

Abstract
Purpose
PIK3CA gene encoding a catalytic subunit of the phosphatidylinositol-3-kinase (PI3K) is mutated and/or amplified in various neoplasia, including lung cancer. Here we investigated PIK3CA gene alterations, the expression of core components of PI3K pathway, and evaluated their clinical importance in non-small cell lung cancer (NSCLC).
Materials and methods
Oncogenic mutations/rearrangements in PIK3CA, EGFR, KRAS, HER2, BRAF, AKT1 and ALK genes were detected in tumors from 1117 patients with NSCLC. PIK3CA gene copy number was examined by fluorescent in situ hybridization and the expression of PI3K p110 subunit alpha (PI3K p110α), p-Akt, mTOR, PTEN was determined by immunohistochemistry in PIK3CA mutant cases and 108 patients without PIK3CA mutation.
Results
PIK3CA mutation was found in 3.9% of squamous cell carcinoma and 2.7% of adenocarcinoma. Among 34 PIK3CA mutant cases, 17 tumors harbored concurrent EGFR mutations and 4 had KRAS mutations. PIK3CA mutation was significantly associated with high expression of PI3K p110α (p<0.0001), p-Akt (p = 0.024) and mTOR (p = 0.001), but not correlated with PIK3CA amplification (p = 0.463). Patients with single PIK3CA mutation had shorter overall survival than those with PIK3CA-EGFR/KRAS co-mutation or wildtype PIK3CA (p = 0.004). A significantly worse survival was also found in patients with PIK3CA mutations than those without PIK3CA mutations in the EGFR/KRAS wildtype subgroup (p = 0.043)
Conclusions
PIK3CA mutations frequently coexist with EGFR/KRAS mutations. The poor prognosis of patients with single PIK3CA mutation in NSCLC and the prognostic value of PIK3CA mutation in EGFR/KRAS wildtype subgroup suggest the distinct mutation status of PIK3CA gene should be determined for individual therapeutic strategies in NSCLC.
Introduction
It has been well established that the phosphatidylinositol-3-kinase (PI3K) pathway is related to carcinogenesis in a variety of human cancers [1], [2], [3]. Upon activation, PI3K initiates events leading to phosphorylation of Akt, which affects additional downstream signaling proteins involved in cell growth, metabolism, proliferation, survival, motility, and invasion [4], [5], [6]. PI3K-dependent activity is frequently elevated due to mutation of PIK3CA, a gene encoding the p110α catalytic subunit of PI3K (PI3K p110α), and the absence of the phosphatase and tensin homolog(PTEN) protein, a tumor suppressor with an important role in regulating the PI3K antiapoptotic and survival pathway [7], [8]. In addition, increased copy number of PIK3CA is also shown to be associated with increased PIK3CA transcription, p110α protein expression and PI3-kinase activity [9].
Aberrations in the components of the PI3K signaling pathway have been reported in many solid tumors, including lung cancer [2], [4], [7], [9]. Multiple mutations of PIK3CA that occur with regularity and in highly conserved regions of the gene lead to amino acid substitutions in the helical binding domain encoded by exon 9 and in the catalytic subunit of p110α encoded by exon 20, which result in upregulating PI3K pathway signaling [10]. In lung cancer, copy number gains of PIK3CA were found to be exclusive to PIK3CA mutation, implying that both alterations may have oncogenic potential to promote carcinogenesis in the lung [11]. The PTEN protein negatively regulates the PI3K pathway [12] and loss of PTEN protein expression has been linked to poor survival in patients with tongue cancer, and with more advanced tumor in esophageal and oral squamous cell cancers, respectively [13], [14]. Furthermore, Akt and mTOR lie downstream of PI3K and increased mTOR phosphorylation is frequently observed alongside with activated Akt in NSCLC and dysregulation of mTOR contributes to lung cancer progression [15], [16].
In our previous study, we reported that 90% of 52 lung adenocarcinoma samples from East Asian never smokers harbored driver mutations in just EGFR, KRAS, HER2 and ALK genes [17]. The frequency of EGFR, KRAS, HER2 mutations and EML4-ALK fusion were 75.3%, 2%, 5.9% and 5%, separately, in recent analysis of 202 lung adenocarcinoma samples from Chinese patients who never smoked [18]. However, no more than 40% of cases of NSCLC which includes squamous cell carcinoma, adenocarcinoma and large cell carcinoma histology would harbor such alterations [18]. It is now evident that even within a clearly identifiable histologic subtype, distinct molecular changes may be associated with a spectrum of clinical characteristics and also correlate with disease outcome and response to treatment [19]. As a result, it will be necessary to clarify different molecular alteration for individual treatment.
To date, there are few studies that constitute a comprehensive picture of the expression of components in PI3K pathway, PIK3CA gene alteration, and their correlation to NSCLC [20], [21]. In the present study, we examined PIK3CA gene mutation, PIK3CA amplification as well as the expression of PI3K p110α, p-Akt, mTOR and PTEN which lie in the PI3K pathway in a consecutive collection of NSCLC tumor samples. This detailed understanding of PI3K pathway alterations in NSCLC might enable a more precise delineation of candidate target populations, facilitating clinical trial design and validation of predictive biomarkers.
Materials and Methods
Patients and samples
From October 2007 to December 2012, we consecutively procured primary tumor samples from NSCLC patients who underwent pulmonary resection at the Department of Thoracic Surgery, Fudan University Shanghai Cancer Centre. Subjects eligible for this study had to meet the following: pathologically confirmed lung adenocarcinoma or lung squamous cell carcinoma, each sample containing sufficient tissue for comprehensive mutational analyses and with no neoadjuvant treatment. Patients were followed up every 3 months for the first 2 years, then biannually thereafter. A contrast-enhanced chest computed tomography (CT) scan was taken every 3 months for the first 2 years and then every 6 months thereafter. If recurrence was suspected either through newly presenting symptoms or through scheduled tests, integrated positron emission tomography/CT (PET/CT) was performed. PET/CT was also taken in patients without symptoms or abnormal findings in the scheduled tests 1 year after the surgical resection. Final diagnosis of recurrence was confirmed by the histopathological examination of samples obtained from surgery or biopsy. If it was impossible to diagnose the recurrence histopathologically, recurrent malignancy was no longer suspected based on the clinical and radiological follow-up period of at least 12 months with no evidence of active malignancy. This research was approved by the Institutional Review Board of the Fudan University Shanghai Cancer Center. Written informed consent was obtained from all patients.
Mutational analyses
Frozen tissue of tumor specimens was grossly dissected into TRIZOL (Invitrogen, Life Technologies, Carlsbad, CA), followed by total RNA extraction using standard protocol. Total RNA samples were reverse transcribed into cDNA. PIK3CA exon 9 includes codons 542 and 545 and PIK3CA exon 20 includes codon 1047, where the majority of mutations occur [22]. So we searched for mutations in PIK3CA exons 9 and 20. EGFR (exons 18–21), HER2 (exons 18–21), KRAS (exons 2–3), BRAF (exons 11–15) and AKT1 (exons 2–3) were also amplified by PCR using cDNA. Amplified products were analyzed by direct dideoxynucleotide sequencing. To identify EML4-ALK fusions, multiple 50 primers were used along with a fixed 30 primer localizing to ALK exon 20 to detect all known EML4 fusion variants as previously described [17]. Primers used to detect fusions between ALK and KIF5b or TFG were as previously reported [23], [24]. ALK FISH was also used to confirm the accuracy of PCR. All mutated cases were confirmed twice with independent PCR reactions. New data was not generated in our study.
Expression of core components of PI3K pathway
Tumoral tissue selection and immunohistochemial evaluation were performed by two pathologists (Yuan L and Lei S). Surgical specimens were fixed in 10% formalin and embedded in paraffin; 4-µm sections from lung tumors were prepared and deparaffinized, and antigen retrieval was done by microwaving. Endogenous peroxidase activity was blocked with 0.3% H2O2. After blocking with normal serum, sections were incubated for 120 minutes with monoclonal antibodies against PI3K p110α (1∶400 dilution, clone: C73F8; Cell Signaling Technology), PTEN (1∶50 dilution, clone: 138G6; Cell Signaling Technology), p-Akt (1∶50 dilution, clone: Ser473; Cell Signaling Technology) and mTOR (1∶50 dilution, clone: 7C10; Cell Signaling Technology). Slides were washed in PBS and detected with horseradish peroxidase conjugated anti-rabbit/mouse Real Envision Detection kit (Gene Tech), followed by counterstaining with hematoxylin.
According to scoring system that has been reported previously in the literature [25], [26], Our PI3K p110α staining scoring was done as follows: The score was 0 if no positive tumor cells were found; 1 if positive tumor cells were <10%; and 2 if positive tumor cells were >10%. Tissues with scores of 0 or 1 were considered low expression; those with scores of 2 were considered high expression. Immunoreactivity of p-Akt, mTOR and PTEN was evaluated semiquantitatively based on staining intensity and proportion, as previously described [27]. Staining intensity was scored as: absent (0); weak (1); moderate (2), or strong staining (3). Staining proportion was scored as: none (0); less than 1/3 (1); 1/3 to 2/3 (2), or more than 2/3 of tumor cells (3). The overall score was calculated as the sum of the intensity score and the proportion score, yielding a score between 0 and 6. An overall score of 0–2 was regarded as low expression while the other scores were regarded as high expression statistical analysis. Immunohistochemical staining was independently evaluated by two pathologists (Yuan L and Lei S). In cases of different overall scores taken from the same tumor, the average score was considered the final overall score.
Assessment of PIK3CA gene amplification
Fluorescent in situ hybridization (FISH) assay for PIK3CA was performed by using PIK3CA probe that hybridizes to the band 3q26.32 with Texas Red (red) and centromere3(CEN3) with FITC(green) (Abbott Molecular, Abbott Park, IL) following routine methods. FISH analyses was interpreted by two experienced evaluators (Lei W and Yunjian P) blinded to the clinical data. At least 100 nuclei per patient were evaluated. Tissue samples with a PIK3CA/CEN3 ratio of 1.0 were classified as normal and those with a PIK3CA/CEN3 ratio between 1.0 and 2.0 were classified as having PIK3CA gains. A PIK3CA/CEN3 ratio of more than 2.0 was considered amplified. A minimum of 50 cells with both centromeric and PIK3CA gene signals were scored to give conclusive data.
Statistical analysis
Difference in proportions was analyzed by X2 or Fisher's exact test. Recurrence-free survival (RFS) duration was defined as the time of surgery to recurrence or last contact. Overall survival (OS) was defined as the time of surgery to death or last contact. Event was stated as recurrence or death since surgery. Patients alive and showing no recurrence at the last follow-up were censored. RFS and OS distributions were estimated using the Kaplan-Meier method. The log-rank test was used to determine survival differences between groups. Regression analyses of survival data, based on the Cox proportional hazards model, were conducted on RFS and OS. A forward stepwise selection procedure was implemented with a P-value threshold of 0.05 for inclusion in the multivariate analysis. Statistical significance was accepted when the P-value was <0.05. All data were analyzed using the Statistical Package for the Social Sciences Version 16.0 Software (SPSS Inc., Chicago, IL).
Results
PIK3CA gene mutation status in NSCLC
A total of 1,117 NSCLC patients including 646 men and 471 women were eligible for mutation analysis in this study. As presented in Table 1 , Figure 1 and Figure S1 in File S1. 3.0% (34/1117) patients harbored mutations in PIK3CA, accounting for 2.7% (22/807) of lung adenocarcinomas, and 3.9% (12/310) of squamous cell carcinomas. No significant correlation was observed between PIK3CA mutations and clinicopathological factors such as gender, age, pathological types, smoking history, tumor differentiation or stage ( Table 1 ).
Table 1. Clinicopathological data of 1117 NSCLC patients.
| Factors | Mutations patients Positive(%) | Wild type patients Positive(%) | p-Value |
| Stage | |||
| I | 13(38.2) | 544(50.2) | 0.168 |
| II∼IV | 21(61.8) | 539(49.8) | |
| Lymph node metastasis | |||
| N0 | 15(44.1) | 613(56.6) | 0.149 |
| N+ | 19(55.9) | 470(43.4) | |
| Smoking history | |||
| Never smoker | 16(47.1) | 582(53.7) | 0.442 |
| Current or former smoker | 18(52.9) | 501(46.3) | |
| Differentiation | |||
| Well | 4(11.8) | 150(13.9) | 0.728 |
| Moderately or poorly | 30(88.2) | 933(86.1) | |
| Pathological types | |||
| AD | 22(64.7) | 785(72.4) | 0.319 |
| SCC | 12(35.3) | 298(27.5) | |
| Age | |||
| ≤60 | 21(61.8) | 599(55.3) | 0.456 |
| >60 | 13(38.2) | 484(44.7) | |
| Gender | |||
| Male | 21(61.8) | 625(57.7) | 0.637 |
| Female | 13(38.2) | 458(42.3) | |
| Chemotherapy* | |||
| Adjuvant chemotherapy | 20(58.8) | 62(57.4) | |
| No chemotherapy | 14(41.2) | 46(42.6) | 0.884 |
N+: lymph node metastasis positive; AD: adenocarcinoma; SCC: squamous cell carcinoma;
Chemotherapeutic analysis based on 34 PIK3CA mutant patients and 108 PIK3CA wildtype patients.
Figure 1. Mutation in PIK3CA.

Boxes represent functional domains (the p85 binding domain, Ras binding domain, C2 domain, helical domain, and kinase domain). Frequency and different types of mutations detected within each region is indicated below and above the box.
Mutations in exon 9 encoding for the helical domain (E545K, E545Q, E545G, E545A, Q546R, E542K, T536I) were found in 21 patients. Exon 20 mutations coding for the kinase domain (H1047R, H1047L, M1043L, G1007R, Y1021C) were found in 13 patients. The most frequent mutations were E545K and H1047R occurring in 16(47.1%, 16/34) of 34 patients with PIK3CA mutations ( Figure 1 , Table 2 ). According to clinicopathologic data identified, analysis of frequency of mutations in the helical vs. kinase domain was carried out. There was a trend that more helical domain PIK3CA mutations were observed in patients with lymph node metastasis staged II∼III, but the difference did not reach statistical significance (Table S1 in File S1).
Table 2. Clinicopathological data about 34 PIK3CA mutant patients.
| PIK3CA | Other | Age | Sex | Tobacco use | pN | pStage | Pathology | Differentiation | PIK3CA amplifivation |
| E545K | EGFR | 50 | F | N | N0 | Ia | AD | Moderate | Not amp |
| E545Q | EGFR | 60 | M | C/F | N1 | IIIa | AD | Poor | Not amp |
| E545A | EGFR | 40 | M | N | N0 | Ia | SCC | Moderate | Not amp |
| E545A | EGFR | 57 | F | N | N0 | Ia | AD | Moderate | Not amp |
| E542K | EGFR | 65 | M | C/F | N2 | IIIa | AD | Moderate | Not amp |
| E545K | EGFR | 63 | M | C/F | N0 | IIa | AD | Moderate | Not amp |
| E545A | EGFR | 56 | M | C/F | N2 | III a | AD | Moderate | Not amp |
| T536I | EGFR | 64 | F | N | N1 | IIb | AD | Well | Not amp |
| E545K | EGFR | 63 | F | N | N2 | IIIa | AD | Moderate | Not amp |
| E545K | EGFR | 65 | F | N | N2 | IIIa | AD | Poor | Not amp |
| E542K | EGFR | 54 | F | N | N2 | IIIa | AD | Moderate | Not amp |
| H1047L | EGFR | 41 | F | N | N2 | IIIa | AD | Moderate | Not amp |
| H1047R | EGFR | 55 | M | N | N2 | IIIa | AD | Moderate | Not amp |
| H1047R | EGFR | 58 | F | N | N0 | Ia | AD | Moderate | Not amp |
| H1047R | EGFR | 34 | F | N | N0 | Ia | AD | Poor | Not amp |
| H1047R | EGFR | 52 | M | C/F | N1 | Ia | AD | Moderate | Not amp |
| H1047L | EGFR | 65 | F | N | N0 | Ia | AD | Well | Not amp |
| E545K | KRAS | 58 | M | C/F | N0 | Ia | SCC | Moderate | Not amp |
| E545K | KRAS | 58 | M | C/F | N2 | IIIa | AD | Moderate | Not amp |
| H1047L | KRAS | 49 | F | N | N0 | Ib | AD | Moderate | Not amp |
| G1007R | KRAS | 59 | M | C/F | N2 | IIIa | AD | Poor | Not amp |
| E545K | 58 | M | C/F | N0 | Ia | SCC | Moderate | Not amp | |
| Q546R | 57 | M | C/F | N2 | IIIa | AD | Poor | Not amp | |
| E542K | 54 | M | C/F | N2 | IIIa | SCC | Moderate | Amp | |
| E545G | 76 | M | N | N2 | IIIa | SCC | Poor | Amp | |
| E545K | 75 | M | C/F | N0 | Ia | SCC | Moderate | Not amp | |
| E545K | 68 | M | C/F | N0 | IIa | SCC | Well | Amp | |
| E542K | 68 | M | C/F | N2 | IIIa | AD | Moderate | Not amp | |
| E545K | 72 | M | N | N1 | IIb | SCC | Moderate | Not amp | |
| Y1021C | 72 | M | C/F | N0 | Ia | SCC | Moderate | Amp | |
| M1043L | 60 | M | C/F | N0 | IIa | SCC | Moderate | Not amp | |
| H1047R | 59 | M | C/F | N1 | IIa | SCC | Well | Not amp | |
| G1007R | 75 | F | C/F | N2 | IIIa | SCC | Moderate | Not amp | |
| H1047R | 38 | F | N | N0 | Ia | AD | Moderate | Amp |
F:female; M:male; N:never smoker; C/F:current or former smoker; AD:adenocarcinoma; SCC:squamous cell carcinoma; Amp:amplification.
PIK3CA mutations frequently coexist with EGFR/KRAS mutations in NSCLC
To explore the coexistence of PIK3CA and other oncogene mutations in non-small cell lung cancer, testing for EGFR, KRAS, HER2, BRAF, AKT1 gene mutations and ALK rearrangement was also arranged. EGFR and KRAS gene mutations were found in 536 (48.0%, 536/1117) and 67(6.0%, 67/1117) of 1117 patients. The occurrence rates of HER2, BRAF, AKT1 mutations and ALK rearrangement were 20(1.8%, 20/1117), 11(1%, 11/1117), 2(0.2%, 2/1117) and 33(3.0%, 33/1117) respectively. All of the identified ALK fusion variants were EML4-ALK. Other fusion variants such as KIF5B-ALK and TFG-ALK were not found in our study. Complete data about ALK rearrangement was shown in Table S2 in File S1. No correlation was found between PIK3CA mutations and other gene alterations neither in lung squamous cell carcinoma nor in adenocarcinoma groups (Table S3–S4 in File S1).
Among 34 PIK3CA mutant cases, twenty one (61.8%, 21/34) harbored concurrent oncogenic mutations — 17(81.0%, 17/21) EGFR mutations and 4(19.0%, 4/21) KRAS mutations ( Figure 2 ), accounting for 3.2% (17/536) of the EGFR-mutant cases and 6.0% (4/67) of the KRAS-mutant cases. Coexistence of PIK3CA with BFAF, HER2, AKT1 gene mutations or ALK rearrangement was not found. This PIK3CA-EGFR/KRAS co-mutation was more common in never smokers than in current or former smokers (p = 0.039), and in adenocarcinoma than in squamous cell carcinoma (p<0.0001). There was also a tendency toward a higher co-mutation ratio in females (p = 0.067) and in patients aged not more than 60 (p = 0.168) (Table S5 in File S1).
Figure 2. Coexisting mutations in patients with PIK3CA-mutant NSCLC.
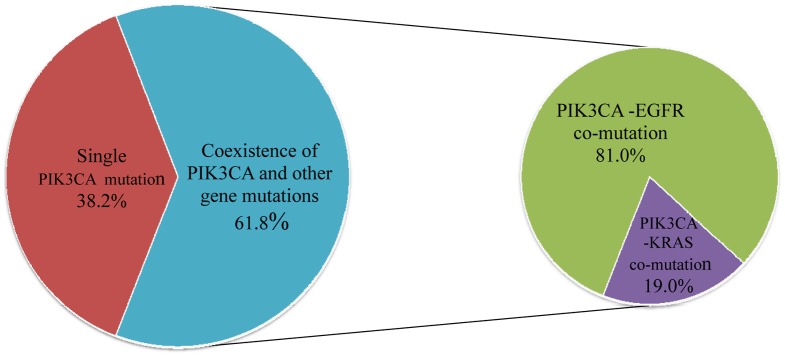
Specific histopathological subtype was also analysed in 807 lung adenocarcinoma and no significant difference in subtype was found between patients with and without PIK3CA mutation (p = 0.082, Table S6 in File S1). In 34 PIK3CA mutant cases, there was no significant correlation in histopathological subtype between patients with PIK3CA-EGFR/KRAS mutations and those only with PIK3CA mutations, either. (p = 0.121, Table S7 in File S1).
Immunohistochemical expression of PI3K p110α, p-AKT, mTOR, PTEN and their correlations
To evaluate the activity of PI3K/AKT pathway both in PIK3CA mutant and PIK3CA wildtype groups, we consecutively selected a smaller series of PIK3CA wildtype patients with primary NSCLC, surgically resected between July 2008 and June 2009 with similar clinical and pathological characteristics from 1117 patients examined above. 108 PIK3CA wildtype patients were collected. We determined PI3K p110α, p-Akt, mTOR and PTEN protein levels in 34 PIK3CA mutant cases and 108 PIK3CA wildtype cases by immunohistochemistry (IHC). Representative immunostaining for each protein was illustrated in Figure S2 in File S1. For PIK3CA mutant group, high cytoplasmic expression of PI3K p110α was detected in 27(79.4%, 27/34) tumors, whereas high expression of p-Akt (mostly in cytoplasmic) and mTOR (in cytoplasmic) was detected in 18(52.9%, 18/34) and 25(73.5%, 25/34) tumors, respectively. Low expression of PTEN (PTEN loss) in the cytoplasm and nucleus was seen in 8(23.5%, 8/34) tumors. For PIK3CA wild type group, high expression of PI3K p110α, p-Akt, mTOR were found in 42(38.9%, 42/108), 34(31.5%, 34/108) and 44(40.7%, 44/108) tumors respectively. Low expression of PTEN was seen in 30(27.8%, 30/108) tumors. As shown in Table S8–S9 in File S1, in either of the two groups, the association between each pair of PI3K p110α, p-Akt, mTOR proteins were statistically significant. However, no significant correlations were found between PTEN and other proteins. In PIK3CA mutant group, high PI3K p110α expression was associated with stage II to IV disease. (p = 0.043) (Table S8 in File S1) and in PIK3CA wild type group more high p-Akt expression was found in older patients (p = 0.037) (Table S9 in File S1). None of other clinicopathologic characteristics showed a significant relationship with the expression of PI3K p110α, p-Akt, mTOR or PTEN.
Analysis of PIK3CA gene amplification
To examine the copy number alterations of PIK3CA, the same panel of 142 frozen NSCLC tumor samples was analyzed by fluorescence in site hybridization including 34 tumors with mutation in the PIK3CA and 108 cases without PIK3CA mutation. PIK3CA amplification was detected in 5 PIK3CA mutant samples (14.7%, 5/34) which was more prevalent in lung squamous cell carcinoma (4/12 for lung squamous cell carcinoma vs. 1/12 for lung adenocarcinomas, p = 0.042). Nevertheless, in PIK3CA wild type group, PIK3CA amplification was detected in 22(20.4%, 20/108) tumors and was significantly associated with male gender (22/90 vs. 0/18, p = 0.021), current/former smoker (22/78 vs. 0/30, p = 0.001) and squamous cell carcinoma pathological type (19/52 vs. 3/56, p<0.0001). (Table S10 and Figure S2 in File S1)
Association among PIK3CA mutation, PIK3CA amplification and the expression of PI3K p110α, p-AKT, mTOR, PTEN
We further determined whether PIK3CA alterations were associated with the activity of PI3K pathway. We observed that PIK3CA mutation was significantly associated with high expression of PI3K p110α (p<0.0001), p-Akt (p = 0.024) and mTOR (p = 0.001) ( Table 3 ). However, no correlations were found between PIK3CA mutation and PTEN expression (p = 0.626) ( Table 3 ). In addition, PIK3CA amplification was not correlated with the expression of PI3K p110α, p-AKT, mTOR, PTEN neither in PIK3CA mutant nor wild type group (Table S10 in File S1). We also compared PIK3CA amplification with PIK3CA mutation status and found that there were five cases harboring PIK3CA amplification in combination with PIK3CA mutations. No evident relation was found between PIK3CA mutation and amplification (p = 0.463) ( Table 3 )
Table 3. Correlation among PIK3CA mutation, amplification and expression of PI3K p110α, p-Akt,m TOR and PTEN.
| Expression | PIK3CA mutation | P | |
| Positive(%) | Negative(%) | ||
| PI3K p110α (+) | 27(79.4) | 42(38.9) | <0.0001 |
| (−) | 7(20.6) | 66(61.1) | |
| PTEN loss (+) | 8(23.5) | 30(27.8) | 0.626 |
| (−) | 26(76.5) | 78(72.2) | |
| p-Akt (+) | 18(52.9) | 34(31.5) | 0.024 |
| (−) | 16(47.1) | 74(68.5) | |
| mTOR (+) | 25(73.5) | 44(40.7) | 0.001 |
| (−) | 9(26.5) | 64(59.3) | |
| PIK3CA amplification (+) | 5(14.7) | 22(20.4) | 0.463 |
| (−) | 29(85.3) | 86(79.6) |
Survival outcomes according to PIK3CA gene mutation and amplification status
We further detect the prognostic value of PIK3CA gene mutation and amplification in the same panel of 142 cases. To clarify whether the therapies influenced over survival (OS) and recurrence-free survival (RFS), we compared the OS and RFS between patients with and without adjuvant chemotherapy, finding no significant difference between two groups neither on OS nor RFS (Figure S3A–S3B in File S1). To identify whether patients with PIK3CA/EGFR co-mutations could benefit from EGFR tyrosine kinase inhibitor, we also investigated the adjuvant therapy of 34 PIK3CA mutant cases. Of these, 2 patients, with L858R and 746-deletion separately, were treated with gefitinib after operation. Partial response was obtained in both of the two patients
The over survival (OS) for whole cohort was 12.0 months with a median follow-up time of 12.2 months. There was no significant difference in median OS between PIK3CA mutant group and PIK3CA wild type group (p = 0.442; Figure 3A ). However, when PIK3CA mutant patients were sub-classified by single PIK3CA mutation or PIK3CA-EGFR/KRAS co-mutation, survival for patients with single PIK3CA mutation was shorter than patients with PIK3CA-EGFR/KRAS co-mutation or PIK3CA wild type group (p = 0.004; Figure 3B ).
Figure 3.

Overall survival curves for patients: with or without PIK3CA mutation (A); with single PIK3CA mutation, coexistence of PIK3CA and other gene mutation, and those in PIK3CA wild-type group (B); with or without PIK3CA mutation in EGFR/KRAS wild-type group (C); with PIK3CA mutation in exon 9 or exon 20(D).
It has been well characterized that EGFR mutation was usually associated with a relatively good prognosis while KRAS mutation was shown to be associated with a poor outcome, leaving an indeterminate prognostic subgroup for the EGFR/KRAS wildtype patients. Therefore, we further investigated the prognostic role of PIK3CA mutations in EGFR/KRAS wildtype NSCLC patients. We found a significantly worse survival in patients with PIK3CA mutations (p = 0.043; Figure 3C ). Besides, after PIK3CA mutant patients were allocated to E9 (PIK3CA mutation in exon 9) and E20 (PIK3CA mutation in exon 20) group, a trend was found that patient in E20 group survived longer than those in E9 group (p = 0.080; Figure 3D ).
The median recurrence-free survival (RFS) was 15.5 months in patients with PIK3CA mutation and 23.3 months in patients without PIK3CA mutations (p = 0.138, Figure 4A ). Like median OS, patients with single PIK3CA mutation had a shorter RFS than patients with PIK3CA-EGFR/KRAS co-mutation (p = 0.014, Figure 4B ). Significant difference on RFS was also found between cases with and without PIK3CA mutation in EGFR/KRAS wildtype subgroup (p = 0.046; Figure 4C ). Patients in E20 group had a longer RFS than those in E9 group (p = 0.003; Figure 4D ). PIK3CA amplification was not associated with overall survival (p = 0.491; Figure S4A in File S1) or recurrence-free survival (p = 0.884; Figure S4B in File S1). We further performed a multivariate analysis (Cox proportional hazards) with PIK3CA mutation status, histological subtype, age, gender, tumor stage, tumor grade as variable, and did not found the independently prognostic value of PIK3CA mutation in these factors.
Figure 4.

Recurrence-free survival curves for patients: with or without PIK3CA mutation (A); with single PIK3CA mutation, coexistence of PIK3CA and other gene mutation, and those in PIK3CA wild-type group (B). with or without PIK3CA mutation in EGFR/KRAS wild-type group (C); with PIK3CA mutation in exon 9 or exon 20 (D).
Discussion
In our study, we investigated the expression of proteins in PI3k pathway, molecular alteration in PIK3CA and its impact on survival in patients with NSCLC. To the best of our knowledge, this is the largest cohort of multiple analysis PIK3CA gene alteration and the activity of PI3K pathway and we found a negative prognostic impact of single PIK3CA mutation in NSCLC. Our study showed frequent overlap of PIK3CA and EGFR/KRAS mutations and demonstrated a poor prognostic value of PIK3CA mutation on EGFR/KRAS wildtype patients.
The frequency of PIK3CA mutation, as determined by direct sequencing was 3.9% in lung squamous cell carcinoma and 2.7% in adenocarcinoma, which is comparable to the value of 2.9% and 2.5% in a previous report which examined a small number of Japanese patients [21]. Interestingly, in contrast to PIK3CA-KRAS co-mutation, which is more prevalent in Western countries [28], half of the PIK3CA mutant patients in our study had simultaneous EGFR mutations. This may be attributed to the higher prevalence EGFR mutations in lung cancer patients from East than KRAS mutations [17], [29]. Moreover, although PI3K could be activated by receptor kinases and Ras, which in turn activated p-Akt, the PI3K/Akt pathway and EGFR signaling pathways interacted closely, PI3K signaling might have additional activators and downstream targets [11], [30]. Our findings on coexistence of PIK3CA and other gene mutations within EGFR signaling pathways are consistent with these observations.
Consistent with previous studies, high expression of PI3K p110α, p-Akt, mTOR and loss of PTEN was found in 79.4%, 52.9%, 73.5%, 23.5% of PIK3CA mutant group and 38.9%, 31.5%, 41.7%, 27.8% of PIK3CA wildtype group [20], [26], [27], [31], [32]. We observed that the presence PIK3CA mutation was associated with high expression of PI3K p110α, p-Akt, mTOR in NSCLC, similar to the results from ovarian clear cell carcinoma [31]. Nonetheless, one recent research on colorectal cancer reported PIK3CA mutation was not in accordance with the expression of PI3K p110α protein, indicating that PIK3CA mutations might not be the unique cause leading to high expression of PI3K p110α and might play diverse roles on the activity of PI3K pathway in distinct types of carcinomas [26]. We also demonstrated that PI3K p110α expression was positively correlated with the expression of p-Akt and mTOR, while p-Akt expression was also positively correlated with mTOR expression both in PIK3CA mutant group and PIK3CA wild type group. These results can be easily understood because that PIK3CA activates p-Akt, and positively regulates mTOR [27], [31]. A study by Marsit et al. suggested that regulation of PTEN is not always at the genetic level but also may occur at the transcriptional or translational level, which might also explain the limited correlation between PTEN loss and PIK3CA mutation observed [12]. Subsequent association analysis demonstrated that PIK3CA mutant tumors with high expression of PI3K p110α were more likely to be stage II to IV disease compared to tumors without high PI3K p110α expression. The expression of PI3K p110α was also found to be correlated with primary and metastatic lesions, suggesting that PI3K p110α might be involved in tumor progression and metastases [25], [26]. The exact molecular mechanism still warrants further study.
To extend our understanding of the mechanism beneath the activation of PI3K pathway, we investigated the correlation between PIK3CA amplification and clinicopathological variables in the same serial of NSCLC patients. Similar to the previous studies, we found that PIK3CA amplification was significantly associated with smoking history and histologic type, which was more prevalent in smokers compared to never smokers, and in squamous cell carcinomas compared to adenocarcinomas in PIK3CA wildtype group [33], [34]. Of note, in the present study, PIK3CA amplification was not associated with expression of proteins in PI3K pathway, which was similar to previous studies [11], [35]. These observations suggested PIK3CA amplification may not lead to activation of PI3K pathway in any types of lung cancer, insofar as it might be only one of the many downstream targets of PI3-kinase [34].
So far, the impact of PIK3CA mutations on survival is still contradictory. Some investigators reported better prognosis in certain cancers such as breast cancer with PIK3CA mutations, whereas others suggested that PIK3CA mutations indicated a worse prognosis in colorectal cancer, endometrial cancer and NSCLC [36], [37]. However, these results might not be able to successfully assess the true prognostic impact of PIK3CA mutation, because survival analysis according to PIK3CA mutation status was performed without consideration of the coexistence of PIK3CA and other oncogene mutation. In our study, patients with single PIK3CA mutation exhibited a shorter OS and RFS than those with PIK3CA-EGFR/KRAS co-mutation or those in PIK3CA wild type group. One plausible explanation was that PIK3CA mutation alone might be a prognostic marker for worse survival, however, patients with other oncogene mutations might be more likely to benefit from tyrosine kinase inhibitor therapy.
Furthermore, although EGFR mutation was found to be associated with good recurrence-free survival and KRAS mutation was shown as a poor prognosis factor, few predictive biomarkers were reported on EGFR/KRAS wildtype subgroup [38], [39]. Our study demonstrated a poor survival of PIK3CA mutation on patients without EGFR or KRAS mutations, confirming the prognostic role of PIK3CA mutation in EGFR/KRAS wildtype subgroup. In addition, as patients in E20 group had a longer OS and RFS than those in E9 group, studies in breast cancer had found clinical significance between patients with mutations in helical (exon 9) and kinase (exon 20) domain with inferior overall survival in those with mutation in the helical domain [40].These observations are also supported by findings in soft tissue sarcoma in which downstream activation level of PI3K is higher in tumors with helical domain mutations than those with kinase domain mutation [41].
Recently, the presence of co-occurrence of EGFR and other gene lesions was reported to reduce the sensitivity of EGFR-TKI [42], and the addition of a constitutively active PI3K mutant has been shown to confer gefitinib resistance in vitro [43]. Furthermore, it was also demonstrated that PIK3CA mutations in exon 20 were associated with resistance to EGFR-targeting monoclonal antibodies [44]. Thus the detection of PIK3CA mutation status and its coexistence with other gene mutation would be helpful to predict response to target therapy.
The strength of this study is the comprehensive analysis of PIK3CA gene alteration and the prognostic value of distinct PIK3CA mutation status in NSCLC. However, we fail to find the independently prognostic role of PIK3CA mutation after Cox regression. The sample size of in this series is relative small to draw definitive conclusions. Long term follow-up and larger studies or combinations of experience of multiple institutions will be helpful in clarifying whether this is a true association.
In conclusion, we demonstrated a high frequency of PIK3CA and EGFR/KRAS co-mutation in NSCLC and a negative prognostic value of PIK3CA mutation in EGFR/KRAS wildtype subgroup, indicating that different PIK3CA mutation status might contribute to distinct therapeutic targets in NSCLC. Further study exploring the PIK3CA alteration in a larger scale of population is warranted.
Supporting Information
Supporting information. Figure S1 Representative PIK3CA mutations by direct sequencing are shown for exon 9(A) and exon 20(B). Figure S2 Representative high expression of PI3K P110α, p-Akt, m TOR and low expression of PTEN protein in lung SCC; original amplification, ×200 (A); Representative high expression of PI3K, p-Akt, m TOR and PTEN protein in lung AD; original amplification, ×200 (B); Representative cell nuclei having normal(C), gain (D), and amplified (E) PIK3CA signals (red) and two centromeric signals (green). Figure S3 Overall survival curves for patients with or without adjuvant chemotherapy (A); Recurrence-free survival curves for patients with or without adjuvant chemotherapy (B). Figure S4 Overall survival curves for patients with or without PIK3CA amplification (A); Recurrence-free survival curves for patients with or without PIK3CA amplification (B). Table S1 Comparison of PIK3CA helical(exon9) and kinase(exon20) domain mutation. Table S2 ALK rearrangement in 1117 NSCLC patients. Table S3 Correlations between PIK3CA mutations and other gene alterations in lung adenocarcinoma. Table S4 Correlations between PIK3CA mutations and other gene alterations in lung squamous cell carcinoma. Table S5 Comparison of patients with single PIK3CA mutation to those with PIK3CA and other oncogene mutation. Table S6 Histopathological subtype in 785 PIK3CA wildtype and 22 PIK3CA mutant patients with lung adenocarcinoma. Table S7 Comparison of histopathological subtype between lung adenocarcinoma patients only with PIK3CA mutation and those co-exited with EGFR/KRAS mutation. Table S8 Associations of PI3K p110 α, p-Akt, mTOR, PTEN expression and PIK3CA amplification with clinicopathologic characteristics of 34 patients in PIK3CA mutant group. Table S9 Associations of PI3K p110 α, p-Akt, mTOR, PTEN expression and PIK3CA amplification with clinicopathologic characteristics of 108 patients in PIK3CA wild-type group. Table S10 Associations of PI3K p110α, p-Akt, mTOR, PTEN expression and PIK3CA amplification with clinicopathologic characteristics.
(PDF)
Acknowledgments
We thank Mr Chenguang Li for scientific review and editing of this article.
Funding Statement
This study was supported by grants from Key Construction Program of the National “985” Project (Grant No. 985III-YFX0102), National Natural Science Foundation of China (Grants No. 81172218 and 81101761), the Science and Technology Commission of Shanghai Municipality (Program of Shanghai Subject Chief Scientist; Grant No. 12XD1402000), the Foundation of Shanghai Health Administration (Grant No. 20114206), and a Grant from Shanghai Hospital Development Center (Grant No. SHDC12012308). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Vogt PK, Bader AG, Kang S (2006) Phosphoinositide 3-kinase: from viral oncoprotein to drug target. Virology 344: 131–138. [DOI] [PubMed] [Google Scholar]
- 2. Stephens P, Edkins S, Davies H, Greenman C, Cox C, et al. (2005) A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nat Genet 37: 590–592. [DOI] [PubMed] [Google Scholar]
- 3. Wang J, Ito T, Udaka N, Okudela K, Yazawa T, et al. (2005) PI3K-AKT pathway mediates growth and survival signals during development of fetal mouse lung. Tissue Cell 37: 25–35. [DOI] [PubMed] [Google Scholar]
- 4. Samuels Y, Ericson K (2006) Oncogenic PI3K and its role in cancer. Curr Opin Oncol 18: 77–82. [DOI] [PubMed] [Google Scholar]
- 5. Dunlap J, Le C, Shukla A, Patterson J, Presnell A, et al. (2010) Phosphatidylinositol-3-kinase and AKT1 mutations occur early in breast carcinoma. Breast Cancer Res Treat 120: 409–418. [DOI] [PubMed] [Google Scholar]
- 6. Paradiso A, Mangia A, Azzariti A, Tommasi S (2007) Phosphatidylinositol 3-kinase in breast cancer: where from here? Clin Cancer Res 13: 5988–5990. [DOI] [PubMed] [Google Scholar]
- 7. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, et al. (2004) High frequency of mutations of the PIK3CA gene in human cancers. Science 304: 554. [DOI] [PubMed] [Google Scholar]
- 8. Sansal I, Sellers WR (2004) The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol 22: 2954–2963. [DOI] [PubMed] [Google Scholar]
- 9. Shayesteh L, Lu Y, Kuo WL, Baldocchi R, Godfrey T, et al. (1999) PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet 21: 99–102. [DOI] [PubMed] [Google Scholar]
- 10. Saal LH, Holm K, Maurer M, Memeo L, Su T, et al. (2005) PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res 65: 2554–2559. [DOI] [PubMed] [Google Scholar]
- 11. Yamamoto H, Shigematsu H, Nomura M, Lockwood WW, Sato M, et al. (2008) PIK3CA mutations and copy number gains in human lung cancers. Cancer Res 68: 6913–6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marsit CJ, Zheng S, Aldape K, Hinds PW, Nelson HH, et al. (2005) PTEN expression in non-small-cell lung cancer: evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum Pathol 36: 768–776. [DOI] [PubMed] [Google Scholar]
- 13. Lee JI, Soria JC, Hassan KA, El-Naggar AK, Tang X, et al. (2001) Loss of PTEN expression as a prognostic marker for tongue cancer. Arch Otolaryngol Head Neck Surg 127: 1441–1445. [DOI] [PubMed] [Google Scholar]
- 14. Tachibana M, Shibakita M, Ohno S, Kinugasa S, Yoshimura H, et al. (2002) Expression and prognostic significance of PTEN product protein in patients with esophageal squamous cell carcinoma. Cancer 94: 1955–1960. [DOI] [PubMed] [Google Scholar]
- 15. Balsara BR, Pei J, Mitsuuchi Y, Page R, Klein-Szanto A, et al. (2004) Frequent activation of AKT in non-small cell lung carcinomas and preneoplastic bronchial lesions. Carcinogenesis 25: 2053–2059. [DOI] [PubMed] [Google Scholar]
- 16. Wislez M, Spencer ML, Izzo JG, Juroske DM, Balhara K, et al. (2005) Inhibition of mammalian target of rapamycin reverses alveolar epithelial neoplasia induced by oncogenic K-ras. Cancer Res 65: 3226–3235. [DOI] [PubMed] [Google Scholar]
- 17. Sun Y, Ren Y, Fang Z, Li C, Fang R, et al. (2010) Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J Clin Oncol 28: 4616–4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li C, Fang R, Sun Y, Han X, Li F, et al. (2011) Spectrum of oncogenic driver mutations in lung adenocarcinomas from East Asian never smokers. PLoS One 6: e28204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, et al. (2011) International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol 6: 244–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spoerke JM, O'Brien C, Huw L, Koeppen H, Fridlyand J, et al. (2012) Phosphoinositide 3-kinase (PI3K) pathway alterations are associated with histologic subtypes and are predictive of sensitivity to PI3K inhibitors in lung cancer preclinical models. Clin Cancer Res 18: 6771–6783. [DOI] [PubMed] [Google Scholar]
- 21. Okudela K, Suzuki M, Kageyama S, Bunai T, Nagura K, et al. (2007) PIK3CA mutation and amplification in human lung cancer. Pathol Int 57: 664–671. [DOI] [PubMed] [Google Scholar]
- 22. Moroni M, Veronese S, Benvenuti S, Marrapese G, Sartore-Bianchi A, et al. (2005) Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol 6: 279–286. [DOI] [PubMed] [Google Scholar]
- 23. Takeuchi K, Choi YL, Togashi Y, Soda M, Hatano S, et al. (2009) KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res 15: 3143–3149. [DOI] [PubMed] [Google Scholar]
- 24. Wong DW, Leung EL, Wong SK, Tin VP, Sihoe AD, et al. (2011) A novel KIF5B-ALK variant in nonsmall cell lung cancer. Cancer [DOI] [PubMed] [Google Scholar]
- 25. Wang Y, Kristensen GB, Helland A, Nesland JM, Borresen-Dale AL, et al. (2005) Protein expression and prognostic value of genes in the erb-b signaling pathway in advanced ovarian carcinomas. Am J Clin Pathol 124: 392–401. [DOI] [PubMed] [Google Scholar]
- 26. Zhu YF, Yu BH, Li DL, Ke HL, Guo XZ, et al. (2012) PI3K expression and PIK3CA mutations are related to colorectal cancer metastases. World J Gastroenterol 18: 3745–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Noh WC, Kim YH, Kim MS, Koh JS, Kim HA, et al. (2008) Activation of the mTOR signaling pathway in breast cancer and its correlation with the clinicopathologic variables. Breast Cancer Res Treat 110: 477–483. [DOI] [PubMed] [Google Scholar]
- 28. Chaft JE, Arcila ME, Paik PK, Lau C, Riely GJ, et al. (2012) Coexistence of PIK3CA and other oncogene mutations in lung adenocarcinoma-rationale for comprehensive mutation profiling. Mol Cancer Ther 11: 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu J, He J, Yang H, Luo X, Liang Z, et al. (2011) Somatic mutation analysis of EGFR, KRAS, BRAF and PIK3CA in 861 patients with non-small cell lung cancer. Cancer Biomark 10: 63–69. [DOI] [PubMed] [Google Scholar]
- 30. Engelman JA, Luo J, Cantley LC (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 7: 606–619. [DOI] [PubMed] [Google Scholar]
- 31. Abe A, Minaguchi T, Ochi H, Onuki M, Okada S, et al. (2013) PIK3CA overexpression is a possible prognostic factor for favorable survival in ovarian clear cell carcinoma. Hum Pathol 44: 199–207. [DOI] [PubMed] [Google Scholar]
- 32. Ali G, Boldrini L, Capodanno A, Pelliccioni S, Servadio A, et al. (2011) Expression of p-AKT and p-mTOR in a large series of bronchopulmonary neuroendocrine tumors. Exp Ther Med 2: 787–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ji M, Guan H, Gao C, Shi B, Hou P (2011) Highly frequent promoter methylation and PIK3CA amplification in non-small cell lung cancer (NSCLC). BMC Cancer 11: 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Massion PP, Taflan PM, Shyr Y, Rahman SM, Yildiz P, et al. (2004) Early involvement of the phosphatidylinositol 3-kinase/Akt pathway in lung cancer progression. Am J Respir Crit Care Med 170: 1088–1094. [DOI] [PubMed] [Google Scholar]
- 35. Jehan Z, Bavi P, Sultana M, Abubaker J, Bu R, et al. (2009) Frequent PIK3CA gene amplification and its clinical significance in colorectal cancer. J Pathol 219: 337–346. [DOI] [PubMed] [Google Scholar]
- 36. Kawano O, Sasaki H, Endo K, Suzuki E, Haneda H, et al. (2006) PIK3CA mutation status in Japanese lung cancer patients. Lung Cancer 54: 209–215. [DOI] [PubMed] [Google Scholar]
- 37. Janku F, Wheler JJ, Naing A, Stepanek VM, Falchook GS, et al. (2012) PIK3CA mutations in advanced cancers: characteristics and outcomes. Oncotarget 3: 1566–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guan J-l, Zhong W-z, An S-j, Yang J-j, Su J, et al. (2012) KRAS Mutation in Patients with Lung Cancer: A Predictor for Poor Prognosis but Not for EGFR-TKIs or Chemotherapy. Annals of Surgical Oncology 20: 1381–1388. [DOI] [PubMed] [Google Scholar]
- 39. Fukuoka M, Wu YL, Thongprasert S, Sunpaweravong P, Leong SS, et al. (2011) Biomarker Analyses and Final Overall Survival Results From a Phase III, Randomized, Open-Label, First-Line Study of Gefitinib Versus Carboplatin/Paclitaxel in Clinically Selected Patients With Advanced Non-Small-Cell Lung Cancer in Asia (IPASS). Journal of Clinical Oncology 29: 2866–2874. [DOI] [PubMed] [Google Scholar]
- 40. Barbareschi M, Buttitta F, Felicioni L, Cotrupi S, Barassi F, et al. (2007) Different prognostic roles of mutations in the helical and kinase domains of the PIK3CA gene in breast carcinomas. Clin Cancer Res 13: 6064–6069. [DOI] [PubMed] [Google Scholar]
- 41. Barretina J, Taylor BS, Banerji S, Ramos AH, Lagos-Quintana M, et al. (2010) Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet 42: 715–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pao W, Chmielecki J (2010) Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer 10: 760–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borras AM, et al. (2006) Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest 116: 2695–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, et al. (2010) Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 11: 753–762. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information. Figure S1 Representative PIK3CA mutations by direct sequencing are shown for exon 9(A) and exon 20(B). Figure S2 Representative high expression of PI3K P110α, p-Akt, m TOR and low expression of PTEN protein in lung SCC; original amplification, ×200 (A); Representative high expression of PI3K, p-Akt, m TOR and PTEN protein in lung AD; original amplification, ×200 (B); Representative cell nuclei having normal(C), gain (D), and amplified (E) PIK3CA signals (red) and two centromeric signals (green). Figure S3 Overall survival curves for patients with or without adjuvant chemotherapy (A); Recurrence-free survival curves for patients with or without adjuvant chemotherapy (B). Figure S4 Overall survival curves for patients with or without PIK3CA amplification (A); Recurrence-free survival curves for patients with or without PIK3CA amplification (B). Table S1 Comparison of PIK3CA helical(exon9) and kinase(exon20) domain mutation. Table S2 ALK rearrangement in 1117 NSCLC patients. Table S3 Correlations between PIK3CA mutations and other gene alterations in lung adenocarcinoma. Table S4 Correlations between PIK3CA mutations and other gene alterations in lung squamous cell carcinoma. Table S5 Comparison of patients with single PIK3CA mutation to those with PIK3CA and other oncogene mutation. Table S6 Histopathological subtype in 785 PIK3CA wildtype and 22 PIK3CA mutant patients with lung adenocarcinoma. Table S7 Comparison of histopathological subtype between lung adenocarcinoma patients only with PIK3CA mutation and those co-exited with EGFR/KRAS mutation. Table S8 Associations of PI3K p110 α, p-Akt, mTOR, PTEN expression and PIK3CA amplification with clinicopathologic characteristics of 34 patients in PIK3CA mutant group. Table S9 Associations of PI3K p110 α, p-Akt, mTOR, PTEN expression and PIK3CA amplification with clinicopathologic characteristics of 108 patients in PIK3CA wild-type group. Table S10 Associations of PI3K p110α, p-Akt, mTOR, PTEN expression and PIK3CA amplification with clinicopathologic characteristics.
(PDF)