

Abstract
Senescence, the state of irreversible cell-cycle arrest, plays paradoxical albeit important roles in vivo: it protects organisms against cancer but also contributes to age-related loss of tissue function. The DNA damage response (DDR) has a central role in cellular senescence. Not only does it contribute to the irreversible loss of replicative capacity but also to the production and secretion of reactive oxygen species (ROS), and bioactive peptides collectively known as the senescence-associated secretory phenotype (SASP). Both ROS and the SASP have been shown to impact on senescence in an autocrine as well as paracrine fashion; however, the underlying mechanisms are not well understood. In this review we describe our current understanding of cellular senescence, examine in detail the intricate pathways linking the DDR, ROS and SASP, and evaluate their impact on the stability of the senescent phenotype.
Keywords: DNA damage response, Telomeres, Senescence, Ageing, Oxidative stress, Inflammation
Review
Introduction
Cellular senescence, the state of irreversible cell cycle arrest described by Hayflick and Moorhead [1] over 50 years ago, remains an intriguing biological process. Senescence is characterised by dramatic changes in cell morphology, including increased cellular volume and flattening of the cytoplasm [2]. The senescent phenotype also results in changes in nuclear structure, gene expression, protein processing and metabolism, and resistance to apoptosis [3-6].
Whether senescence exists to any significant extent in vivo has been the subject of a longstanding debate [7]. In the past decade, remarkable advances have been made demonstrating that senescence plays an important role in vivo. Several studies suggest that senescence can act as a tumour suppressor mechanism [8,9]. On the other hand, numerous lines of evidence indicate that senescence can, in the long run, have adverse effects, by impairing organ regeneration and releasing a host of bioactive molecules, including reactive oxygen species (ROS) and a wide variety of pro-inflammatory cytokines, chemokines and growth factors (collectively referred to as the senescence-associated secretory phenotype (SASP)).
Senescent cells containing telomere-induced foci have been shown to increase with age in the skin of baboons, which have similar telomere length to humans and absence of telomerase activity [10]. In mice, cells bearing senescent markers have been reported to increase with age in a variety of tissues [11-13], including post-mitotic neurons [14]. Moreover, senescent cells have been associated with several age-related diseases, such as diabetes [15] and atherosclerosis [16]. While noteworthy, these data do not provide causality. A major challenge in the field has been to determine if and how senescent cells contribute to age-related tissue dysfunction, or if they merely correlate with it.
Mounting evidence indicates that activation of pathways involved in cellular senescence impacts on mammalian lifespan [17-19]. Recently, the van Deursen group has shown that inducible elimination of p16Ink4a-positive senescent cells from the eye, adipose and skeletal tissues in the BubR1 progeroid mouse model delayed acquisition of age-related pathologies in these tissues. They showed that elimination of p16Ink4a-positive cells also attenuated the progression of already established age-related disorders, suggesting that cellular senescence may have a causal role in age-related tissue impairment [20].
Though several mechanisms responsible for the activation of senescence have been identified, it is still unclear how a cell “commits” to becoming irreversibly arrested. Recent studies have revealed that the SASP, as well as mitochondrial/metabolic alterations, may contribute to the reinforcement of the growth arrest via a series of positive feedback loops involving a persistent activation of the DNA damage response (DDR) [21-23].
The aim of this review is to describe the current understanding of cellular senescence, providing special focus on the intricate pathways that link the nucleus, mitochondria and secreted proteins, and contribute to the stability of the senescent phenotype.
Telomeres and the stabilisation of cellular senescence
Telomeres are regions of DNA and associated proteins present at the end of linear chromosomes; in vertebrates they are tandem repeats of the sequence TTAGGG [24].
Telomeres are bound by a group of telomere-associated proteins known as the “shelterin” complex [25]. These proteins are thought to arrange telomeric DNA into a loop structure known as the T-loop [26]. This structure was first visualised in purified telomere restriction fragments using electron microscopy, and it is proposed to prevent the activation of a DDR by hiding the exposed DNA ends. The shelterin complex is comprised of six proteins: TRF1, TRF2 and POT1, which recognise the telomeric repeat sequence, and additional proteins TIN2, TPP1 and Rap1 [25].
Telomere shortening is probably the best studied mechanism driving cellular senescence. It mainly occurs during cell division due to the inability of the DNA replication machinery, specifically DNA polymerase, to synthesise in a 3′-5′ direction leading to the incomplete replication of the lagging strand. It has been shown that telomere shortening contributes causally to cellular senescence, since overexpression of telomerase, an enzyme able to maintain telomere length, resulted in cell immortalisation [27]. Mouse models, where telomere function has been compromised, strongly support a role for senescence (and telomeres) in the ageing process. Telomerase knock-out (mTERC-/-) mice which carry a homozygous deletion of the RNA component of telomerase [28] show a progressive generation-dependent telomere shortening, which results in both cell-cycle arrest and apoptosis [29]. Telomere dysfunction in mTERC-/- mice has been shown to limit stem cell function, regeneration, organ homeostasis and lifespan [30].
It is believed that the progressive loss of telomere repeats destabilises T-loops [26] and, as a consequence, increases the probability of telomere uncapping (that is, loss of “shelterin”). Uncapping of telomeres, whether by inhibition of TRF2 or telomere shortening, has been shown to activate the DDR in a manner similar to DNA double strand breaks (DSBs) [31,32]. The DDR can elicit a transient cell-cycle arrest, allowing sufficient time for the cellular repair machinery to act and repair the DNA damage [33]. However, if the damage is irreparable, the arrest can become permanent. This response is initiated by the phosphatidylinositol 3-kinase-like protein kinases ATM and ATR, which phosphorylate proteins such as H2A.X and NBS1, and downstream kinases CHK1 and CHK2, which ultimately activate p53 and p21 proteins [34]. Several groups have reported that senescence is characterised by a persistent activation of the DDR, which is necessary for both the development and stability of the phenotype [21,35].
One important question is: what contributes to a persistent DDR during cellular senescence? Recent work has highlighted the importance of telomeres in the maintenance of senescence. It has been demonstrated that DNA damage at telomeres can occur as a consequence of genotoxic and oxidative stress, and that this damage is mostly irreparable [13,36]. In order to establish whether a telomeric location is necessary for foci to persist, using live-cell imaging, our group has tracked the lifespan of DNA damage foci using a AcGFP-53BP1c fusion protein in combination with a fluorescently labelled PNA probe that specifically tags telomere repeats. Using this method it was found that the majority of long-lived foci in stress-induced senescent cells co-localise with telomeres [13], which suggests that they are major contributors to a persistent DDR.
These findings raise questions regarding how the cellular repair machinery distinguishes telomeres and DSBs. Non-homologous end joining (NHEJ) is strongly inhibited in telomeric regions, perhaps as a mechanism to prevent end-to-end fusions [37]. NHEJ is the major pathway for the repair of DSBs. Moreover, displacement of TRF2 from telomeres by overexpression of TRF2ΔBΔM, or conditional deletion of TRF2, has been shown to result in telomere fusions [37-39]. It has also been demonstrated in vitro that TRF2 and its binding partner RAP1 are required to prevent NHEJ-dependent telomeric DNA fusions by inhibiting DNA-PK and ligase IV mediated end-joining [40]. Consistent with these data, Fumagalli and colleagues have shown in budding yeast that induction of a DNA DSB adjacent to a telomeric sequence impairs the recruitment of ligase IV to the site of damage [36]. This suggests that damage at telomeres, occurring in the presence of sufficient shelterin components including TRF2, may elicit a persistent DDR due to inhibition of repair. In accordance with this hypothesis, it has been shown recently that during replicative senescence of human fibroblasts, telomeres positive for DDR retain both TRF2 and RAP1 and are not associated with end-to-end fusions [41].
Recent studies have shown that the role of telomeres in senescence may extend beyond attrition due to replication. A recent study has shown that oncogenic signals cause replication fork stalling, resulting in telomeric DNA damage accumulation, activation of a DDR and consequently senescence [42]. However, it has been reported that in both replicative and stress-induced senescent cells, 50% of DNA damage foci can be found in non-telomeric regions of the genome and are short-lived. Live-cell imaging studies have shown that these short-lived foci are maintained in relatively constant numbers per cell and that new foci are regularly being created during senescence [13,21]. Moreover, data indicate that these foci are mainly the result of ROS production during senescence and contribute to some degree to the stability and development of the phenotype. Consistently, following the activation of a DDR, inhibition of ROS production results in a small fraction of cells being able to resume proliferation [21].
Therefore, it is highly likely that both telomeric and non-telomeric regions are contributors to the senescent phenotype (Figure 1); however, their relative contribution towards senescence signalling is experimentally very difficult to dissect.
Figure 1.

Both telomeric and non-telomeric DNA damage contribute to the stabilisation of cellular senescence. DNA damage at telomeres is distinct from that throughout the genome; it is irreparable due to the repression of DNA repair pathways by telomere bound proteins, known as the “shelterin” complex. This contributes to a permanent DNA damage response (DDR). However, continuous generation of short-lived DDR foci by elevated reactive oxygen species (ROS) may equally contribute to the maintenance of the phenotype, as long as a dynamic equilibrium between damage induction and repair can be maintained.
Importantly, mechanisms other than the DDR have been shown to impact on the stability of the senescent phenotype. In several types of cells, senescence is accompanied by drastic changes in chromatin organisation, such as formation of senescence-associated heterochromatic foci, which are dependent on the p16/Rb pathway [6]. Senescence-associated heterochromatic foci have been shown to accumulate on the promoters of cell-cycle genes during senescence, and their occurrence has been shown to correlate with the irreversibility of the senescent phenotype [6,43].
Involvement of reactive oxygen species in the stabilisation of cellular senescence
ROS are likely to be involved in both the induction and stabilisation of cellular senescence: several studies have shown that ROS can accelerate telomere shortening [44], and can damage DNA directly and thus induce a DDR and senescence [45-47] (Figure 2a). ROS have been implicated in organismal ageing, with countless reports of associations between oxidative damage and the ageing process [48-50]; however, genetically manipulated animal models where mitochondrial function and oxidative stress were targeted have generated conflicting results [51].
Figure 2.
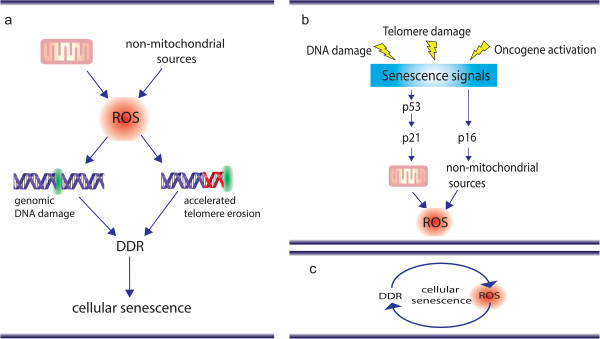
Two different models by which reactive oxygen species can impact on cellular senescence. (a) Reactive oxygen species (ROS) produced via mitochondrial and non-mitochondrial sources can induce genomic DNA damage and accelerate telomere erosion/damage, both of which contribute to activation of a DNA damage response (DDR). (b) ROS can act as signalling molecules in senescence: activation of “senescence signals” has been shown to result in increased ROS generation (mitochondrial and non-mitochondrial). ROS has been shown to impact on a variety of pathways which may help stabilise the senescence growth arrest. (c) Simplified feedback loop model involving ROS and DNA damage. Telomere uncapping or general DNA damage triggers a DDR which culminates through yet unidentified processes to ROS generation. ROS generation leads to additional DNA damage to the genome, stabilising the DDR and leading to a stable senescence arrest.
Several studies have shown that cellular senescence is characterised by mitochondrial dysfunction contributing to metabolic inefficiency and elevated ROS [52-56]. Elevated ROS levels have been associated with replicative, stress- and oncogene-induced senescence [8,45,55,57].
Evidence indicates that activation of major downstream effectors of the DDR in senescence result in elevated ROS. Activation of a DDR by genotoxic stress or telomere uncapping [21], over-expression of activated RAS [58], BRAFV600E[59], p53 [60], p21 [61] and p16 [62] all resulted in elevated ROS generation. In most of the above reported cases treatment with antioxidants, such as N-acetyl cysteine, were able to prevent the cell-cycle arrest supporting a causal role for ROS in the process (Figure 2b).
These data indicate that elevated ROS are a consequence of the activation of the senescence programme and has led to the suggestion that ROS may act as signalling molecules during cellular senescence [63]. However, mechanistically it is still unclear how these pathways contribute to mitochondrial dysfunction and ROS generation. Takahashi and colleagues, using human fibroblasts expressing a temperature-sensitive simian virus 40 large T antigen, connected p16 with ROS production via protein kinase Cδ signalling [62]. Protein kinase Cδ has been shown to activate a non-mitochondrial source of ROS, generated by NADPH-oxidase through phosphorylation of p47phox, an essential component of NADPH oxidase [64]. Consistent with this study, NADPH oxidases have been shown to limit the replicative lifespan of human endothelial cells in culture via ROS generation [65].
Oncogene-induced senescence has been associated with mitochondrial dysfunction and ROS production, which is dependent on intact p53 and Rb tumour suppression pathways. Mitochondrial dysfunction resulted in the loss of ATP and activation of AMPK; in addition, mitochondrial-derived ROS were shown to contribute to the oxidation of DNA [66]. In a recent study, it was shown that BRAFV600E-induced senescence was accompanied by the activation of pyruvate dehydrogenase, which resulted in the enhanced use of pyruvate by the tricarboxylic acid cycle followed by increased respiration and ROS generation [59].
The role of p53 and p21 in ROS generation during senescence is still not well understood. An association between p53 and transcriptional activation of genes involved in mitochondrial apoptosis has been demonstrated [67], as well as a stress-induced translocation of p53 to mitochondria resulting in increased outer membrane permeabilisation [68]; however, a direct role of mitochondrial p53 in cellular senescence has not yet been demonstrated. In contrast, transcriptional regulation of mitochondrial genes by p53 has been reported to impact on mitochondrial function and contribute to ageing. p53 knock-out mice exhibited reduced expression of the Sco2 gene, which is required for the assembly of the mitochondrial DNA-encoded COX II subunit [69]. In late generation telomerase knock-out mice that have critically short telomeres, activation of p53 has been shown to repress the promoters of PGC-1α and PGC-1β genes, master regulators of mitochondrial biogenesis and function, thereby contributing to decreased mitochondrial function [70].
Knockdown of both p53 and p21 by RNA-mediated interference has been shown to reduce ROS generation in both telomere-dependent and -independent senescence [21]. Our group has found that ROS levels increase in senescent cells as a result of signalling through p21, and feed back into DNA damage induction and the DDR, generating a stable, self-sustaining feedback loop (Figure 2c). This feedback loop persists even in irreversibly deep senescence. Moreover, p21 appears to be the critical mediator between the DDR and MAPK and transforming growth factor (TGF)-β stress-induced signalling cascades, which have been shown to contribute to ROS generation [21,71,72]. Consistently, a p21 knock-out rescued at least some accelerated ageing phenotypes in telomerase (mTERC) knock-out mice [17], as well as markers of oxidative stress and DNA damage foci [21]. ROS has also been shown to impact on the DDR and ultimately senescence in a non-cell-autonomous fashion. A recent study has shown that senescent cells can induce a DDR in neighbouring cells via a gap junction-mediated cell-cell contact and processes involving ROS [73].
Synergistic interactions between the senescence-associated secretory phenotype and reactive oxygen species during senescence
During senescence, another major contributor to the stabilisation of the growth arrest is mediated by autocrine signalling involving the secretion of bioactive, frequently pro-inflammatory peptides, known as the SASP [74] or senescence-messaging secretome [75]. The SASP includes several families of soluble and insoluble factors. The soluble factors include signalling molecules such as growth factors, inflammatory and immune-modulatory cytokines and chemokines, whereas the insoluble factors mainly comprise extracellular matrix components [76]. It has long been recognised that the primary function of secreted factors is to allow inter- and intra-cellular communication. However, the SASP has been found to play a series of somewhat contradictory roles, with important consequences for ageing and cancer. First, it can contribute to the surveillance and elimination of senescent cells by the immune system [77,78]. Second, it can be pro-tumorigenic [74,79,80]; both cell culture experiments and studies involving the co-transplantation of senescent and cancer cells into recipient mice have shown that senescent fibroblasts can stimulate the hyperproliferation of cancer cells, neoplastic progression and tissue damage. Third, it can contribute to the reinforcement of oncogene- or stress-induced senescence in a cell-autonomous fashion [22,23]. Fourth, it can induce senescence in neighbouring cells via a bystander effect both in vitro and in vivo[81].
Mechanistically, it is still not entirely understood how the SASP contributes to the reinforcement of senescence; however, several lines of evidence suggest the existence of synergistic interactions between the DDR, ROS and inflammatory signals (Figure 3a). Kinetic analysis has shown that ROS levels increase 2 to 3 days following activation of a DDR [21], while the SASP occurs 7 to 10 days later [76]. Induction of both ROS and the SASP in X-ray irradiation-induced senescence has been shown to be dependent on activation of the DDR [21,35].
Figure 3.

Senescence is a multi-layered process involving interactions between the DNA damage response, reactive oxygen species and senescence-associated secretory phenotype. (a) Initially, stressors such as telomeric and non-telomeric DNA damage can lead to activation of a DNA damage response (DDR) and cell cycle arrest. Following activation of the DDR, p53, p21 and p38MAPK pathways have been shown to enhance nuclear factor (NF)-κB transcriptional activity. NF-κB activation is both responsible for the senescence-associated secretory phenotype (SASP) and can induce (and be activated) by reactive oxygen species (ROS). p16 has been shown to induce ROS generation via NADPH oxidases [62]; however, it has been shown to be unrelated to the SASP [88]. Secretion of bioactive molecules such as ROS and SASP factors contribute not only to reinforce senescence in an autocrine fashion, but also to induce senescence in neighbouring cells. (b) Components of the SASP (such as IL-8, β-IFN and transforming growth factor (TGF)-β) have been shown to reinforce the senescence arrest via ROS through yet unidentified mechanisms [21,22,89]. (c) NF-κB transcriptional activity has been shown to be dependent on the DDR and ROS. However, NF-κB activation has been shown to increase ROS generation (via regulating expression of mitochondrial genes or antioxidant, pro-oxidant genes) [96,97]. DDF - DNA Damage Foci.
The nuclear factor (NF)-κB family of transcriptional factors regulate expression of numerous genes involved in a variety of cellular processes including stress response and inflammation [82]. Importantly, activation of NF-κB has been considered critical in chronic inflammatory diseases by increasing the expression of the genes for many cytokines, enzymes, and adhesion molecules [83]. Increased NF-κB activity has been shown to play an important role in senescence [84] and the SASP [85].
Recent investigations using progeroid mouse models (models of premature ageing) driven by DNA damage have reported that these mice have increased activation of NF-κB driven chronic inflammation and senescence [86,87]. Interestingly, in a murine model of XFE (xeroderma pigmentosum F–excision repair) progeroid syndrome, Ercc1–/Δ mice, inhibition of NF-κB signalling not only reduced the onset of several age-related pathologies, but also both DNA and protein oxidation [87], suggesting a potential link between inflammation and ROS pathways.
Another link between ROS and the SASP during senescence involves the p38 mitogen-activated protein kinase (p38MAPK). p38MAPK has been shown to regulate the SASP in senescence mainly through NF-κB transcriptional activity [85]. Similarly, the p38MAPK pathway has been shown to be important for ROS generation in both stress-induced and replicative senescence and for the stability of the DDR [21]. p16, an important tumour suppressor gene which can be induced by stresses other than DNA damage, has been linked to increased ROS production [62]; however, less is known about its impact on the SASP. The Campisi laboratory has shown that ionising radiation or oncogenic RAS-induced senescence developed a SASP regardless of expression of p16, suggesting that these are two separate pathways. However, the mechanisms behind it are not yet understood [88].
A few studies connect the SASP with reinforcement of senescence via increased ROS (Figure 3b). Acosta and colleagues have shown that inhibition of CXCR2, a promiscuous receptor that transmits signals from several CXC chemokine family members (CXCLs), including IL-8, delayed the onset of both replicative and oncogene-induced senescence and led to decreased activation of a DDR [22]. Mechanistically, the authors proposed that inhibition of CXCR2 reduced the DDR potentially by reducing ROS. β-IFN has been shown to induce senescence through ROS production and subsequent activation of the DDR, which could be inhibited with the antioxidant N-acetyl cysteine [89]. TGF-β, a family of secreted peptides that regulate a variety of processes such as proliferation, adhesion, migration, and differentiation in several cell types, has also been implicated in senescence. Inactivation of TGF-β1 secretion in mouse keratinocytes was sufficient to prevent oncogene-induced senescence [90]. In human fibroblasts, blocking TGF-β1 type II receptor (TGFBR2) activity has been shown to prevent Ultraviolet B-induced senescence and hydrogen peroxide-induced senescence [91,92]. Recently, it was demonstrated that the TGF-β induced senescence in a paracrine fashion [81]. Interestingly, neutralising antibodies or chemical inhibitors against the TGFBR2 have been shown to decrease ROS production downstream of the DDR induced in a telomere-dependent and -independent fashion [21].
Another potential link between the SASP and ROS is the fact that several studies indicate that NF-κB, the main regulator of the SASP, is also a major player in the regulation of mitochondrial function and oxidative stress (Figure 3c). Firstly, NF-κB is localised in mitochondria from yeast [93] and mammalian cells and contributes to the regulation of mitochondrial encoded genes [94]. Bakkar and colleagues reported that activation of the RelB subunit of NF-κB during myogenesis is important for mitochondrial biogenesis [95]. More recently it was demonstrated that IKKα and RelB regulate the transcription co-activator PGC-1β, a master regulator of mitochondrial function, to promote oxidative muscle metabolism [96]. Secondly, it has also been reported that NF-κB is involved in the transcriptional regulation of both nuclear-encoded anti-oxidant and pro-oxidant genes [97]. A recent study in a mouse model of type II diabetes-induced cardiac dysfunction has shown that enhanced NF-κB activity is associated with increased oxidative stress. The authors demonstrated that chemical inhibition of NF-κB alleviated oxidative stress, improved mitochondrial structural integrity, and ultimately restored cardiac function in type II diabetes [98].
In contrast, numerous reports have implicated ROS in the activation of NF-κB [99]. Both DNA binding and transactivation by NF-κB have been shown to be strongly activated by H2O2[100]. Mechanistically, evidence suggests that ROS are both cause and consequence of NF-κB pathway activation during senescence, making it challenging to establish which process occurs first. Further work is needed in order to understand the kinetics of activation of these pathways during senescence.
Conclusions
In addition to its previously documented role as a tumour suppressive mechanism, recent evidence strongly implicates cellular senescence in ageing and age-related diseases. Both telomeric and non-telomeric DNA damage has been shown to contribute to the phenotype, with ROS playing an important role in both the induction and stabilisation of senescence. Moreover, the activation of the DDR, and the MAPK and NF-κB pathways has been shown to contribute to the regulation of both ROS and the SASP. Despite accumulating evidence suggesting that ROS and the SASP cooperate to induce and stabilise the senescent phenotype, further research is necessary to mechanistically delineate their interactions in regulating their response, and their contributions to modulating the surrounding tissue micro-environment.
Abbreviations
DDF: DNA damage foci; DDR: DNA damage response; DSB: double strand break; IFN: interferon; IL: interleukin; NF: nuclear factor; NHEJ: non-homologous end joining; p38MAPK: p38 mitogen-activated protein kinase; ROS: reactive oxygen species; SASP: senescence-associated secretory phenotype; TGF: transforming growth factor.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CCM and JFP wrote the majority of the manuscript. GH wrote the section about telomeres and made figure schemes. All authors read and approved the final manuscript.
Contributor Information
Clara Correia-Melo, Email: c.correia-melo@ncl.ac.uk.
Graeme Hewitt, Email: Graeme.hewitt@ncl.ac.uk.
João F Passos, Email: joao.passos@ncl.ac.uk.
Acknowledgements
We would like to thank Rhys Anderson for critically reading the manuscript. GH is supported by a case studentship from the BBSRC; CCM is supported by a FCT (Foundation for Science and Technology, Portugal) studentship through the GABBA program, University of Porto; JFP is supported by a David Phillips Fellowship provided by the BBSRC.
References
- Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- Bayreuther K, Rodemann HP, Hommel R, Dittmann K, Albiez M, Francz PI. Human skin fibroblasts in vitro differentiate along a terminal cell lineage. Proc Natl Acad Sci U S A. 1988;85:5112–5116. doi: 10.1073/pnas.85.14.5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J. Cancer, aging and cellular senescence. In Vivo. 2000;14:183–188. [PubMed] [Google Scholar]
- Sitte N, Merker K, von Zglinicki T, Grune T. Protein oxidation and degradation during proliferative senescence of human MRC-5 fibroblasts. Free Radic Biol Med. 2000;28:701–708. doi: 10.1016/S0891-5849(99)00279-8. [DOI] [PubMed] [Google Scholar]
- von Zglinicki T, Pilger R, Sitte N. Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic Biol Med. 2000;28:64–74. doi: 10.1016/S0891-5849(99)00207-5. [DOI] [PubMed] [Google Scholar]
- Narita M, Nunez S, Heard E, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/S0092-8674(03)00401-X. [DOI] [PubMed] [Google Scholar]
- Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37:961–976. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Ramsey M, Sharpless N. ROS as a tumour suppressor? Nat Cell Biol. 2006;8:1213–1215. doi: 10.1038/ncb1106-1213. [DOI] [PubMed] [Google Scholar]
- Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene. 2007;26:7773–7779. doi: 10.1038/sj.onc.1210881. [DOI] [PubMed] [Google Scholar]
- Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006;311:1257. doi: 10.1126/science.1122446. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy J, Torrice C, Ramsey M, Kovalev G, Al-Regaiey K, Su L, Sharpless N. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell. 2009;8:311–323. doi: 10.1111/j.1474-9726.2009.00481.x. [DOI] [PubMed] [Google Scholar]
- Hewitt G, Jurk D, Marques F, Correia-Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, Passos J. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun. 2012;3:708. doi: 10.1038/ncomms1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurk D, Wang C, Miwa S, Maddick M, Korolchuk V, Tsolou A, Gonos E, Thrasivoulou C, Saffrey M, Cameron K, von Zglinicki T. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging cell. 2012;11:996–1004. doi: 10.1111/j.1474-9726.2012.00870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sone H, Kagawa Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia. 2005;48:58–67. doi: 10.1007/s00125-004-1605-2. [DOI] [PubMed] [Google Scholar]
- Minamino T, Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ Res. 2007;100:15–26. doi: 10.1161/01.RES.0000256837.40544.4a. [DOI] [PubMed] [Google Scholar]
- Choudhury AR, Ju Z, Djojosubroto MW, Schienke A, Lechel A, Schaetzlein S, Jiang H, Stepczynska A, Wang C, Buer J, Lee HW, von Zglinicki T, Ganser A, Schirmacher P, Nakauchi H, Rudolph KL. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat Genet. 2007;39:99–105. doi: 10.1038/ng1937. [DOI] [PubMed] [Google Scholar]
- Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999;96:701–712. doi: 10.1016/S0092-8674(00)80580-2. [DOI] [PubMed] [Google Scholar]
- Tyner S, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, Park S, Thompson T, Karsenty G, Bradley A, Donehower L. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- Baker D, Wijshake T, Tchkonia T, LeBrasseur N, Childs B, van de Sluis B, Kirkland J, van Deursen J. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, Miwa S, Olijslagers S, Hallinan J, Wipat A, Saretzki G, Rudolph KL, Kirkwood TB, von Zglinicki T. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol. 2010;6:347. doi: 10.1038/msb.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d'Adda di Fagagna F, Bernard D, Hernando E, Gil J. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- d'Adda di Fagagna F, Teo SH, Jackson SP. Functional links between telomeres and proteins of the DNA-damage response. Genes Dev. 2004;18:1781–1799. doi: 10.1101/gad.1214504. [DOI] [PubMed] [Google Scholar]
- de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T. Mammalian Telomeres End in a Large Duplex Loop. Cell. 1999;97:503–514. doi: 10.1016/S0092-8674(00)80760-6. [DOI] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu C-P, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of Life-Span by Introduction of Telomerase into Normal Human Cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Blasco MA, Lee H-W, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW. Telomere Shortening and Tumor Formation by Mouse Cells Lacking Telomerase RNA. Cell. 1997;91:25–34. doi: 10.1016/S0092-8674(01)80006-4. [DOI] [PubMed] [Google Scholar]
- Lee H-W, Blasco MA, Gottlieb GJ, Horner JW, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392:569–574. doi: 10.1038/33345. [DOI] [PubMed] [Google Scholar]
- Wong K-K, Maser RS, Bachoo RM, Menon J, Carrasco DR, Gu Y, Alt FW, DePinho RA. Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature. 2003;421:643–648. doi: 10.1038/nature01385. [DOI] [PubMed] [Google Scholar]
- d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- Takai H, Smogorzewska A, de Lange T. DNA Damage Foci at Dysfunctional Telomeres. Curr Biol. 2003;13:1549–1556. doi: 10.1016/S0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci. 2006;31:402–410. doi: 10.1016/j.tibs.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov J, Bucci G, Dobreva M, Matti V, Beausejour C, Herbig U, Longhese M, d'Adda di Fagagna F. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol. 2012;14:355–365. doi: 10.1038/ncb2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, Karlseder J, Holtgreve-Grez H, Jauch A, de Lange T. DNA Ligase IV-Dependent NHEJ of Deprotected Mammalian Telomeres in G1 and G2. Curr Biol. 2002;12:1635–1644. doi: 10.1016/S0960-9822(02)01179-X. [DOI] [PubMed] [Google Scholar]
- van Steensel B, Smogorzewska A, de Lange T. TRF2 Protects Human Telomeres from End-to-End Fusions. Cell. 1998;92:401–413. doi: 10.1016/S0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- Celli GB, de Lange T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol. 2005;7:712–718. doi: 10.1038/ncb1275. [DOI] [PubMed] [Google Scholar]
- Bae N, Baumann P. A RAP1/TRF2 complex inhibits nonhomologous end-joining at human telomeric DNA ends. Mol Cell. 2007;26:323–334. doi: 10.1016/j.molcel.2007.03.023. [DOI] [PubMed] [Google Scholar]
- Kaul Z, Cesare A, Huschtscha L, Neumann A, Reddel R. Five dysfunctional telomeres predict onset of senescence in human cells. EMBO Rep. 2012;13:52–59. doi: 10.1038/embor.2011.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suram A, Kaplunov J, Patel P, Ruan H, Cerutti A, Boccardi V, Fumagalli M, Di Micco R, Mirani N, Gurung R, Hande M, d'Adda di FagagnaHerbig U F, Herbig U. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J. 2012;31:2839–2851. doi: 10.1038/emboj.2012.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastogi S, Joshi B, Dasgupta P, Morris M, Wright K, Chellappan S. Prohibitin facilitates cellular senescence by recruiting specific corepressors to inhibit E2F target genes. Mol Cell Biol. 2006;26:4161–4171. doi: 10.1128/MCB.02142-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. 2002;27:339–344. doi: 10.1016/S0968-0004(02)02110-2. [DOI] [PubMed] [Google Scholar]
- Lu T, Finkel T. Free radicals and senescence. Exp Cell Res. 2008;314:1918–1922. doi: 10.1016/j.yexcr.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Fischer A, Reagan J, Yan L, Ames B. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc Natl Acad Sci U S A. 1995;92:4337–4341. doi: 10.1073/pnas.92.10.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai P, Onder T, Young J, McFaline J, Pang B, Dedon P, Weinberg R. Continuous elimination of oxidized nucleotides is necessary to prevent rapid onset of cellular senescence. Proc Natl Acad Sci U S A. 2009;106:169–174. doi: 10.1073/pnas.0809834106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga CG, Shigenaga MK, Park JW, Degan P, Ames BN. Oxidative damage to DNA during aging: 8-hydroxy-2′-deoxyguanosine in rat organ DNA and urine. Proc Natl Acad Sci U S A. 1990;87:4533–4537. doi: 10.1073/pnas.87.12.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver CN, Ahn BW, Moerman EJ, Goldstein S, Stadtman ER. Age-related changes in oxidized proteins. J Biol Chem. 1987;262:5488–5491. [PubMed] [Google Scholar]
- Hamilton ML, Van Remmen H, Drake JA, Yang H, Guo ZM, Kewitt K, Walter CA, Richardson A. Does oxidative damage to DNA increase with age? Proc Natl Acad Sci U S A. 2001;98:10469–10474. doi: 10.1073/pnas.171202698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H. Trends in oxidative aging theories. Free Radical Biol Med. 2007;43:477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- Allen RG, Tresini M, Keogh BP, Doggett DL, Cristofalo VJ. Differences in electron transport potential, antioxidant defenses, and oxidant generation in young and senescent fetal lung fibroblasts (WI-38) J Cell Physiol. 1999;180:114–122. doi: 10.1002/(SICI)1097-4652(199907)180:1<114::AID-JCP13>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Hutter E, Unterluggauer H, Uberall F, Schramek H, Jansen-Durr P. Replicative senescence of human fibroblasts: the role of Ras-dependent signaling and oxidative stress. Exp Gerontol. 2002;37:1165–1174. doi: 10.1016/S0531-5565(02)00136-5. [DOI] [PubMed] [Google Scholar]
- Hutter E, Renner K, Pfister G, Stockl P, Jansen-Durr P, Gnaiger E. Senescence-associated changes in respiration and oxidative phosphorylation in primary human fibroblasts. Biochem J. 2004;380:919–928. doi: 10.1042/BJ20040095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, Wappler I, Birket MJ, Harold G, Schaeuble K, Birch-Machin MA, Kirkwood TB, von Zglinicki T. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007;5:e110. doi: 10.1371/journal.pbio.0050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwerschke W, Mazurek S, Stockl P, Hutter E, Eigenbrodt E, Jansen-Durr P. Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem J. 2003;376:403–411. doi: 10.1042/BJ20030816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saretzki G, Murphy MP, von Zglinicki T. MitoQ counteracts telomere shortening and elongates lifespan of fibroblasts under mild oxidative stress. Aging Cell. 2003;2:141–143. doi: 10.1046/j.1474-9728.2003.00040.x. [DOI] [PubMed] [Google Scholar]
- Lee AC, Fenster BE, Ito H, Takeda K, Bae NS, Hirai T, Yu Z-X, Ferrans VJ, Howard BH, Finkel T. Ras Proteins Induce Senescence by Altering the Intracellular Levels of Reactive Oxygen Species. J Biol Chem. 1999;274:7936–7940. doi: 10.1074/jbc.274.12.7936. [DOI] [PubMed] [Google Scholar]
- Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov V, Mackay G, van der Burg S, Verdegaal E, Cascante M, Shlomi T, Gottlieb E, Peeper D. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. 2013;498:109–112. doi: 10.1038/nature12154. [DOI] [PubMed] [Google Scholar]
- Macip S, Igarashi M, Berggren P, Yu J, Lee SW, Aaronson SA. Influence of Induced Reactive Oxygen Species in p53-Mediated Cell Fate Decisions. Mol Cell Biol. 2003;23:8576–8585. doi: 10.1128/MCB.23.23.8576-8585.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macip S, Igarashi M, Fang L, Chen A, Pan Z, Lee S, Aaronson S. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 2002;21:2180–2188. doi: 10.1093/emboj/21.9.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi A, Ohtani N, Yamakoshi K, Iida S, Tahara H, Nakayama K, Nakayama KI, Ide T, Saya H, Hara E. Mitogenic signalling and the p16INK4A-RB pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol. 2006;8:1291–1297. doi: 10.1038/ncb1491. [DOI] [PubMed] [Google Scholar]
- Passos JF, von Zglinicki T. Oxygen free radicals in cell senescence: are they signal transducers? Free Radic Res. 2006;40:1277–1283. doi: 10.1080/10715760600917151. [DOI] [PubMed] [Google Scholar]
- Talior I, Tennenbaum T, Kuroki T, Eldar-Finkelman H. PKC-δ-dependent activation of oxidative stress in adipocytes of obese and insulin-resistant mice: role for NADPH oxidase. Am J Physiol - Endocrinol Metab. 2005;288:E405–E411. doi: 10.1152/ajpendo.00378.2004. [DOI] [PubMed] [Google Scholar]
- Lener B, Kozieł R, Pircher H, Hütter E, Greussing R, Herndler-Brandstetter D, Hermann M, Unterluggauer H, Jansen-Dürr P. The NADPH oxidase Nox4 restricts the replicative lifespan of human endothelial cells. Biochem J. 2009;423:363–374. doi: 10.1042/BJ20090666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseeva O, Bourdeau V, Roux A, Deschemes-Simard X, Ferbeyre G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol Cell Biol. 2009;29:4495–4507. doi: 10.1128/MCB.01868-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- Moll U, Marchenko N, X-k Z. p53 and Nur77/TR3 – transcription factors that directly target mitochondria for cell death induction. Oncogene. 2006;25:4725–4743. doi: 10.1038/sj.onc.1209601. [DOI] [PubMed] [Google Scholar]
- Matoba S, Kang J-G, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 Regulates Mitochondrial Respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, Maser RS, Tonon G, Foerster F, Xiong R, Wang YA, Shukla SA, Jaskelioff M, Martin ES, Heffernan TP, Protopopov A, Ivanova E, Mahoney JE, Kost-Alimova M, Perry SR, Bronson R, Liao R, Mulligan R, Shirihai OS, Chin L, DePinho RA. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koli K, Myllarniemi M, Keski-Oja J, Kinnula VL. Transforming growth factor-beta activation in the lung: focus on fibrosis and reactive oxygen species. Antioxid Redox Signalling. 2008;10:333–342. doi: 10.1089/ars.2007.1914. [DOI] [PubMed] [Google Scholar]
- Torres M, Forman HJ. Redox signalling and MAP kinase pathways. Biofactors. 2003;17:287–296. doi: 10.1002/biof.5520170128. [DOI] [PubMed] [Google Scholar]
- Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, von Zglinicki T. A senescent cell bystander effect: senescence-induced senescence. Aging Cell. 2012;11:345–349. doi: 10.1111/j.1474-9726.2012.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé J-P, Patil C, Rodier F, Sun Y, Muñoz D, Goldstein J, Nelson P, Desprez P-Y, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009;9:81–94. doi: 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- Coppé J-P, Desprez P-Y, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Ann Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang T-W, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, Iken M, Vucur M, Weiss S, Heikenwalder M, Khan S, Gil J, Bruder D, Manns M, Schirmacher P, Tacke F, Ott M, Luedde T, Longerich T, Kubicka S, Zender L. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–551. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Hornsby P. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007;67:3117–3126. doi: 10.1158/0008-5472.CAN-06-3452. [DOI] [PubMed] [Google Scholar]
- Krtolica A, Parrinello S, Lockett S, Desprez P, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Nat Acad Sci U S A. 2001;98:12072–12077. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang T-W, Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, Jin H, Dharmalingam G, Snijders AP, Carroll T, Capper D, Pritchard C, Inman GJ, Longerich T, Sansom OJ, Benitah SA, Zender L, Js G. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978–990. doi: 10.1038/ncb2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl H. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- Barnes P, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. New Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- Rovillain E, Mansfield L, Caetano C, Alvarez-Fernandez M, Caballero OL, Medema RH, Hummerich H, Jat PS. Activation of nuclear factor-kappa B signalling promotes cellular senescence. Oncogene. 2011;30:2356–2366. doi: 10.1038/onc.2010.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30:1536–1548. doi: 10.1038/emboj.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio FG, Barcena C, Soria-Valles C, Ramsay AJ, de Carlos F, Cobo J, Fueyo A, Freije JMP, Lopez-Otin C. Nuclear lamina defects cause ATM-dependent NF-κB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012;26:2311–2324. doi: 10.1101/gad.197954.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilstra JS, Robinson AR, Wang J, Gregg S, Clauson CL, Reay DP, Nasto LA, St Croix CM, Usas A, Vo N, Huard J, Clemens PR, Stolz DB, Guttridge DC, Watkins SC, Garinis GA, Wang Y, Niedernhofer LJ, Robbins PD. NF-kB inhibition delays DNA damage induced senescence and aging in mice. J Clin Invest. 2012;122:2601–2612. doi: 10.1172/JCI45785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé J-P, Rodier F, Patil CK, Freund A, Desprez P-Y, Campisi J. Tumor Suppressor and Aging Biomarker p16INK4a Induces Cellular Senescence without the Associated Inflammatory Secretory Phenotype. J Biol Chem. 2011;286:36396–36403. doi: 10.1074/jbc.M111.257071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseeva O, Mallette F, Mukhopadhyay U, Moores A, Ferbeyre G. DNA damage signaling and p53-dependent senescence after prolonged beta-interferon stimulation. Mol Biol Cell. 2006;17:1583–1592. doi: 10.1091/mbc.E05-09-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremain R, Marko M, Kinnimulki V, Ueno H, Bottinger E, Glick A. Defects in TGF-beta signaling overcome senescence of mouse keratinocytes expressing v-Ha-ras. Oncogene. 2000;19:1698–1709. doi: 10.1038/sj.onc.1203471. [DOI] [PubMed] [Google Scholar]
- Debacq-Chainiaux F, Borlon C, Pascal T, Royer V, Eliaers F, Ninane N, Carrard G, Friguet B, de Longueville F, Boffe S, Remacle J, Toussaint O. Repeated exposure of human skin fibroblasts to UVB at subcytotoxic level triggers premature senescence through the TGF-beta1 signaling pathway. J Cell Sci. 2005;118:743–758. doi: 10.1242/jcs.01651. [DOI] [PubMed] [Google Scholar]
- Frippiat C, Chen Q, Zdanov S, Magalhaes J, Remacle J, Toussaint O. Subcytotoxic H2O2 stress triggers a release of transforming growth factor-beta 1, which induces biomarkers of cellular senescence of human diploid fibroblasts. J Biol Chem. 2001;276:2531–2537. doi: 10.1074/jbc.M006809200. [DOI] [PubMed] [Google Scholar]
- Bottero V, Rossi F, Samson M, Mari M, Hofman P, Peyron J. Ikappa b-alpha, the NF-kappa B inhibitory subunit, interacts with ANT, the mitochondrial ATP/ADP translocator. J Biol Chem. 2001;276:21317–21324. doi: 10.1074/jbc.M005850200. [DOI] [PubMed] [Google Scholar]
- Cogswell P, Kashatus D, Keifer J, Guttridge D, Reuther J, Bristow C, Roy S, Nicholson D, Baldwin A. NF-kappa B and I kappa B alpha are found in the mitochondria. Evidence for regulation of mitochondrial gene expression by NF-kappa B. J Biol Chem. 2003;278:2963–2968. doi: 10.1074/jbc.M209995200. [DOI] [PubMed] [Google Scholar]
- Bakkar N, Wang J, Ladner K, Wang H, Dahlman J, Carathers M, Acharyya S, Rudnicki M, Hollenbach A, Guttridge D. IKK/NF-kappaB regulates skeletal myogenesis via a signaling switch to inhibit differentiation and promote mitochondrial biogenesis. J Cell Biol. 2008;180:787–802. doi: 10.1083/jcb.200707179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkar N, Ladner K, Canan B, Liyanarachchi S, Bal N, Pant M, Periasamy M, Li Q, Janssen P, Guttridge D. IKKα and alternative NF-κB regulate PGC-1β to promote oxidative muscle metabolism. J Cell Biol. 2012;196:497–511. doi: 10.1083/jcb.201108118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan M, Z-g L. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariappan N, Elks C, Sriramula S, Guggilam A, Liu Z, Borkhsenious O, Francis J. NF-kappaB-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type II diabetes. Cardiovasc Res. 2010;85:473–483. doi: 10.1093/cvr/cvp305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72:1493–1505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Meyer M, Schreck R, Baeuerle P. H2O2 and antioxidants have opposite effects on activation of NF-kappa B and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor. EMBO J. 1993;12:2005–2015. doi: 10.1002/j.1460-2075.1993.tb05850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]