Pizarro TT. Interleukin-1 and interleukin-1 receptor antagonist in inflammatory bowel disease. Aliment Pharmacol Ther. 1996;2:49–53. doi: 10.1046/j.1365-2036.1996.22164020.x. [DOI] [PubMed] [Google Scholar]
- 7.Heresbach D, Alizadeh M, Dabadie A, et al. Significance of interleukin-1beta and interleukin-1 receptor antagonist genetic polymorphism in inflammatory bowel diseases. Am J Gastroenterol. 1997;92:1164–9. [PubMed] [Google Scholar]
- 8.Hyams JS, Fitzgerald JE, Wyzga N, et al. Relationship of interleukin-1 receptor antagonist to mucosal inflammation in inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 1995;21:419–25. doi: 10.1097/00005176-199511000-00008. [DOI] [PubMed] [Google Scholar]
- 9.Nemetz A, Nosti-Escanilla MP, Molnar T, et al. IL1B gene polymorphisms influence the course and severity of inflammatory bowel disease. Immunogenetics. 1999;49:527–31. doi: 10.1007/s002510050530. [DOI] [PubMed] [Google Scholar]
- 10.Manchanda PK, Bid HK, Mittal RD. Ethnicity greatly influences the interleukin-1 gene cluster(IL-1b promoter, exon-5 and IL-1Ra) polymorphisms: a pilot study of a north Indian population. Asian Pac J Cancer Prev. 2005;6:541–6. [PubMed] [Google Scholar]
- 11.Ma TY, Anderson JM. Tight junctions and the intestinal barrier. In: Johnson LR, editor. Physiology of the Gastrointestinal Tract. Burlington: MA; 2006. pp. 1559–94. [Google Scholar]
- 12.Utech M, Bruwer M, Nusrat A. Tight junctions and cell-cell interactions. Methods Mol Biol. 2006;341:185–95. doi: 10.1385/1-59745-113-4:185. [DOI] [PubMed] [Google Scholar]
- 13.Hollander D. Crohn’s disease–a permeability disorder of the tight junction. Gut. 1988;29:1621–4. doi: 10.1136/gut.29.12.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hollander D, Vadheim CM, Brettholz E, et al. Increased intestinal permeability in patients with Crohn’s disease and their relatives. A possible etiologic factor. Ann Intern Med. 1986;105:883–5. doi: 10.7326/0003-4819-105-6-883. [DOI] [PubMed] [Google Scholar]
- 15.May GR, Sutherland LR, Meddings JB. Is small intestinal permeability really increased in relatives of patients with Crohn’s disease. Gastroenterology. 1993;104:1627–32. doi: 10.1016/0016-5085(93)90638-s. [DOI] [PubMed] [Google Scholar]
- 16.Miehsler W, Puspok A, Oberhuber T, et al. Impact of different therapeutic regimens on the outcome of patients with Crohn’s disease of the upper gastrointestinal tract. Inflamm Bowel Dis. 2001;7:99–105. doi: 10.1097/00054725-200105000-00004. [DOI] [PubMed] [Google Scholar]
- 17.Wild GE, Waschke KA, Bitton A, et al. The mechanisms of prednisone inhibition of inflammation in Crohn’s disease involve changes in intestinal permeability, mucosal TNFalpha production and nuclear factor kappa B expression. Aliment Pharmacol Ther. 2003;18:309–17. doi: 10.1046/j.1365-2036.2003.01611.x. [DOI] [PubMed] [Google Scholar]
- 18.Wyatt J, Vogelsang H, Hubl W, et al. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet. 1993;341:1437–9. doi: 10.1016/0140-6736(93)90882-h. [DOI] [PubMed] [Google Scholar]
- 19.Arnott ID, Watts D, Ghosh S. Review article: is clinical remission the optimum therapeutic goal in the treatment of Crohn’s disease. Aliment Pharmacol Ther. 2002;16:857–67. doi: 10.1046/j.1365-2036.2002.01242.x. [DOI] [PubMed] [Google Scholar]
- 20.Al-Sadi R, Ye D, Dokladny K, et al. Mechanism of IL-1beta-induced increase in intestinal epithelial tight junction permeability. J Immunol. 2008;180:5653–61. doi: 10.4049/jimmunol.180.8.5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brun P, Castagliuolo I, Di Leo V, et al. Increased intestinal permeability in obese mice: new evidence in the pathogenesis of nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2007;292:G518–25. doi: 10.1152/ajpgi.00024.2006. [DOI] [PubMed] [Google Scholar]
- 22.Tadros T, Traber DL, Heggers JP, et al. Effects of interleukin-1alpha administration on intestinal ischemia and reperfusion injury, mucosal permeability, and bacterial translocation in burn and sepsis. Ann Surg. 2003;237:101–9. doi: 10.1097/00000658-200301000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walsh SV, Hopkins AM, Chen J, et al. Rho kinase regulates tight junction function and is necessary for tight junction assembly in polarized intestinal epithelia. Gastroenterology. 2001;121:566–79. doi: 10.1053/gast.2001.27060. [DOI] [PubMed] [Google Scholar]
- 24.Ieda Y, Waguri-Nagaya Y, Iwahasi T, et al. IL-1beta-induced expression of matrix metalloproteinases and gliostatin/platelet-derived endothelial cell growth factor (GLS/PD-ECGF) in a chondrosarcoma cell line (OUMS-27) Rheumatol Int. 2001;21:45–52. doi: 10.1007/s002960100129. [DOI] [PubMed] [Google Scholar]
- 25.Fyfe M, Bergstrom M, Aspengren S, et al. PAR-2 activation in intestinal epithelial cells potentiates interleukin-1beta-induced chemokine secretion via MAP kinase signaling pathways. Cytokine. 2005;31:358–67. doi: 10.1016/j.cyto.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 26.Xie Z, Singh M, Singh K. ERK1/2 and JNKs, but not p38 kinase, are involved in reactive oxygen species-mediated induction of osteopontin gene expression by angiotensin II and interleukin-1beta in adult rat cardiac fibroblasts. J Cell Physiol. 2004;198:399–407. doi: 10.1002/jcp.10419. [DOI] [PubMed] [Google Scholar]
- 27.Nadjar A, Combe C, Busquet P, et al. Signaling pathways of interleukin-1 actions in the brain: anatomical distribution of phospho-ERK1/2 in the brain of rat treated systemically with interleukin-1beta. Neuroscience. 2005;134:921–32. doi: 10.1016/j.neuroscience.2005.04.035. [DOI] [PubMed] [Google Scholar]
- 28.Rosell A, Arai K, Lok J, et al. Interleukin-1beta augments angiogenic responses of murine endothelial progenitor cells in vitro. J Cereb Blood Flow Metab. 2009;29:933–43. doi: 10.1038/jcbfm.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strle K, McCusker RH, Johnson RW, et al. Prototypical anti-inflammatory cytokine IL-10 prevents loss of IGF-I-induced myogenin protein expression caused by IL-1beta. Am J Physiol Endocrinol Metab. 2008;294:E709–18. doi: 10.1152/ajpendo.00662.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang HT, Cohen P, Rousseau S. IL-1beta-stimulated activation of ERK1/2 and p38alpha MAPK mediates the transcriptional up-regulation of IL-6, IL-8 and GRO-alpha in HeLa cells. Cell Signal. 2008;20:375–80. doi: 10.1016/j.cellsig.2007.10.025. [DOI] [PubMed] [Google Scholar]
- 31.Clayburgh DR, Rosen S, Witkowski ED, et al. A differentiation-dependent splice variant of myosin light chain kinase, MLCK1, regulates epithelial tight junction permeability. J Biol Chem. 2004;279:55506–13. doi: 10.1074/jbc.M408822200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Al-Sadi RM, Ma TY. IL-1beta causes an increase in intestinal epithelial tight junction permeability. J Immunol. 2007;178:4641–9. doi: 10.4049/jimmunol.178.7.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Al-Sadi R, Boivin M, Ma T. Mechanism of cytokine modulation of epithelial tight junction barrier. Front Biosci. 2009;14:2765–78. doi: 10.2741/3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ye D, Ma I, Ma TY. Molecular mechanism of tumor necrosis factor-alpha modulation of intestinal epithelial tight junction barrier. Am J Physiol Gastrointest Liver Physiol. 2006;290:G496–504. doi: 10.1152/ajpgi.00318.2005. [DOI] [PubMed] [Google Scholar]
- 35.Larsen CM, Wadt KA, Juhl LF, et al. Interleukin-1beta-induced rat pancreatic islet nitric oxide synthesis requires both the p38 and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinases. J Biol Chem. 1998;273:15294–300. doi: 10.1074/jbc.273.24.15294. [DOI] [PubMed] [Google Scholar]
- 36.Boivin MA, Ye D, Kennedy JC, et al. Mechanism of glucocorticoid regulation of the intestinal tight junction barrier. Am J Physiol Gastrointest Liver Physiol. 2007;292:G590–8. doi: 10.1152/ajpgi.00252.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma TY, Iwamoto GK, Hoa NT, et al. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–76. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- 38.Madara JL, Stafford J. Interferon-gamma directly affects barrier function of cultured intestinal epithelial monolayers. J Clin Invest. 1989;83:724–7. doi: 10.1172/JCI113938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Madsen KL, Malfair D, Gray D, et al. Interleukin-10 gene-deficient mice develop a primary intestinal permeability defect in response to enteric microflora. Inflamm Bowel Dis. 1999;5:262–70. doi: 10.1097/00054725-199911000-00004. [DOI] [PubMed] [Google Scholar]
- 40.Wang Q, Hasselgren PO. Heat shock response reduces intestinal permeability in septic mice: potential role of interleukin-10. Am J Physiol Regul Integr Comp Physiol. 2002;282:R669–76. doi: 10.1152/ajpregu.00606.2001. [DOI] [PubMed] [Google Scholar]
- 41.Arrieta MC, Madsen K, Doyle J, et al. Reducing small intestinal permeability attenuates colitis in the IL10 gene-deficient mouse. Gut. 2009;58:41–8. doi: 10.1136/gut.2008.150888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clayburgh DR, Barrett TA, Tang Y, et al. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest. 2005;115:2702–15. doi: 10.1172/JCI24970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clayburgh DR, Musch MW, Leitges M, et al. Coordinated epithelial NHE3 inhibition and barrier dysfunction are required for TNF-mediated diarrhea in vivo. J Clin Invest. 2006;116:2682–94. doi: 10.1172/JCI29218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferrier L, Mazelin L, Cenac N, et al. Stress-induced disruption of colonic epithelial barrier: role of interferon-gamma and myosin light chain kinase in mice. Gastroenterology. 2003;125:795–804. doi: 10.1016/s0016-5085(03)01057-6. [DOI] [PubMed] [Google Scholar]
- 45.Reardon C, McKay DM. TGF-beta suppresses IFN-gamma-STAT1-dependent gene transcription by enhancing STAT1-PIAS1 interactions in epithelia but not monocytes/macrophages. J Immunol. 2007;178:4284–95. doi: 10.4049/jimmunol.178.7.4284. [DOI] [PubMed] [Google Scholar]
- 46.Savkovic SD, Ramaswamy A, Koutsouris A, et al. EPEC-activated ERK1/2 participate in inflammatory response but not tight junction barrier disruption. Am J Physiol Gastrointest Liver Physiol. 2001;281:G890–8. doi: 10.1152/ajpgi.2001.281.4.G890. [DOI] [PubMed] [Google Scholar]
- 47.Ma TY, Hoa NT, Tran DD, et al. Cytochalasin B modulation of Caco-2 tight junction barrier: role of myosin light chain kinase. Am J Physiol Gastrointest Liver Physiol. 2000;279:G875–85. doi: 10.1152/ajpgi.2000.279.5.G875. [DOI] [PubMed] [Google Scholar]
- 48.Madara JL, Barenberg D, Carlson S. Effects of cytochalasin D on occluding junctions of intestinal absorptive cells: further evidence that the cytoskeleton may influence paracellular permeability and junctional charge selectivity. J Cell Biol. 1986;102:2125–36. doi: 10.1083/jcb.102.6.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Madara JL, Moore R, Carlson S. Alteration of intestinal tight junction structure and permeability by cytoskeletal contraction. Am J Physiol. 1987;253:C854–61. doi: 10.1152/ajpcell.1987.253.6.C854. [DOI] [PubMed] [Google Scholar]
- 50.Sharrocks AD. Complexities in ETS-domain transcription factor function and regulation: lessons from the TCF (ternary complex factor) subfamily. The Colworth Medal Lecture. Biochem Soc Trans. 2002;30:1–9. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]