

Abstract
Many neurodegenerative disorders are characterized by synaptic dysfunction preceding general neuronal loss and subsequent cognitive or behavioral anomalies. Much recent research has been aimed at understanding the early underlying processes leading to dysfunction at the synapse, as this knowledge would likely inform interventions that could potentially slow progression and delay onset of disease. We have recently reported that synaptic dysfunction in a Drosophila melanogaster model of Huntington’s disease (HD) can be prevented by enhanced neuronal expression of Rab11, a Rab family GTPase involved in endosomal recycling, which complements studies that have found disrupted Rab11 activity in several models of this disorder. Indeed, inhibition of Rab11 function in fibroblasts of HD patients has been observed to perturb vesicle formation from recycling endosomes. Therefore, our study investigated a potential role of Rab11 in synaptic dysfunction prior to the onset of HD symptoms, with the aim of finding a possible early intervention to disease progression. We found that Rab11 ameliorates synaptic dysfunction due to expression of mutant huntingtin—the causative protein in HD—by normalizing synaptic vesicle size, which consequently ameliorates locomotor deficits in Drosophila larvae. Here we further consider these results and the implications this work has on potential therapeutic intervention in HD and other neurodegenerative disorders.
Keywords: Huntington’s disease, Rab family GTPases, Rab11, huntingtin, neurodegeneration, synapse, synaptic dysfunction
Many, if not all, neurodegenerative diseases manifest themselves with early synaptic dysfunction before any neuronal loss or behavioral abnormalities can be detected.1-4 Much research is now focused on unravelling the molecular mechanisms that lead to dysregulation of synaptic transmission by studying several synaptic proteins involved in neurotransmission. The neuronal network relies on plasticity mechanisms where reversible formation and disassembling of synaptic connections occurs in a controlled manner. It is generally accepted that in neurodegenerative conditions an early decline of genes responsible for expression of synaptic proteins occurs, opening the possibility of intervention to manipulate neuroprotective pathways that balance between degenerative and survival signaling. In our studies, we have employed fruit fly models of Huntington’s disease (HD) to explore the potential of Rab11 for modulating synaptic dysfunction in neurodegenerative disease.
HD is caused by the expansion of a polyglutamine stretch near the N-terminus of the huntingtin (HTT) protein, which exerts cellular toxicity by perturbing a number of cellular processes, including vesicle trafficking and transcription, as well as by overwhelming antioxidant systems.5 Rab family GTPases play critical roles in intracellular membrane trafficking and it is clear that alterations in these cellular processes contribute to impaired neuronal trafficking in HD, as well as other neurodegenerative diseases.6
More than 60 Rab proteins are expressed in mammalian cells,7 most of which participate in the directed transport and tethering of vesicles to their target membranes.8,9 Much recent data suggests that modulation of Rab family GTPases may be therapeutically relevant in HD. For example, Rab11, which plays a key role in endosomal recycling,10-12 is functionally perturbed in models of HD.13-17 We have also found that Rab11 abrogates loss of dendritic spines in primary neurons expressing mutant HTT, and ameliorates neuron loss and shortened lifespan in adult HD flies.17
Rab5 and Rab8 have also been implicated in HD pathogenesis. Mutant HTT alters the localization of the Rab8/optineuron complex from the Golgi apparatus, perturbing post-Golgi trafficking and thereby impairing lysosomal function.18 Furthermore, overexpression of Rab5 reduces mutant HTT toxicity—while Rab5 inhibition exacerbates this phenotype—apparently via regulation of macroautophagy.19 In related work, we have found an enrichment in vesicle trafficking genes in genetic screens for both gene deletion and overexpression suppressors of mutant HTT toxicity in yeast.20,21 These data support the notion of interrogating the therapeutic promise of Rab GTPases and other vesicle trafficking genes in neurodegenerative disease.
Drosophila is a robust model system for studying HD pathology and other disease pathologies and has been widely used to identify novel mechanisms and potential therapeutic strategies.22 Expression of mutant HTT constructs in different neuronal populations leads to a number of disease-relevant phenotypes, including degeneration of photoreceptor neurons, reduced lifespan, and impaired locomotion.21,23,24
Work in Drosophila has demonstrated that mutant HTT can exert pathology in the absence of nuclear localization, providing evidence for cytoplasmic dysfunction.25 This study found that mutant HTT is differentially distributed in neurons, implying that HTT aggregates may associate with cytoskeletal machinery to undergo directed transport, a process highly relevant for axonal function. A particularly striking feature of HD pathology and genetic models is the aggregation of mutant HTT protein, but it remains unknown how aggregates contribute to toxicity in this diseases. Indeed, it is likely that these aggregates, as well as soluble oligomeric forms of mutant HTT, contribute to disease pathogenesis in a combinatorial fashion.26
A great amount of data supports the role of mutant HTT in disrupting axonal transport. Drosophila larval motor neurons transport mutant HTT along axons, leading to accumulations in axon termini at NMJs.25 Visualizing mutant HTT in motor neurons showed that large aggregates caused axons to swell, indicating that they might physically compromise axonal transport. In this context, it was found that synaptic vesicle proteins accumulate in large amounts at sites of mutant HTT aggregation, suggesting a role for cytoplasmic toxicity in HD pathogenesis that might be mediated through alterations in axonal transport.27,28
We recently showed that mutant HTT is linked to synaptic pathology in Drosophila models of HD, in particular affecting synaptic vesicle homeostasis,29 which could represent an early physiological deficit before onset of disease pathology. The reduction in presynaptic quantal size detected at the Drosophila neuromuscular junction (NMJ) was due to smaller vesicular size as observed by electron microscopy. As a result, evoked synaptic transmission was compromised by the presynaptic expression of mutant HTT, leading to behavioral deficits. We found that 2 different mutant HTT transgenes (Htt93Q, which expresses an exon 1 fragment of human HTT,23 and Htt128QFL, which expresses full-length HTT4) have similar defects on synaptic physiology. Interestingly, of these models only the Htt93Q flies exhibit mutant HTT aggregation, while the Htt128QFL flies do not. This suggests that toxic oligomeric species of mutant HTT may play a critical role in synaptic dysfunction in HD. Indeed, toxic oligomers of additional amyloidogenic proteins may similarly impair synaptic function in other neurodegenerative disorders, such as Alzheimer and Parkinson.30
In our study we asked the question whether the Rab11 subfamily of GTPases—which is crucially involved in cellular trafficking, cytoskeletal regulation, and endosomal recycling—could counteract the synaptic deficits induced by mutant HTT. Strikingly, we found that overexpression of Rab11 reversed the synaptic neurotransmission and vesicle deficits, and restored normal locomotor behavior. Several GTPases have been implicated in mammalian systems to regulate neurotransmission,31,32 and Rab11, an evolutionarily conserved, ubiquitously expressed subfamily of GTPases regulates diverse cellular and developmental events such as exocytotic and transcytotic events.33 In Drosophila, Rab11 has been shown to be involved in embryonic nervous system development34 or post-Golgi trafficking.35 Furthermore, at the Drosophila NMJ, presynaptic Rab3 [and its interaction partner Rab3 GTPase Activating Protein (Rab3-GAP)] is required for synaptic homeostasis,36 illustrating the broad and conserved functions of Rab-GTPase signaling.
But how can Rab activities modulate transmission or even reverse early mutant HTT-induced synaptic deficits? Rab11 has been shown not only to regulate dendritic morphology,37 but more importantly several Rab isoforms are also present at the synaptic vesicle, pointing toward direct interactions between Rabs and vesicular signaling. In particular, Rab3, Rab5, and Rab11 are present at synaptic vesicles,31 providing a potential mechanism for direct interaction with the vesicle recycling machinery (see Figure 1). Rab GTPases also participate in vesicle tethering, docking, and fusion events via association with v-SNARE and/or t-SNARE proteins forming trans-SNARE complexes, thereby organizing fusion competent microdomains.38 However, as we detected Rab11-mediated rescue of synaptic vesicles sizes prior to fusion, it is unlikely that Rab11 modulates vesicle fusion in our model.
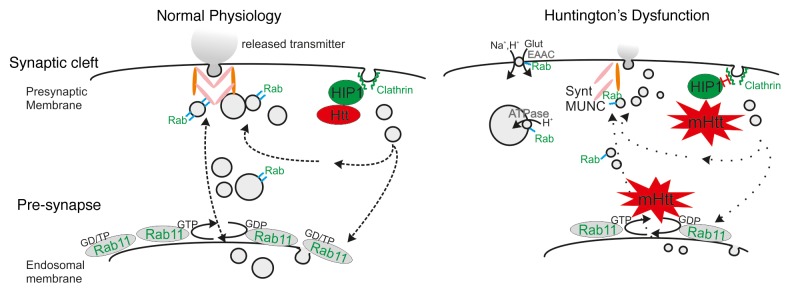
Figure 1. Model of impaired neurotransmission caused by mutant HTT. Mutant HTT leads to reduced synaptic vesicle size (right) via impaired Rab11 activity and altered interactions with HIP1, causing reduced neurotransmitter release and synaptic dysfunction. Overexpression of Rab11 may reverse these defects by several mechanisms: 1) Rab11 modulates vesicle size via enhancing proton (H+) ATPase trafficking. Rab11 has been shown to interact with the ε subunit of the vacuolar-type H+-ATPase and a possible enhanced interaction with the vesicular H+-ATPase could alleviate the mutant HTT-induced synaptic deficiency. 2) Huntington’s disease (HD) has been associated with lower cell surface expression of the glutamate/cysteine transporter EAAC141 that regulates glutamate uptake, potentially leaving the neuron with limited glutamate resources. Expression of a dominant-active Rab11 mutant in primary HD mouse neurons rectified this deficit. 3) Rab3, Rab5, and Rab11 are present at synaptic vesicles, providing a potential mechanism for direct interaction with the vesicle recycling machinery. Rab GTPases also participate in vesicle tethering, docking, and fusion events via association with v-SNARE and/or t-SNARE proteins forming trans-SNARE complexes, thereby organizing fusion competent microdomains. EAAC, glutamate/cysteine transporter; HIP, Huntingtin Interacting Protein; Glut, Glutamate; Synt, Syntaxin; Rab/Rab11, possible functions of various Rab proteins
Alternatively, enhanced Rab11 activity could increase the endosomal recycling rate and thereby deliver more vesicle-required proteins to the membrane, ultimately leading to improved Rab-regulated trafficking and endosomal signaling. One tempting speculation of how Rab11 modulates vesicle size is via enhancing proton (H+) ATPase trafficking. Rab11 has been shown to interact with the ε subunit of the vacuolar-type H+-ATPase39,40 and a possible enhanced interaction with the vesicular H+-ATPase could alleviate the mutant HTT-induced synaptic deficiency (Fig. 1). In addition, HD has been associated with lower cell surface expression of the glutamate/cysteine transporter EAAC1,41 which regulates glutamate uptake, potentially leaving the neuron with limited glutamate resources. Expression of a dominant-active Rab11 mutant in primary HD mouse neurons rectified this deficit,41 highlighting Rab11 as a promising candidate for modulating HD pathologies. Both these mechanisms are potentially involved in regulating vesicular glutamate content and may thereby indirectly modulate vesicular size.
Finally, other members of the Rab family have been reported to be involved in increased secretion of α-synuclein,42 likely reflecting the reported role of Rabs in autophagy initiation. A similar mechanism could explain our observations whereby mutant HTT is secreted in an enhanced manner leading to reduced toxicity. As mutant HTT has been implicated in disrupting axonal transport in Drosophila neurons,27 enhanced extrusion/secretion of mutant HTT could improve axonal transport of proteins necessary for vesicle formation. In this context, it would be interesting to explore whether Rab11 as a recycling endosomal marker molecule co-localizes with mutant HTT protein, or perhaps even directly interacts with HTT.
In conclusion, with the known involvement of Rab11 in neurodegenerative disorders such as Alzheimer and HD,41,43,44 we propose a new potential use of Rab11 as a therapeutic strategy in neurodegeneration. Supporting this concept, approaches aimed at increasing Rab11 activity are already being developed for targeting disease.45 Reduction in synapse number is a consistent early feature of neurodegenerative diseases, preceding neuronal loss, and correlating with cognitive deficits. As this is a reversible stage of disease, it permits intervention prior to neuron loss, making an attractive therapeutic strategy in neurodegenerative disorders.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank David Dinsdale (Imaging and Pathology Group at the MRC Toxicology Unit) and Natalie Allcock (Electron Microscopy Lab, University of Leicester) for assistance with electron microscopy. The original study was primarily funded by a research grant from the Huntington's Disease Association and supported by the Medical Research Council (MRC). Funding to pay the Open Access publication charges for the original article was provided by the MRC.
Footnotes
Previously published online: www.landesbioscience.com/journals/cib/article/26807
References
- 1.Picconi B, Piccoli G, Calabresi P. Synaptic dysfunction in Parkinson’s disease. Adv Exp Med Biol. 2012;970:553–72. doi: 10.1007/978-3-7091-0932-8_24. [DOI] [PubMed] [Google Scholar]
- 2.Milnerwood AJ, Raymond LA. Early synaptic pathophysiology in neurodegeneration: insights from Huntington’s disease. Trends Neurosci. 2010;33:513–23. doi: 10.1016/j.tins.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 3.Marcello E, Epis R, Saraceno C, Di Luca M. Synaptic dysfunction in Alzheimer’s disease. Adv Exp Med Biol. 2012;970:573–601. doi: 10.1007/978-3-7091-0932-8_25. [DOI] [PubMed] [Google Scholar]
- 4.Mallucci GR. Prion neurodegeneration: starts and stops at the synapse. Prion. 2009;3:195–201. doi: 10.4161/pri.3.4.9981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 6.Li X, DiFiglia M. The recycling endosome and its role in neurological disorders. Prog Neurobiol. 2012;97:127–41. doi: 10.1016/j.pneurobio.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Pereira-Leal JB, Seabra MC. Evolution of the Rab family of small GTP-binding proteins. J Mol Biol. 2001;313:889–901. doi: 10.1006/jmbi.2001.5072. [DOI] [PubMed] [Google Scholar]
- 8.Pfeffer S. Membrane domains in the secretory and endocytic pathways. Cell. 2003;112:507–17. doi: 10.1016/S0092-8674(03)00118-1. [DOI] [PubMed] [Google Scholar]
- 9.Khvotchev MV, Ren M, Takamori S, Jahn R, Südhof TC. Divergent functions of neuronal Rab11b in Ca2+-regulated versus constitutive exocytosis. J Neurosci. 2003;23:10531–9. doi: 10.1523/JNEUROSCI.23-33-10531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simons K, Zerial M. Rab proteins and the road maps for intracellular transport. Neuron. 1993;11:789–99. doi: 10.1016/0896-6273(93)90109-5. [DOI] [PubMed] [Google Scholar]
- 11.Ullrich O, Reinsch S, Urbé S, Zerial M, Parton RG. Rab11 regulates recycling through the pericentriolar recycling endosome. J Cell Biol. 1996;135:913–24. doi: 10.1083/jcb.135.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hales CM, Vaerman JP, Goldenring JR. Rab11 family interacting protein 2 associates with Myosin Vb and regulates plasma membrane recycling. J Biol Chem. 2002;277:50415–21. doi: 10.1074/jbc.M209270200. [DOI] [PubMed] [Google Scholar]
- 13.Li X, Valencia A, McClory H, Sapp E, Kegel KB, Difiglia M. Deficient Rab11 activity underlies glucose hypometabolism in primary neurons of Huntington’s disease mice. Biochem Biophys Res Commun. 2012;421:727–30. doi: 10.1016/j.bbrc.2012.04.070. [DOI] [PubMed] [Google Scholar]
- 14.Li X, Sapp E, Valencia A, Kegel KB, Qin ZH, Alexander J, Masso N, Reeves P, Ritch JJ, Zeitlin S, et al. A function of huntingtin in guanine nucleotide exchange on Rab11. Neuroreport. 2008;19:1643–7. doi: 10.1097/WNR.0b013e328315cd4c. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Standley C, Sapp E, Valencia A, Qin ZH, Kegel KB, Yoder J, Comer-Tierney LA, Esteves M, Chase K, et al. Mutant huntingtin impairs vesicle formation from recycling endosomes by interfering with Rab11 activity. Mol Cell Biol. 2009;29:6106–16. doi: 10.1128/MCB.00420-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, Sapp E, Chase K, Comer-Tierney LA, Masso N, Alexander J, Reeves P, Kegel KB, Valencia A, Esteves M, et al. Disruption of Rab11 activity in a knock-in mouse model of Huntington’s disease. Neurobiol Dis. 2009;36:374–83. doi: 10.1016/j.nbd.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richards P, Didszun C, Campesan S, Simpson A, Horley B, Young KW, Glynn P, Cain K, Kyriacou CP, Giorgini F, et al. Dendritic spine loss and neurodegeneration is rescued by Rab11 in models of Huntington’s disease. Cell Death Differ. 2011;18:191–200. doi: 10.1038/cdd.2010.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.del Toro D, Alberch J, Lázaro-Diéguez F, Martín-Ibáñez R, Xifró X, Egea G, Canals JM. Mutant huntingtin impairs post-Golgi trafficking to lysosomes by delocalizing optineurin/Rab8 complex from the Golgi apparatus. Mol Biol Cell. 2009;20:1478–92. doi: 10.1091/mbc.E08-07-0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ravikumar B, Imarisio S, Sarkar S, O’Kane CJ, Rubinsztein DC. Rab5 modulates aggregation and toxicity of mutant huntingtin through macroautophagy in cell and fly models of Huntington disease. J Cell Sci. 2008;121:1649–60. doi: 10.1242/jcs.025726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giorgini F, Guidetti P, Nguyen Q, Bennett SC, Muchowski PJ. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat Genet. 2005;37:526–31. doi: 10.1038/ng1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mason RP, Casu M, Butler N, Breda C, Campesan S, Clapp J, Green EW, Dhulkhed D, Kyriacou CP, Giorgini F. Glutathione peroxidase activity is neuroprotective in models of Huntington’s disease. Nat Genet. 2013;45:1249–54. doi: 10.1038/ng.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Green EW, Giorgini F. Choosing and using Drosophila models to characterize modifiers of Huntington’s disease. Biochem Soc Trans. 2012;40:739–45. doi: 10.1042/BST20120072. [DOI] [PubMed] [Google Scholar]
- 23.Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–43. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- 24.Romero E, Cha GH, Verstreken P, Ly CV, Hughes RE, Bellen HJ, Botas J. Suppression of neurodegeneration and increased neurotransmission caused by expanded full-length huntingtin accumulating in the cytoplasm. Neuron. 2008;57:27–40. doi: 10.1016/j.neuron.2007.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee WC, Yoshihara M, Littleton JT. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington’s disease. Proc Natl Acad Sci U S A. 2004;101:3224–9. doi: 10.1073/pnas.0400243101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Labbadia J, Morimoto RI. Huntington’s disease: underlying molecular mechanisms and emerging concepts. Trends Biochem Sci. 2013;38:378–85. doi: 10.1016/j.tibs.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krench M, Littleton JT. Modeling Huntington disease in Drosophila: Insights into axonal transport defects and modifiers of toxicity. Fly (Austin) 2013;7:7. doi: 10.4161/fly.26279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sinadinos C, Burbidge-King T, Soh D, Thompson LM, Marsh JL, Wyttenbach A, Mudher AK. Live axonal transport disruption by mutant huntingtin fragments in Drosophila motor neuron axons. Neurobiol Dis. 2009;34:389–95. doi: 10.1016/j.nbd.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 29.Steinert JR, Campesan S, Richards P, Kyriacou CP, Forsythe ID, Giorgini F. Rab11 rescues synaptic dysfunction and behavioural deficits in a Drosophila model of Huntington’s disease. Hum Mol Genet. 2012;21:2912–22. doi: 10.1093/hmg/dds117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gadad BS, Britton GB, Rao KS. Targeting oligomers in neurodegenerative disorders: lessons from α-synuclein, tau, and amyloid-β peptide. J Alzheimers Dis. 2011;24(Suppl 2):223–32. doi: 10.3233/JAD-2011-110182. [DOI] [PubMed] [Google Scholar]
- 31.Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–47. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- 32.Sakane A, Manabe S, Ishizaki H, Tanaka-Okamoto M, Kiyokage E, Toida K, Yoshida T, Miyoshi J, Kamiya H, Takai Y, et al. Rab3 GTPase-activating protein regulates synaptic transmission and plasticity through the inactivation of Rab3. Proc Natl Acad Sci U S A. 2006;103:10029–34. doi: 10.1073/pnas.0600304103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kelly EE, Horgan CP, McCaffrey MW. Rab11 proteins in health and disease. Biochem Soc Trans. 2012;40:1360–7. doi: 10.1042/BST20120157. [DOI] [PubMed] [Google Scholar]
- 34.Bhuin T, Roy JK. Rab11 is required for embryonic nervous system development in Drosophila. Cell Tissue Res. 2009;335:349–56. doi: 10.1007/s00441-008-0711-8. [DOI] [PubMed] [Google Scholar]
- 35.Satoh AK, O’Tousa JE, Ozaki K, Ready DF. Rab11 mediates post-Golgi trafficking of rhodopsin to the photosensitive apical membrane of Drosophila photoreceptors. Development. 2005;132:1487–97. doi: 10.1242/dev.01704. [DOI] [PubMed] [Google Scholar]
- 36.Müller M, Pym EC, Tong A, Davis GW. Rab3-GAP controls the progression of synaptic homeostasis at a late stage of vesicle release. Neuron. 2011;69:749–62. doi: 10.1016/j.neuron.2011.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawauchi T, Sekine K, Shikanai M, Chihama K, Tomita K, Kubo K, Nakajima K, Nabeshima Y, Hoshino M. Rab GTPases-dependent endocytic pathways regulate neuronal migration and maturation through N-cadherin trafficking. Neuron. 2010;67:588–602. doi: 10.1016/j.neuron.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 38.Wickner W, Schekman R. Membrane fusion. Nat Struct Mol Biol. 2008;15:658–64. doi: 10.1038/nsmb.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oehlke O, Schlosshardt C, Feuerstein M, Roussa E. Acidosis-induced V-ATPase trafficking in salivary ducts is initiated by cAMP/PKA/CREB pathway via regulation of Rab11b expression. Int J Biochem Cell Biol. 2012;44:1254–65. doi: 10.1016/j.biocel.2012.04.018. [DOI] [PubMed] [Google Scholar]
- 40.Oehlke O, Martin HW, Osterberg N, Roussa E. Rab11b and its effector Rip11 regulate the acidosis-induced traffic of V-ATPase in salivary ducts. J Cell Physiol. 2011;226:638–51. doi: 10.1002/jcp.22388. [DOI] [PubMed] [Google Scholar]
- 41.Li X, Valencia A, Sapp E, Masso N, Alexander J, Reeves P, Kegel KB, Aronin N, Difiglia M. Aberrant Rab11-dependent trafficking of the neuronal glutamate transporter EAAC1 causes oxidative stress and cell death in Huntington’s disease. J Neurosci. 2010;30:4552–61. doi: 10.1523/JNEUROSCI.5865-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, et al. α-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol. 2010;190:1023–37. doi: 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Greenfield JP, Leung LW, Cai D, Kaasik K, Gross RS, Rodriguez-Boulan E, Greengard P, Xu H. Estrogen lowers Alzheimer beta-amyloid generation by stimulating trans-Golgi network vesicle biogenesis. J Biol Chem. 2002;277:12128–36. doi: 10.1074/jbc.M110009200. [DOI] [PubMed] [Google Scholar]
- 44.Dumanchin C, Czech C, Campion D, Cuif MH, Poyot T, Martin C, Charbonnier F, Goud B, Pradier L, Frebourg T. Presenilins interact with Rab11, a small GTPase involved in the regulation of vesicular transport. Hum Mol Genet. 1999;8:1263–9. doi: 10.1093/hmg/8.7.1263. [DOI] [PubMed] [Google Scholar]
- 45.Agola JO, Jim PA, Ward HH, Basuray S, Wandinger-Ness A. Rab GTPases as regulators of endocytosis, targets of disease and therapeutic opportunities. Clin Genet. 2011;80:305–18. doi: 10.1111/j.1399-0004.2011.01724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]