

Abstract
Clobazam (CLB), a 1,5-benzodiazepine (BZD), was FDA-approved in October 2011 for the adjunctive treatment of seizures associated with Lennox-Gastaut syndrome (LGS) in patients 2 years and older. BZDs exert various CNS effects through allosteric modulation of GABAA receptors. The structurally distinct, 1,4-BZD clonazepam (CLN) is also approved to treat LGS. The precise mechanisms of action and clinical efficacy of both are unknown. Data show that the GABAA α1-subunit–selective compound zolpidem [ZOL] exhibits hypnotic/sedative effects. Conversely, data from knock-in mice carrying BZD binding site mutations suggest that the α2 subunit mediates anticonvulsant effects, without sedative actions. Hence, the specific pattern of interactions across the GABAA receptor complexes of BZDs might be reflected in their clinical efficacies and adverse effect profiles. In this study, GABAA-receptor binding affinities of CLB, N-desmethylclobazam (N-CLB, the major metabolite of CLB), CLN, and ZOL were characterized with native receptors from rat-brain homogenates and on cloned receptors from HEK293 cells transfected with combinations of α (α1, α2, α3, or α5), β2, and γ2 subtypes. Our results demonstrate that CLB and N-CLB have significantly greater binding affinities for α2- vs. α1-receptor complexes, a difference not observed for CLN, for which no distinction between α2 and α1 receptors was observed. Our experiments with ZOL confirmed the high preference for α1 receptors. These results provide potential clues to a new understanding of the pharmacologic modes of action of CLB and N-CLB.
Introduction
GABAA receptors are the principal inhibitory neurotransmitter-receptor system in the mammalian brain. GABAA receptors are hetero-oligopentameric complexes that are members of the pentameric, ligand-gated family (also known as cys-loop family). Upon activation by the endogenous ligand GABA, receptors become permeable to chloride ions, thereby triggering chloride ion influx, which hyperpolarizes the cell and dampens activity. Most GABAA receptors consist of α, β, and γ subunits, and the most prominently expressed combination is the α1β2γ2 receptor complex [1].
Pharmacologic modulation of GABAA receptors has long been used to treat a range of conditions such as epilepsy, anxiety and panic disorders, muscle spasms, and sleep disorders. A subset of GABAA-receptor modulators bind to the allosteric site situated at the α- and γ-subunit interface, termed the benzodiazepine (BZD) site. The variety of possible GABAA-receptor subunit combinations suggests that subunit-specific compounds may allow for clinical tuning of desired therapeutic effects [2], [3], [4], [5]. Such subtype selectivity toward the GABAA α1 subunit is linked to the clinical effectiveness of zolpidem as a sedative [6], [7]. Preclinical studies with knock-in mice carrying single amino acid point–mutations in the BZD binding site demonstrate that BZD modulation of the GABAA α1-subunit receptors confers hypnotic/sedative effects, in addition to anticonvulsant effects. In contrast, BZD modulation of GABAA α2–containing receptors appears to mediate anxiolytic effects and potentially may also determine anticonvulsant effect, but with no effect on sedation [2], [3], [4], [8], [9]. However, the anticonvulsant role of the GABAA α2–containing receptors is described in the literature with divergent results.
For example, a comparative study by Fradley et al. [4] using identical drug dosages, pre-treatment times, and scorings across different mouse strains (α1, α2, and α5 H→R knock-in mice) found that, apparently, the α2 subtype plays a greater role than α1-containing GABAA receptors in determining anticonvulsant effect following GABAA receptor modulation. They address this through two models using pentylenetetrazole (PTZ)-induced seizures or tonic seizures induced by electroshock. Wild-type mice were dosage-dependently protected by diazepam in the PZT model scored using the Racine scale from 6 reversed to seizure level 1 at greatest diazepam dosage. In contrast, with the α2(H101R) mice, the greatest diazepam dosage produced less protection, to a scoring of approximately 3.5 on the Racine scale. No difference was observed in diazepam protection of the electroshock-induced seizures, for which a single 20-mg/kg dosage was evaluated. The authors conclude that experiments of anticonvulsant effects in rodents indicate that effect on more than one subtype is required, and that these can act synergistically [4]. Earlier work showed that the anticonvulsant action of diazepam is partially but not completely reduced in α1(H101R) mice [2], indicating that other GABAA receptor subtypes also mediate anticonvulsant actions. Further, a study by Low et al. [10] examining diazepam-induced behavior in α2(H101R) and α3(H126R) knock-in mice revealed that the anxiolytic-like action of diazepam is absent in the α2(H101R), but preserved in the α3(H126R) mice. This suggests that the α2 subunit mediates this action of BZDs. In addition, the authors showed that diazepam-induced sedation and motor impairment is preserved in the two lines of mice, suggesting that other GABAA subtypes mediate these effects. The anticonvulsive effects of 3-, 10-, and 30-mg/kg diazepam were assessed by percentage of mice developing tonic convulsions following PTZ administration. This yielded 100% protection at 10 and 30 mg/kg in both wild-type and α2(H101R) mice, while the lower dosage of 3 mg/kg resulted in convulsing in approximately 20% of the wild-type mice, but convulsing in approximately 50% of the α2(H101R) mice. Moreover, the α1 inactive compound L-838,417 that binds to α1 receptors but does not potentiate GABA responses retains anticonvulsant activity to PTZ and audiogenic-induced seizures in mice [3]. Collectively, the precise roles of the α1 and α2 subunits in drug-induced efficacy across preclinical models of human disease continue to be debated, except for the clear involvement of α1 in sedation.
Beyond the α1 and α2 subunits, the roles of BZD site modulation of the α3- and α5-containing receptor complexes are less established. Together with the α2 subunit, the α3 subunit has been implicated in BZD modulation of inflammatory and neuropathic pain responses, as well as in having some role in anticonvulsant activity [4], [11]. Negative modulation of α5-containing receptors has been pursued in disorders with cognitive deficits such as Down syndrome (e.g., Roche's RG-1662 currently in Phase I trials [NCT01436955 and NCT01667367]). In addition, development of tolerance to the sedative effects of diazepam (DZP) has been coupled to continued activation of α5 receptors [12]. The data suggest that conclusions regarding the clinical impact of significant differences in binding related to the α3- and α5-containing receptors cannot be drawn with certainty.
Clobazam (Onfi®; CLB), a structurally unique 1,5-BZD, was approved by the US Food and Drug Administration (FDA) in October 2011 for the adjunctive treatment of seizures associated with Lennox-Gastaut syndrome (LGS) in patients 2 years and older, based on results from a Phase III randomized controlled study [13]. CLB was first approved and used as an anxiolytic agent in the early 1970s [14]. Since then, the efficacy of CLB for patients with treatment-refractory epilepsy has been well-documented in a retrospective study [15]. Moreover, Lennox-Gastaut syndrome is well-known for its highly refractory nature [16]. CLB's primary active metabolite, N-desmethylclobazam (N-CLB), has a much longer half-life than the parent compound (79 h vs. 36 h), resulting in greater metabolite than CLB concentrations following prolonged dosing in humans [17]. The ratio of CLB to N-CLB is dependent on CYP2C19 genotypes and shows considerable correlation to polymorphisms in the CYP2C19 gene [18], [19]. Clonazepam (Klonopin®) is a 1,4-BZD used in the United States either alone or as an adjunctive treatment for LGS (petit mal variant), akinetic and myoclonic seizures [20]. Zolpidem (Ambien®), on the other hand, is approved in the United States for the short-term treatment of insomnia characterized by difficulties with sleep initiation [21].
Although drawing clinical conclusions from in vitro and in vivo animal data is not feasible, elucidating possible mechanisms of action may be an important first step in providing potential explanations of and links between preclinical and clinical findings. To examine and compare the modes of action of CLB, N-CLB, and CLN, we here present the results of radio-ligand binding assays to the BZD site on native receptors from the rat brain, as well as human receptors of known composition.
Materials and Methods
Animals
Male Lister Hooded rats from Charles River, Germany (250–300 g, 7–8 weeks) were used in these studies. All animals were housed two per cage under a 12 h light/dark cycle in a temperature- and humidity-controlled environment. Food and water were available ad libitum. Rats were used 1 week after arrival. The experiments were carried out at H. Lundbeck A/S, Denmark, and ethical permissions were granted by The Danish Animal Experiments Inspectorate. All animal procedures for these studies were conducted in compliance with the EC Directive 86/609/EEC and Danish law regulating experiments on animals.
Rat-Brain Membrane Preparation
Rats were killed by rapid decapitation. Brains were rapidly removed and kept briefly on ice. The cerebellum was removed and the remaining brain tissue was homogenized in 10 volumes of buffered solution (10 mM Tris-citrate buffer [pH 7.4] and 0.32 M sucrose, 4°C) using a glass-teflon homogenizer. The homogenate was centrifuged for 10 minutes at 1,000g at 4°C, the supernatant collected, and the pellet re-suspended and centrifuged again. The combined supernatants were centrifuged at 13,000g for 20 minutes at 4°C. The pellet was re-suspended in 20–40 volumes of 5 mM Tris-citrate buffer (pH 7.4) and 5 mM EDTA solution at 4°C using an ULTRA-TURRAX® (24,000 RPM, 10–30 sec). Following 15–30 minutes of incubation on ice and 3 subsequent rounds of centrifugation (48,000g for 10 min at 4°C) and re-suspension (20–40 volumes of 5 mM Tris-citrate buffer [pH 7.4] and 5 mM EDTA solution at 4°C), the pellet was re-suspended in 50 mM Tris-citrate buffer (pH 7.4) and stored overnight at −20°C. On the day of the experiment, the homogenate was centrifuged (48,000g for 10 min at 4°C) and re-suspended (20–40 volumes of 50 mM Tris-citrate [pH 7.4] at 4°C) for a total of 4 times. For each batch of brain homogenate, the total protein content, tracer equilibrium dissociation constant (Kd), and maximum density of receptors corrected for protein concentration (Bmax) were determined.
Transfection and Expression of Recombinant Human GABAA Receptors
Human embryonic kidney cells (i.e., HEK293) served as the host for overexpression of recombinant human GABAA receptors. Specifically, α1, α2, α3, and α5 subunits were individually co-expressed with β2 and γ2 subunits in a 1∶1∶3 ratio (constructs were in the pcDNA3 vector). Batches of HEK293 cells were transiently transfected with GABAA-receptor cDNAs complemented with enhanced green fluorescent protein (EGFP) cDNA serving as an indicator for successful transfection. All transfections (4–6 h) were accomplished with PolyFect Transfection Reagent (QIAGEN, Denmark) in a 1∶5 weight/volume ratio (µg total cDNA to µL transfection reagent), according to the manufacturer's instructions. Cells were harvested and used for binding experiments 2–3 days after transfection.
Cell Homogenate Preparation
The cell media was first removed followed by 3 washing steps with phosphate-buffered saline (PBS) without Ca2+/Mg2+. The cells were then harvested by scraping in PBS without Ca2+/Mg2+ and pelleted by centrifugation (3,000g for 5 min at 4°C). The supernatant was removed and the cell pellet was either stored at −80°C for later use or homogenized using an ULTRA-TURRAX® (24,000 rpm for 10–24 sec) in ice-cold buffer (5 mM Tris-citrate [pH 7.4] and 5 mM EDTA solution) and Protease Inhibitor Cocktail (Sigma Aldrich, Denmark). The cell-homogenate was centrifuged again (50,000g for 60 min at 4°C), the supernatant was discarded, and the pellet was re-suspended in buffer (50 mM Tris-citrate [pH 7.4] at 4°C). Protein content was determined using the BCA Protein Assay Reagent (Pierce/Thermo Scientific, Denmark) according to the manufacturer's instructions; and the homogenates were either stored at −80°C or used immediately for binding experiments. For each batch of cell homogenates, we determined total protein content, Kd, and Bmax.
Radio-Ligand Binding
All experiments utilizing cloned GABAA receptors were conducted using 3H-flumazenil (Ro15-1788, NET 757250UC, Perkin Elmer, Denmark) and those conducted with native receptors from brain homogenates employed 3H-flunitrazepam (NET 567250UC, Perkin Elmer, Denmark). 3H-flunitrazepam was not used with cloned GABAA receptors as it provided substantial non-related binding, likely from the presence of the peripheral BZD receptors in HEK293 cells (data not shown).
For each experiment, a suitable amount of cell or brain homogenate was thawed and mixed with diluted 3H-flumazenil or 3H-flunitrazepam, and the test compound of choice in a 4∶1∶1 volume ratio (typically 100 µL homogenate, 25 µL radio ligand, and 25 µL test compound). Dimethyl sulfoxide (DMSO) stock solutions were made of compounds, and these were diluted into assay buffer, with the final DMSO content kept below 0.3%. Dilutions were made using assay buffer at 4°C (for cloned receptors: 50 mM Tris-citrate [pH 7.4] and 150 mM NaCl solution; for brain homogenates: 50 mM Tris-citrate [pH 7.4]). Binding experiments were allowed to equilibrate for 90 minutes at 4°C with slow plate rotation and then harvested using a Tomtec harvester (Tomtec, Inc., CT, USA) with harvest buffer (50 mM Tris-citrate [pH 7.4] at 4°C) onto 96-well format glass-fiber filters (B-size filters) pre-wetted with 0.1% polyethyleneimine. The degree of retained radioactivity was quantified on a Wallac MicroBeta counter (Wallac/Perkin Elmer, Denmark). On each 96-well plate, controls for total binding (assay buffer) and non-specific binding (100 µM and 20 µM DZP for cloned receptors and brain homogenates, respectively) were included, allowing for calculations of the relative percentage inhibition. The time of radioactivity counting was set so the total counts per well were >10,000.
Saturation experiments to determine Kd and Bmax were performed with 12 different concentrations of tracer (up to 10 nM). Specific binding at the Kd concentration was >80% and depletion of tracer <10% in all cases. Half-maximum inhibitory concentrations (IC50) for test compounds were determined through 10-point, serial 5-fold dilutions covering at least 7 log scales surrounding the experimentally determined IC50 values. Individual determinations for each compound on each GABAA-receptor subtype or brain homogenate were made on independent experimental days and were conducted with a tracer concentration close to the Kd value.
Data Analysis
Kd and Bmax values were determined through saturation binding experiments and fitting the specific binding signal (total binding minus the background binding obtained with DZP) to a hyperbolic function (Equation 1).
| (1) |
where L is the concentration of the radio-ligand. Kd and Bmax were presented as the average values from several experiments. The experimentally determined concentration-inhibition data were fitted to the 4 Parameter Logistic (4PL) model (Equation 2) using non-linear regression (Microsoft Excel and XLfit) to yield the IC50 values.
 |
(2) |
The IC50 values were transformed to Ki (binding affinity) values using the Cheng-Prusoff correction (Equation 3).
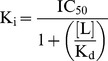 |
(3) |
where L is the concentration of the radio-ligand. The individual Ki values were transformed into pKi (pKi = −log[Ki]) and presented as mean values with standard errors of the mean (SEM). The mean of the pKi values were evaluated for statistical differences across the receptor subtypes per compound by one-way ANOVA with Tukey's Multiple-Comparison post-hoc Test (P<0.05 considered significant). Testing the hypothesis of same mean across subtypes was done using GraphPad Prism 4 (GraphPad Software, Inc., La Jolla, CA).
Results
Kd and Bmax values for 3H-flunitrazepam (used as a tracer for binding) on rat-brain homogenates were 1.5±0.2 nM and 1,906±166 fmol/mg, respectively (Figure 1A). Specific binding at the Kd value was determined to be 85%±3.3% (N = 6). CLN was determined to have subnanomolar binding affinity (0.26 nM), whereas CLB and N-CLB exhibited submicromolar binding affinities (151 and 133 nM, respectively, Table 1).
Figure 1. Saturation binding experiments of equilibrium binding of various concentrations of radio-ligand.

(A) Rat brain homogenate with 3H-flunitrazepam, and (B) recombinant human GABAA receptors with 3H-flumazenil.Total binding, non-specific binding, and specific binding are shown.
Table 1. Clonazepam, clobazam, and N-desmethylclobazam binding affinities for rat-brain homogenates using 3H-flunitrazepam as tracer.
| pKi (mean ±SEM) | Ki (nM) | N | |
| Clonazepam | 9.58 (±0.08) | 0.26 | 4 |
| Clobazam | 6.82 (±0.10) | 151 | 4 |
| N -desmethylclobazam | 6.88 (±0.17) | 133 | 4 |
Ki = binding affinity; SEM = standard error of the mean.
Transfected HEK293 cell homogenates were probed with 3H-flumazenil as the tracer; Kd values were 0.43–1.24 nM and Bmax values were 188–300 fmol/mg across the 4 GABAA-receptor complexes (Figure 1B and Table 2). These Kd values are in agreement with previously published data for combinations of α1, α2, α3, and α5 subunits with β3 and γ2 expressed in mouse fibroblast L(tk−) cells [22].
Table 2. Saturation equilibrium binding results of 3H-flumazenil on four different GABAA-receptor complexes overexpressed in HEK293 cells.
| α1β2γ2 | α2β2γ2 | α3β2γ2 | α5β2γ2 | |
| 3H-flumazenil | Mean (±SEM) | Mean (±SEM) | Mean (±SEM) | Mean (±SEM) |
| Bmax (fmol/mg) | 300 (±22) | 225 (±11) | 188 (±18) | 227 (±24) |
| Kd (nM) | 0.50 (±0.05) | 1.11 (±0.12) | 1.24 (±0.14) | 0.43 (±0.05) |
Bmax = maximum density of receptor binding sites; Kd: tracer equilibrium dissociation constant; SEM = standard error of the mean.
The number of experiments ranged from 9 to 12.
All 4 compounds fully displaced 3H-flumazenil in a concentration-dependent manner, consistent with competitive binding to the BZD binding site on GABAA receptors. CLB displaced 3H-flumazenil at submicromolar Ki values across all α-subunit subtypes tested in the range of 205 nM to 753 nM (α2<α5<α1<α3). In addition, submicromolar Ki values in the interval of 147 nM to 668 nM were determined for N-CLB (α2<α5<α1<α3, Table 3). CLN and ZOL were found to have Ki values ranging from 0.65–2.20 nM (α1<α2<α3<α5) and 30–5,431 nM (α1<α2<α5<α3), respectively (Table 3), with ZOL showing preferential binding at α1. These results for ZOL and CLN are in agreement with published data [23], [24]. The range of pKi values for all 4 compounds are depicted in Figure 2, and the results (i.e., P-values) of pair-wise comparisons of the mean pKi values between receptor subtypes performed with Tukey's Multiple-Comparison Test are provided in Table 4. The binding profiles across the receptor subtypes are different for these compounds (i.e., both CLB and N-CLB show significantly greater binding affinities for α2 over α1, whereas CLN does not show this particular subtype difference). In line with its use as a sedative, ZOL has the greatest affinity for α1 versus all other receptor subtypes.
Table 3. Binding affinities of clobazam, N-desmethylclobazam, clonazepam, and zolpidem to four different human GABAA-receptors complexes expressed in HEK293 cells obtained by displacement of 3H-flumazenil.
| α1β2γ2 | α2β2γ2 | α3β2γ2 | α5β2γ2 | |||||||||
| pKi | pKi | pKi | pKi | |||||||||
| Drug | Mean (±SEM) | N | Ki (nM) | Mean (±SEM) | N | Ki (nM) | Mean (±SEM) | N | Ki (nM) | Mean (±SEM) | N | Ki (nM) |
| Clobazam | 6.28 (±0.05) | 7 | 519 | 6.69 (±0.11) | 6 | 205 | 6.12 (±0.03) | 5 | 753 | 6.48 (±0.08) | 5 | 331 |
| N-desmethylclobazam | 6.20 (±0.06) | 5 | 634 | 6.83 (±0.10) | 6 | 147 | 6.18 (±0.07) | 5 | 668 | 6.53 (±0.09) | 6 | 292 |
| Clonazepam | 9.19 (±0.03) | 7 | 0.65 | 9.14 (±0.10) | 6 | 0.72 | 8.65 (±0.04) | 6 | 2.2 | 9.11 (±0.08) | 6 | 0.78 |
| Zolpidem | 7.52 (±0.05) | 7 | 30 | 6.78 (±0.04) | 5 | 165 | 6.35 (±0.05) | 5 | 442 | 5.27 (±0.04) | 6 | 5431 |
Ki = binding affinity; SEM = standard error of the mean.
Figure 2.

Distribution of individually determined pKi values for (A) clobazam, (B) N-desmethylclobazam, (C) clonazepam, and (D) zolpidem across GABAA-receptor subtypes.
Table 4. Binding affinities normalized to GABAA α2 (ratios of Ki values, show in gray) and pair-wise comparison of the mean pKi values of clobazam, N-desmethylclobazam, clonazepam, and zolpidem across GABAA receptor subtypes.
| Clobazam | α1β2γ2 | α2β2γ2 | α3β2γ2 | α5β2γ2 |
| α1β2γ2 | 2.5 | P<0.01 | NS | NS |
| α2β2γ2 | – | 1 | P<0.001 | NS |
| α3β2γ2 | – | – | 3.7 | P<0.05 |
| α5β2γ2 | – | – | – | 1.6 |
NS = not significant.
The degree of significance from the comparison of the mean of the pKi values following one-way ANOVA with Tukey's multiple-comparison post-hoc test is presented across the subtypes per compound.
Discussion
This report describes the binding affinities of a set of compounds (clobazam [CLB], N-desmethylclobazam [N-CLB], clonazepam [CLN], and zolpidem [ZOL]) to native GABAA-receptor complexes overall (obtained from rat-brain homogenates) and to specific human GABAA-receptor subunits (obtained from HEK293 cells transiently transfected with human cDNA encoding GABAA-receptors, namely α1β2γ2, α2β2γ2, α3β2γ2, or α5β2γ2). CLB and N-CLB were found to have similar submicromolar binding affinities for native receptors, and CLN was found to display subnanomolar binding affinity. Data from our head-to-head comparisons on human receptors demonstrate that the 1,5-BZD CLB, and its active metabolite N-CLB, have significantly greater affinities for the α2-receptor subtype versus the α1-receptor subtype, whereas the 1,4-BZD CLN does not. This subtype preference was slightly more marked for N-CLB than for CLB. With the same experimental conditions, the sedative agent ZOL displayed the greatest affinity for α1-containing receptor complexes. The results from these experiments confirm that our assay is sensitive in determining differences in subtype affinity. Importantly, the binding affinities observed for the 2 different sources of GABAA receptors (rat-brain homogenates and transfected HEK293 cells) were in the same concentration range for the tested compounds. Therefore, the recombinant receptors are representative of the native receptors. The Ki values for CLB on native receptors are in the same range as previously published data [25], whereas our determined affinity for N-CLB is markedly different than what this previous group has published. We can only conjecture as to what may have caused this difference in binding affinity for N-CLB. Arendt et al. [25] unfortunately do not present information on the variation of their results and nor on the Kd or Bmax values obtained for 3H-flunitrazepam. Hence, evaluation of the variation and significance of their results is not possible. We note that the affinities we found using native receptors are in the same range as those we found using recombinant receptors. Interestingly, Haigh et al. [26] reported that N-CLB is effective both in a preclinical convulsion model in mice, but also in human patients when administered at dosages leading to concentrations similar to those obtained following dosing of clobazam (i.e., 8 of 9 patients responded favorably to N-CLB).
Upon initiation of CLB or CLN in clinical settings, drug concentrations in the human brain and plasma rise gradually. Extrapolating our preclinical binding data to such a clinical setting would imply that CLB (and resulting N-CLB) will initially bind to the α2 receptor before interacting with α1 receptor because of the difference in binding affinities. CLN, on the other hand, will bind simultaneously to both the α1 receptor and the α2 receptor by virtue of identical binding affinities for the two receptors. With the assumption that similar percentages of binding to α2 receptors are obtained during antiepileptic treatment with CLB and CLN, our data suggest that a separation to α1-receptor binding may be present for CLB but not for CLN.
During prolonged clinical use of CLB, concentrations of N-CLB build up (a direct result of its longer half-life) to more than 2-fold of the parent compound [27]. In this situation, given their similar Ki values, both compounds potentially would interact with GABAA receptors and act together to produce a combined modulatory effect. In addition, the significant separation between α1 and α2 for CLB/N-CLB could increase during N-CLB accumulation following prolonged dosing, as N-CLB presents the largest Ki-value ratio of α1 to α2 (Table 4).
Our results are interesting in the light of data from published studies comparing the clinical effects of orally administered CLB and CLN [28], [29], [30] for use for acute conditions. These independent double-blind, placebo-controlled (in the case of [30], an active placebo was used), cross-over studies of healthy volunteers show a greater incidence of sedation following oral dosing with CLN (0.5, 1 or 2 mg) relative to CLB (10 or 20 mg) [28], [29], [30]. The FDA-recommended starting oral dosages of CLB for patients with LGS weighing more than 30 kg are 10 mg/day [31]. The FDA-recommended initial oral dosage of CLN for adult patients with seizure disorders is 1.5 mg/day [20]. While the clinical data are very limited, preclinical data from a mice study suggest greater specificity of CLB and N-CLB for anticonvulsive/antiepileptic over sedative effects relative to 1,4-BZDs [32]. Findings from the spontaneously epileptic Ihara rat also suggest differential effects of CLB relative to CLN, with respect to differences in antiepileptic and sedative effects [33].
In summary, our binding studies with GABAA-receptor complexes expressing different α-subunit subtypes show that CLB and N-CLB have significantly greater affinities for the α2-containing receptors over α1-containing receptors. On the other hand, CLN has similar affinities for both α1- and α2-containing receptors, and, as previously shown, ZOL has greatest affinity for α1-containing receptors. This data set presents information on one important aspect of BZD interaction with GABAA-receptors (i.e., affinity), but our work did not investigate another important aspect of BZD function (i.e., efficacy — the degree of potentiation of the GABA response across the subtypes) [5]. To gain a full understanding of the modulatory effect of these compounds and the differences across subtypes we report here, such experiments should be conducted.
Acknowledgments
The authors would like to thank Charlotte Fredsøe Hundahl for expert technical assistance. The authors also thank Jouko Isojarvi, Niels Plath, and Tine Bryan Stensbøl, all of Lundbeck, for their critical scientific reviews of this manuscript. Writing assistance and editorial support during manuscript preparation were provided by Apurva Davé, PhD, of Prescott Medical Communications Group (Chicago, IL), and Michael A. Nissen, ELS, Lundbeck LLC (Deerfield, IL).
Funding Statement
The authors have no support or funding to report.
References
- 1.Berezhnoy D, Gravielle MC, Farb DH (2007) Pharmacology of the GABAA Receptor, in Handbook of Contemporary Neuropharmacology, John Wiley & Sons, Inc. [Google Scholar]
- 2. Rudolph U, Crestani F, Benke D, Brunig I, Benson JA, et al. (1999) Benzodiazepine actions mediated by specific GABAA receptor subtypes. Nature 401: 796–800. [DOI] [PubMed] [Google Scholar]
- 3. McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, et al. (2000) Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABAA receptor alpha1 subtype. Nat Neurosci 3: 587–592. [DOI] [PubMed] [Google Scholar]
- 4. Fradley RL, Guscott MR, Bull S, Hallett DJ, Goodacre SC, et al. (2007) Differential contribution of GABAA receptor subtypes to the anticonvulsant efficacy of benzodiazepine site ligands. J Psychopharmacol 21: 384–391. [DOI] [PubMed] [Google Scholar]
- 5. Rudolph U, Knoflach F (2011) Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. Nat Rev Drug Discov 10: 685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nutt DJ, Stahl SM (2010) Searching for perfect sleep: the continuing evolution of GABAA receptor modulators as hypnotics. Psychopharmacol 11: 1601–1612. [DOI] [PubMed] [Google Scholar]
- 7. Möhler H, Fritschy JM, Crestani F, Hensch T, Rudolph U (2004) Specific GABA(A) circuits in brain development and therapy. Biochem Pharmacol 68: 1685–1690. [DOI] [PubMed] [Google Scholar]
- 8. Sanger DJ, Benavides J, Perrault G, Morel E, Cohen C, et al. (1994) Recent developments in the behavioral pharmacology of benzodiazepine (ω) receptors: evidence for the functional significance of receptor subtypes. Neurosci Biobehav Rev 18: 355–372. [DOI] [PubMed] [Google Scholar]
- 9. Rowlett JK, Platt DM, Lelas S, Atack JR, Dawson GR (2005) Different GABAA receptor subtypes mediate the anxiolytic, abuse-related, and motor effects of benzodiazepine-like drugs in primates. Proc Natl Acad Sci U S A 102: 915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Löw K, Crestani F, Keist R, Benke D, Brünig I, et al. (2000) Molecular and neuronal substrate for the selective attenuation of anxiety. Science 6: 131–134. [DOI] [PubMed] [Google Scholar]
- 11. Knabl J, Witschi R, Hosl K, Reinold H, Zeilhofer UB, et al. (2008) Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature 451: 330–334. [DOI] [PubMed] [Google Scholar]
- 12. van Rijnsoever C, Tauber M, Choulli MK, Keist R, Rudolph U, et al. (2004) Requirement of α5-GABAA receptors for the development of tolerance to the sedative action of diazepam in mice. J Neurosci 24: 6785–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ng YT, Conry JA, Drummond R, Stolle J, Weinberg M (2011) on behalf of the OV-1012 Study Investigators (2011) Randomized, phase III study results of clobazam in Lennox-Gastaut Syndrome. Neurology 77: 1473–1481. [DOI] [PubMed] [Google Scholar]
- 14. Sankar R (2012) GABAA receptor physiology and its relationship to the mechanism of action of the 1,5-benzodiazepine clobazam. CNS Drugs 26: 229–244. [DOI] [PubMed] [Google Scholar]
- 15. Canadian Clobazam Cooperative Group (1991) Clobazam in treatment of refractory epilepsy: the Canadian experience. A retrospective study. Epilepsia 32: 407–416. [PubMed] [Google Scholar]
- 16. Arzimanoglou A, French J, Blume WT, Cross JH, Ernst JP, et al. (2009) Lennox-Gastaut syndrome: a consensus approach on diagnosis, assessment, management, and trial methodology. Lancet Neurol 8: 82–93. [DOI] [PubMed] [Google Scholar]
- 17. Tolbert D, Bekersky I (2013) Pharmacokinetics of N-desmethylclobazam, the Active and Primary Metabolite of Clobazam. 66th Annual Meeting of the American Epilepsy Society. San Diego, California. Epilepsy Currents 13 1 Suppl 1: Abstract #2.226 [Google Scholar]
- 18. Kosaki K, Tamura K, Sato R, Samejima H, Tanigawara Y, et al. (2004) A major influence of CYP2C19 genotype on the steady-state concentration of N-desmethylclobazam. Brain Dev 26: 530–534. [DOI] [PubMed] [Google Scholar]
- 19. Yamamoto Y, Takahashi Y, Imai K, Miyakawa K, Nishimura S, et al. (2013) Influence of CYP2C19 polymorphism and concomitant antiepileptic drugs on serum clobazam and N-desmethylclobazam concentrations in patients with epilepsy. Ther Drug Monit 35: 305–312. [DOI] [PubMed] [Google Scholar]
- 20.Klonopin (clonazepam) (2010) [prescribing information]. South San Francisco, CA: Genentech. [Google Scholar]
- 21.Ambien (zolpidem) (2013) [prescribing information]. Bridgewater, NJ: Sanofi-Aventis US LLC. [Google Scholar]
- 22. Atack JR, Wafford KA, Tye SJ, Cook SM, Sohal B, et al. (2006) TPA023 [7-(1,1-dimethylethyl)-6-(2-ethyl-2H-1,2,4-triazol-3-ylmethoxy)-3-(2-fluorophenyl)-1,2,4-triazolo[4,3-b]pyridazine], an agonist selective for α2- and α3-containing GABAA receptors, is a nonsedating anxiolytic in rodents and primates. J Pharmacol Exp Ther 316: 410–422. [DOI] [PubMed] [Google Scholar]
- 23. Pritchett DB, Luddens H, Seeburg PH (1989) Type I and type II GABAA-benzodiazepine receptors produced in transfected cells. Science 245: 1389–1392. [DOI] [PubMed] [Google Scholar]
- 24. Huang Q, He X, Ma C, Liu R, Yu S, et al. (2000) Pharmacophore/receptor models for GABAA/BzR subtypes (α1β3γ2, α5 β3γ2, and α6β3γ2) via a comprehensive ligand-mapping approach. J Med Chem 43: 71–95. [DOI] [PubMed] [Google Scholar]
- 25. Arendt RM, Greenblatt DJ, Liebisch DC, Luu MD, Paul SM (1987) Determinants of benzodiazepine brain uptake: lipophilicity versus binding affinity. Psychopharmacology (Berl) 93: 72–76. [DOI] [PubMed] [Google Scholar]
- 26. Haigh JR, Pullar T, Gent JP, Dailley C, Feely M (1987) N-desmethylclobazam: a possible alternative to clobazam in the treatment of refractory epilepsy? Br J Clin Pharmacol 23: 213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walzer M, Bekersky I, Blum RA, Tolbert D (2012) Pharmacokinetic Drug Interactions Between Clobazam and Drugs Metabolized by Cytochrome P450 Isoenzymes. Pharmacotherapy 32: 340–353. [DOI] [PubMed] [Google Scholar]
- 28. van der Meyden CH, Bartel PR, Sommers DK, Blom M, Pretorius LC (1989) Effects of clobazam and clonazepam on saccadic eye movements and other parameters of psychomotor performance. Eur J Clin Pharmacol 37: 365–369. [DOI] [PubMed] [Google Scholar]
- 29. Wildin JD, Pleuvry BJ, Mawer GE, Onon T, Millington L (1990) Respiratory and sedative effects of clobazam and clonazepam in volunteers. Br J Clin Pharmacol 29: 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vuilleumier PH, Besson M, Desmeules J, Arendt-Nielsen L, Curatolo M (2013) Evaluation of anti-hyperalgesic and analgesic effects of two benzodiazepines in human experimental pain: a randomized placebo-controlled study. PLoS One 8 3: e43896 doi: 10.1371/journal.pone.0043896. E-pub 2013 Mar 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Onfi (clobazam) (2013) [prescribing information]. Deerfield, IL: Lundbeck LLC. [Google Scholar]
- 32. Steru L, Chermat R, Millet B, Mico JA, Simon P (1986) Comparative study in mice of ten 1,4-benzodiazepines and of clobazam: anticonvulsant, anxiolytic, sedative, and myorelaxant effects. Epilepsia Suppl 1: S14–S17. [DOI] [PubMed] [Google Scholar]
- 33. Miura Y, Amano S, Torii R, Ihara N (2002) Clobazam shows a different antiepileptic action profile from clonazepam and zonisamide in Ihara epileptic rats. Epilepsy Res 49: 189–202. [DOI] [PubMed] [Google Scholar]