

Abstract
Dimebon (Latrepirdine) was initially developed in Russia as an over-the-counter antihistamine for allergy treatment. In early 1990s Dimebon was identified as a low affinity NMDA receptor antagonist, patented for treatment of neurodegenerative disorders and licensed by Medivation Inc. Extremely promising results were obtained by double-blinded placebo-controlled phase II AD trial (NCT00377715). However, a phase II trial of Dimebon in Huntington’s disease (HD) patients (NCT00497159) was much less successful. A phase III AD trial of Dimebon (NCT00838110, CONNECTION trial) failed to result in any significant improvement in primary or secondary outcomes. The failure of Dimebon may in large part be explianed by insufficient understanding of its mechanism of action. The NMDA receptor blocking activity of Dimebon is too weak to be physiologically relevant. The “novel mitochondrial mechanism of action” proposed by Medivation lacked credible scientific evidence or a molecular target. Studies by our laboratory and other independent laboratories (Avineuro Pharmaceuticlas and Cephalon Inc) indicate that the clinical effects of Dimebon are most likely result from inhibition of H1 histamine and 5-HT6 serotonin receptors. It would appear that careful preclinical and mechanistic studies of novel potential therapies are needed to minimize chances of making similar costly mistakes in the future.
The birth of Dimebon
Dimebon is a trade name for dimebolin (2,3,4,5-tetrahydro-2,8-dimethyl-5-(2-(6-methyl-3-pyridyl)ethyl)-1H-pyrido(4,3-b)indole) (Fig 1). Dimebon was initially developed and launched in Russia (USSR at the time) in 1983 as an over-the-counter oral antihistamine for allergy treatment. In the same time it was demonstrated that Dimebon acted by blocking H1 histamine receptors with high affinity [1]. As anti-allergic medicine Dimebon was used in doses 10–20 mg 2–3 times per day. When better antihistamines came along, and the Russian market opened to Western medicines, Dimebon was discontinued, and taken off the Russian market.
Figure 1. Chemical structure of Dimebon (Latrepirdine).
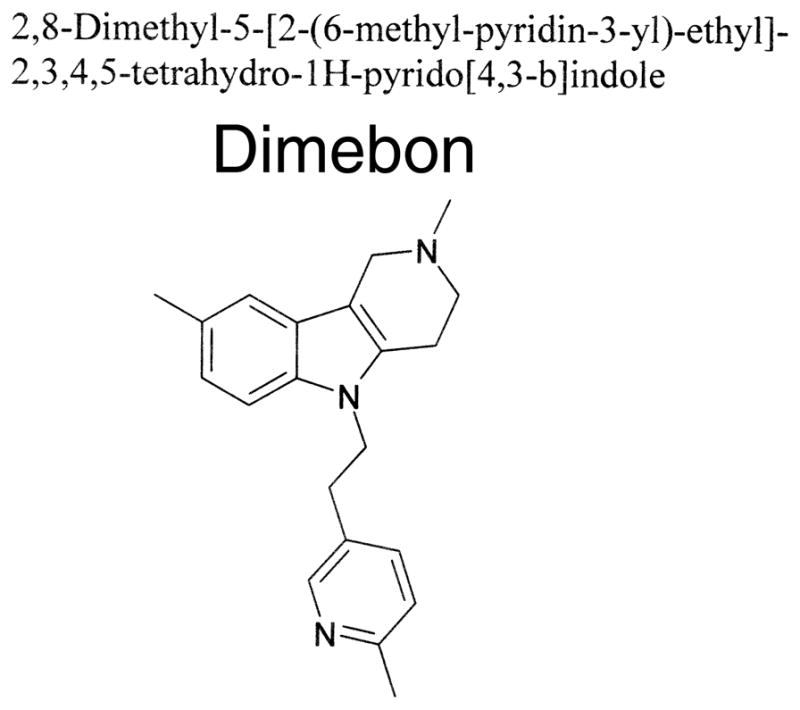
Chemical structure of 2,3,4,5-Tetrahydro-2,8-dimethyl-5-(2-(6-methyl-3-pyridyl)ethyl)-1H-pyrido(4,3-b)indole (CAS 3613-73-8)
The abrupt end of socialism in 1990 resulted in dramatically changing economic landscape in Russia. As a result, in the early 1990s Russian Academy of Science became much more interested in translational research. One of the groups who initiated such a drug discovery program was headed by Dr Sergey Bachurin at the Institute of Physiologically Active Compounds, Chernogolovka, Moscow Region, Russia. This group was interested in developing novel Alzheimer’s disease therapeutics. Specifically, this group focused on isolating novel compounds with ability to inhibit NMDA receptors. NMDA receptors are targets for memantine (trade name Namenda), which is widely used for Alzheimer’s disease treatment. Therefore, novel NMDAR antagonists have potential utility for AD treatment. A whole animal screen for novel compounds with anti-NMDAR activity was performed in mice to identify such compounds [2]. A solution of test agents was injected intraperitoneally 40 minutes prior to the injection of 0.1 μg NMDA, directly into the lateral vehicle of the brain. In the control group of mice, injection of the NMDA caused mice to run and jump, induced convulsions, and eventually caused death of animals within several hours following the injection. The test substances that prevented convulsions and death of animals were considered to possess anti-NMDAR activity. Using the bioassay described above, it was found that hydrogenated pyrido[4,3-b]indole derivatives had NMDA antagonist properties. The anti-NMDA activity of all these compounds was characterized by an ED50 in the range 16–45 mg/kg, by using an intraperitoneal route of delivery [2]. Dimebon was a member of this group of compounds, with an ED50 equal to 42 mg/kg, i.p. [2,3]. Dimebon was selected for evaluation in an animal model of AD. In this model, cerebral cholinergic functions aree impaired by injecting an intracerebroventricular toxin, ethylcholine aziridinium ion (AF64A), resulting in learning deficits. It was demonstrated that systemic administration of Dimebon improved active avoidance conditioning in AF64A-injected rats, suggesting that Dimebon might be useful for treating cognitive disorders [3,4].
Based on apparent anti-NMDA and pro-cognitive activities of Dimebon, a small scale pilot trial of Dimebon in AD patients was carried out in the Moscow Center of Gerontology, under direct supervision of Drs E. Bukatina and V. Grigorieva [2,3]. Dimebon was tested in open label study with 14 moderate AD patients, with ages ranging from 64 to 88 years old. No placebo group was included in the design of this study. Dimebon was tested in concentration of 20 mg, three times per day, using exactly the same dose and regimen as used for allergy treatments. The trial ran for 8 weeks, with patients evaluated after 4 and 8 weeks of therapy, and 8 weeks after drug was discontinued. Patients were evaluated using the Hazegawa scale and the Bukatina scale. The Hazegawa scale is similar to Mini-Mental State Exam (MMSE) scale used in USA. Bukatina scale was developed by Dr E. Bukatina who conducted the Dimebon trial, and is not used internationally. Using the Bukatina scale, it was discovered that after 8 weeks of treatment the patients on Dimebon displayed significant improvement in cognitive function (+ 2.5 units). After the drug was discontinued, patients quickly deteriorated (−3.28 units after 8 weeks) [2,3]. It was also found that treatment with Dimebon significantly reduced symptoms of depression in these patients [3].
Based on results obtained with Dimebon in the NMDA-treated animal model, and the clinical trial of Dimebon with 14 AD patients, a use-patent application was filed in Russia on October 23, 1995. This application claimed Dimebon as a useful agent for treating neurodegenerative disorders, based on ability of Dimebon to block NMDAR (Russian Federation application No 95118252). The initial Russian patent for Dimebon served as a basis for filing a US patent application with the same claims (US patent 6,187,785 issued on February 13, 2001). Dr Sergey Sablin, one of the students of Dr Bachrin [3], moved to San Francisco and started a company, Selena Pharmaceuticals in an effort to commercialize the Dimebon patent.
Phase II AD trial of Dimebon
In October 2003, San Francisco-based Medivation Inc (CEO Dr David Hung) acquired the rights to Dimebon from Selena. Medivation went public in December 2004, with Dimebon as the only drug in its pipeline. Medivation used its financial resources to organize a phase II trial of Dimebon with 183 AD patients (NCT00377715). The trial was performed in 11 sites in Russia. The study director of the trial was Dr Lynn Seely, the Chief Medical Officer of Medivation. Dr Rachelle Doody (Baylor University College of Medicine, Houston, Texas) was the primary and corresponding author for the resulting publication [5]. The Russian investigators conducting the trial were trained on Good Clinical Practice (GCP) assessment measures by their US counterparts. In this double-blinded trial, patients were randomized to treatment (89 patients) and placebo (94 patients) treatment arms. Patients in the treatment arm received 20 mg of Dimebon 3 times per day. This was exactly the same dosing regimen as in the first trial. The treatment continued for 26 weeks, with an additional 26 weeks blinded extension of the study. The primary outcome measure was the ADAS-cog (Alzheimer’s Disease Assessment Scale-cognition) scale. ADAS-cog scale is a standard way to evaluate potential AD therapeutics in the USA and acceptable by FDA. Secondary outcome measures included MMSE and Clinician Interview-based Impression of Change plus Caregiver Input (CIBIC-plus).
The results of this trial were spectacular [5]. Dimebon was well tolerated. Common side effects included dry mouth (14% of patients on Dimebon, 1% in placebo group), depression (14% of patients on Dimebon, 5% in placebo group), and insomnia (9% of patients on Dimebon, 4% in placebo group). It was not clear why Dimebon acted as anti-depressant in the previous clinical trial [3], but induced depression in the second trial [5]. At week 26, treatment with Dimebon resulted in significant improvement in the ADAS-cog score compare to placebo (mean drug-placebo difference −4.0, p < 0.0001). Similar efficacy was observed for MMSE (mean drug-placebo difference 2.2, p < 0.0001) and CIBIC-plus (mean drug-placebo difference 0.6, p < 0.0001). Moreover, the ADAS-cog difference between drug and placebo increased substantially during additional 6 months extension phase, indicating that benefits of Dimebon continue to increase over time [5]. The number of patients was relatively small and the trial was designed as a phase II trial. However, results of the trial were so strong that FDA agreed to treat this trial as a first pivotal trial. Moreover, results of this trial prompted Pfizer to enter into a partnership deal with Medivation in September 2008. Pfizer paid Medivation $225 million in upfront payments, and agreed to pickup 60% of the costs for further development of the drug. In addition, Pfizer committed to additional $500 million in milestone-based payments. In return Pfizer received 60% of profits from future Dimebon sales. In preparation for launching sales, Medivation and Pfizer assigned Dimebon a new trade name - Latrepirdine.
Phase II HD trial of Dimebon
Using transgenic fly model of Huntington’s disease [6], Medivation-sponsored investigators demonstrated that feeding the flies with 100 μM of Dimebon partially reduced photoreceptor degeneration in this model [7]. No significant rescue was observed at concentrations below or above 100 μM (1 μM, 10 μM, 30 μM, 300 μM, or 1 mM) [7]. There was also a positive effect in the climbing assay with transgenic HD flies. The significant positive effect in the climbing assay was observed in a concentration of 1 mM, but not at concentrations 10 μM or 100 μM [7]. These results were never published, but were sufficient for Medivation to receive a patent claiming use of Dimebon for treatment of Huntington’s disease (HD) [7], and to initiate a phase II trial of Dimebon in HD patients (NCT00497159). This was a multi-center trial conducted in the USA with Dr Karl Kieburtz (University of Rochester, Rochester, New York) as the principle investigator. Dr Kieburtz was also the primary author of the resulting publication [8]. The study was organized and managed through the Huntington’s Study Group (HSG). The study enrolled 91 participants, randomized to treatment (46 patients) and placebo (45 patients) arms. Dimebon was used in the same dose of 20 mg, 3 times per day, as in the two previous trials. The study drug was formulated as a tablet and encapsulated to maintain masking. The duration of the treatment was 90 days, with patients evaluated at 60 and 90 days, using the Unified Huntington’s Disease Rating Scale (UHDRS), ADAS-cog, and MMSE outcome measures.
In contrast to the phase II AD trial described above, the results of phase II HD trial were much more modest. After 90 days treatment with Dimebon, there was no significant change in UHDRS score, although “qualitative improvement” in the drug treated group was noted [8]. There was no significant difference in ADAS-cog (mean drug-placebo difference −0.31, p = 0.79). There was no significant effect on MMSE at 60 days time point. However, there was a small effect at the 90 day time point (mean drug-placebo difference 0.97, p = 0.03) [8]. The spectrum of side effects reported in the HD trial differed from the AD trial. Dry mouth and depression were not reported, and most common side effects were headache and somnolence [8]. It is not clear why the side effects and efficacy of Dimebon tested in phase II HD trial performed in the USA differed so dramatically from the phase II AD trial performed in Russia.
Mitochondrial mechanism of action?
As discussed above, the initial discovery of Dimebon was based on it’s NMDAR blocking activity [2,3]. The same mechanism of action was further investigated by Sergey Bachurin and his colleagues in a side-by-side comparison between Dimebon and memantine [9]. In experiments with cortical neuronal cultures it was discovered that Dimebon inhibited NMDA-induced currents in group 1 neurons with an IC50 of 7.7 μM, and in group 2 neurons with an IC50 of 73 μM [9]. Memantine was approximately 5 times more effective in the same experiments – with an IC50 of 1.4 μM for group 1 neurons and IC50 of 15 μM for group 2 neurons [9]. In a search for additional targets for Dimebon, Sergey Bachurin and his colleagues also evaluated effects of Dimebon on voltage-gated Ca2+ channels. They found that Dimebon acted as a low affinity inhibitor of L-type voltage-gated Ca2+ channels, with an IC50 equal to 57 μM [3,10]. In biochemical experiments they also demonstrated that Dimebon inhibited acetylcholine esterase with an IC50 of 42 μM, and butyrylcholine esterase with an IC50 7.9 μM [3]. However, because the activity of Dimebon against all these targets was in the range of 10–50 μM, it was not clear whether any of these activities were relevant to the effects of Dimebon observed in the AD clinical trials.
Because of these discrepancies a number of investigators raised questions about Dimebon’s mechanism of action at scientific meetings. In response to these questions, on May 5, 2008 Medivation issued a regulation FD disclosure (form 8-K) with securities and exchange commission (http://biz.yahoo.com/e/080505/mdvn8-k.html). In this disclosure Medivation included the following paragraph: “The fact that Dimebon shows low activity against acetylcholinesterase and the N-methyl-D-aspartate (NMDA) receptor in laboratory testing is completely expected. While Dimebon does in fact hit those target, it does so with very low affinity (IC50 = 72.3 μM for acetylcholinesterase; IC50 = 83.7 for the NMDA-receptor). Thus, very high concentrations of Dimebon are required to show even minimal effects against these targets. This is a primary reason Medivation does not believe that either of these targets is responsible for Dimebon’s clinical effects”.
If NMDAR is not the target of Dimebon, then what is? In the same disclosure Medivation stated that “based on data generated in independent outside laboratories, Medivation believes that Dimebon is achieving its clinical effects through a novel mitochondrial mechanism of action”. So, what are the data that Medivation invoked here? In experiments with mitochondria isolated from rat liver, Sergey Bachrin and his colleagues previously found that Dimebon inhibited Ca2+-induced opening of mitochondria permeability transition pore, with an IC50 in the 100 μM range [11]. However, this is many orders above concentration levels of the drug obtained physiologically. It now appears that the main support for the “novel mitochondrial mechanism of action” comes from the experiments performed by Dr Maria Ankarcrona (Karolinska Institute, Sweden). These data have never been published, but were presented by Dr Ankarcrona or by Dr Andy Potter, the chief scientific officer of Medivation, at multiple scientific and investor conferences during 2008 and 2009. From these presentations it became apparent that Dr Ankarcrona performed a series of experiments with cortical neuronal cultures. In these experiments, cortical neurons were challenged by the Ca2+ ionophore ionomycin, and the mitochondrial membrane potential was measured with fluorescent indicators. It was claimed that incubation with 0.1 nM of Dimebon was sufficient to reduce mitochondrial potential changes in response to ionomycin. However, the mechanism of Dimebon’s action on mitochondria was not clarified. Futhermore, the dose dependence of Dimebon’s effects were problematic, since similar magnitudes of the effect were observed at 0.1 nM, 1 nM, 10 nM and 100 nM concentrations. Also, experiments performed by Dr Ankarcrona did not appear to include appropriate positive or negative controls.
Netheless, based on these unpublished and low quality results Medivation repeatedly stated that Dimebon acts by mitochondrial mechanism of action. The same statement was repeated in the review by Dr. Rachelle Doody that discussed Dimebon as potential therapy for AD [12]. Similar statements were repeated by Pfizer. For example, in their November 3, 2009 news release Pfizer stated that “in preclinical studies, Dimebon has been shown to protect brain cells from damage and enhance brain cell survival, potentially by stabilizing and improving mitochondrial function. The dimebon mechanism is distinct from currently available AD medications" (http://finance.yahoo.com/news/Pfizer-And-Medivation-bw-1010812008.html).
Although Medivation and Pfizer were convinced about the “novel mitochondrial mechanism of action”, not all scientists were. Experiments performed by Dr Ankarcrona were performed using a non-physiological ionomycin challenge, lacked appropriate controls or dose-dependence, and did not identify Dimebon’s molecular target. Despite being continuously presented at scientific and investors meetings, these results have never been published in a peer-reviewed journal. Not convinced by Dr Ankarcrona’s results, a number of scientists attempted to investigate Dimebon’s mechanism of action in their own laboratories. Unfortunately, Dimebon was not made available for independent experimental studies by Medivation. When our request for a sample of Dimebon was not satisfied by Medivation, we hired a contract research organization Nanosyn Inc (http://www.nanosyn.com/) to synthesize a sample of Dimebon based on publicly available structural information. An identity of generated compound was independently confirmed by NMR [13]. Using generated sample, in experiments with primary neuronal cultures from an HD transgenic mouse model we demonstrated that concentrations of at least 10 μM were required to inhibit VGCC and NMDAR [13]. Our results were in excellent agreement with previous results published by Sergey Bachurin and his colleagues for the same targets [3,9–11]. However, using physiological glutamate toxicity model we did not observe any evidence of mitochondrial protective effects in nanomolar concentration range as claimed by Dr Ankarcrona. In our experiments at least 50 μM of Dimebon was needed to exert neuroprotective effects in glutamate excitotoxicity model with striatal cultures from HD transgenic mice [13], consistent with the results obtained in transgenic HD fly model [7]. Based on these results we concluded that Dimebon is not likely to act on mitochondria.
To identify potential targets of Dimebon, we performed an unbiased search for its physiologically relevant targets. In the unbiased screen performed by MDS Pharma Services (http://discovery.mdsps.com/), we discovered that Dimebon potently inhibits α1B adrenergic receptors, histamine H1 receptors and serotonin 5-HT6 receptors, as well as number of additional receptors [13]. These findings were recently confirmed in an independent study by Avineuro Pharmaceuticlas [14]. We argued that the cognitive effects of Dimebon observed in AD and HD clinical trials [5,8] might be due to interaction with these receptors. In particular, the ability of Dimebon to inhibit 5-HT6 serotonin receptors with high affinity (Ki=34 nM) [14] is of interest. Serotonin 5-HT6 receptors are known targets for cognitive enhancement, which has been previously considered as a target for AD treatment [15]. A recently published evaluation of Dimebon in an animal model confirmed the ability of Dimebon to interact with 5-HT6 receptors in vivo and to exert acute behavioral effects similar to the specific 5-HT6 receptor antagonist SB-399885 [16]. These studies, performed by researchers at Cephalon, Inc., support the hypothesis that cognitive effects of Dimebon are most likely due to its ability to inhibit 5-HT6 serotonin receptors. In additional studies, other potential activities of Dimebon such as its effects on amyloid metabolism [17] and protein aggregation [18] have also been reported. These activities may have also contributed to some of the results observed in the clinic.
Phase III trial of Dimebon in AD
A large Phase III clinical trial of Dimebon in AD patients has been recently completed (NCT00838110, CONNECTION trial). This trial was sponsored by Medivation and Pfizer and lasted 26 weeks. The CONNECTION trial was a multi-national, double-blind, placebo-controlled, safety and efficacy trial, involving 598 patients with mild-to-moderate AD, at 63 sites in North America, Europe, and South America. The study director of the trial was Dr Lynn Seely, a Chief Medical Officer of Medivation. More than 40 percent of the patients enrolled were in the United States. In the study, patients were randomized to one of three treatment groups: those receiving Dimebon 20 mg three times a day; those receiving 5 mg Dimebon three times per day; or those receiving placebo. The primary outcome measures were ADAS-cog and CIBIC-plus. The secondary outcome measure was MMSE.
The results of the trial were released by Medivation on March 3, 2010 (http://investors.medivation.com/releasedetail.cfm?ReleaseID=448818). It was reported that no statistically significant improvements for the 20 mg treatment group relative to placebo were achieved on any of the primary endpoints. There was no significant difference for Dimebon-treated patients when compared to the patients receiving placebo on ADAS-cog measure (p=0.86) or CIBIC-plus (p = 0.81). On the MMSE measure, both Dimebon and placebo-treated groups improved significantly over baseline (dimebon 0.7; placebo 1.2). The difference favoring placebo was not significant (p=0.10). Responding to the failure of the CONNECTION trial, Medivation’s CEO Dr Huang stated in the same news release: “The results from the CONNECTION study are unexpected, and we are disappointed for the Alzheimer’s community".
Lessons learned
The spectacular rise and dramatic fall of Dimebon can be best illustrated by a plot of the Medivation (MDVN) closing share price on the NASDAQ stock exchange between January 1, 2006 and May 5, 2010 (Fig 2). Release of the phase II AD trial data (NCT00377715) and the initiation of phase II HD trial (NCT00497159) resulted in an increase in the share price from the $2–3 range to $15–20 range in late 2006 (Fig 2). The partnership with Pfizer, announced in September 2008, resulted in a further increase in the share price to above $ 30 (Fig 2). The runup during anticipation of results of the phase III AD trial (NCT00838110, CONNECTION trial) pushed the share price to above $40 (Fig 2). Announcement of CONNECTION trial data on March 3, 2010 resulted in collapse of the share price to the $10–12 range (Fig 2), wiping out $1B in shareholders value in one day.
Figure 2. Daily closing price of MDVN shares on NASDAQ stock exchange.
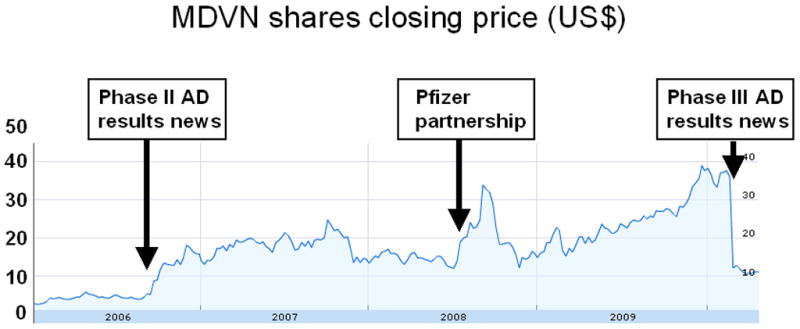
The share closing price is shown for the period between January 1, 2006 – May 5, 2010. Also shown are times of the key events - announcement of phase II AD trial data, announcement of partnership with Pfizer and announcement of phase III AD trial data.
There are a number of lessons to be learned from the Dimebon story. In my opinion the most important lesson is the importance of preclinical studies aimed at understanding mechanism of action of a novel therapeutic compound. In my opinion failure of Dimebon was not “unexpected”, but in large part it was a result of neglect to investigate the mechanism of its action or to find its real target. It appeared that the very weak “novel mitochondrial mechanism of action” narrative was supported by Medivation at the expense of proper scientific study of Dimebon’s real mechanism of action. As a result, the most informative preclinical investigations of Dimebon were performed not by Medivation and its contractors, but by laboratories independently generating their own samples of compound for studies – such as our laboratory [13], Avineuro Pharmaceuticlas [14] and Cephalon Inc [16]. All of these three laboratories have agreed that Dimebon is basically a broad specificity anti-histamine with additional ability to engage serotonin receptors [13,14,16]. All these laboratories also agree that the clinical effects of Dimebon are most likely can result from inhibition of H1 histamine and 5-HT6 serotonin receptors [13,14,16]. By contrast,. no support for the “mitochondrial mechanism of action” has been provided by these independent studies.
Second lesson is that small scale AD clinical trials can be very misleading, and should not be given so much weight in the absence of supporting preclinical data. The main excitement about Dimebon was based on a single phase II clinical trial performed in Russia (NCT00377715) [5]. Obviously, there are many factors that can influence the outcome of a clinical trial. It is particularly the case for AD trials which are based on cognitive outcomes and lack objective biomarkers. Performing clinical trials with patient population not previously exposed to modern medicine and with physicians who had to be trained in modern good clinical practices introduces additional complications. I am sure that careful comparison of phase III and phase II datasets will reveal reasons for the apparent discrepancies in the clinical results. But in my opinion the whole story of Dimebon demonstrates that sometimes clinical data can be very misleading, and that the only way to prevent mistakes is to perform proper preclinical evaluation of the compound in question. Although drug approval process in the FDA is mostly focused on clinical outcomes, and not on mechanistic studies and on the target identification, cutting corners at preclinical stage increases chances of making a very costly mistake, as the example of Dimebon illustrates.
Acknowledgments
IB is a holder of Carla Cooke Francis Professorship in Alzheimer’s Research and supported by the McKnight Neuroscience of Brain Disorders Award, Alzheimer’s Disease Drug Discovery Foundation, Carl and Florence King Foundation, and NIH grant R01AG030746.
References
- 1.Matveeva IA. Action of dimebon on histamine receptors. Farmakol Toksikol. 1983;46:27–9. [PubMed] [Google Scholar]
- 2.Zefirov NS, Afanasiev AZ, Afanasieva SV, Bachurin SO, Tkachenko SE, Grigoriev VV, Jurovskaya MA, Chetverikov VP, Bukatina EE, Grigorieva IV. Agents for treating neurodegenerative disorders. 7,071,206. US Patent. 2006 Jul 4;
- 3.Bachurin S, Bukatina E, Lermontova N, Tkachenko S, Afanasiev A, Grigoriev V, Grigorieva I, Ivanov Y, Sablin S, Zefirov N. Antihistamine agent Dimebon as a novel neuroprotector and a cognition enhancer. Ann N Y Acad Sci. 2001;939:425–35. doi: 10.1111/j.1749-6632.2001.tb03654.x. [DOI] [PubMed] [Google Scholar]
- 4.Lermontova NN, Lukoyanov NV, Serkova TP, Lukoyanova EA, Bachurin SO. Dimebon improves learning in animals with experimental Alzheimer’s disease. Bull Exp Biol Med. 2000;129:544–6. doi: 10.1007/BF02434871. [DOI] [PubMed] [Google Scholar]
- 5.Doody RS, Gavrilova SI, Sano M, Thomas RG, Aisen PS, Bachurin SO, Seely L, Hung D. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: a randomised, double-blind, placebo-controlled study. Lancet. 2008;372:207–15. doi: 10.1016/S0140-6736(08)61074-0. [DOI] [PubMed] [Google Scholar]
- 6.Marsh JL, Lukacsovich T, Thompson LM. Animal models of polyglutamine diseases and therapeutic approaches. J Biol Chem. 2009;284:7431–5. doi: 10.1074/jbc.R800065200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hung D. HYDROGENATED PYRIDO-INDOLE COMPOUNDS FOR THE TREATMENT OF HUNTINGTON ‘ S DISEASE. WO/2007/041697. International Patent. 2007
- 8.Kieburtz K, McDermott MP, Voss TS, Corey-Bloom J, Deuel LM, Dorsey ER, Factor S, Geschwind MD, Hodgeman K, Kayson E, Noonberg S, Pourfar M, Rabinowitz K, Ravina B, Sanchez-Ramos J, Seely L, Walker F, Feigin A. A randomized, placebo-controlled trial of latrepirdine in Huntington disease. Arch Neurol. 2010;67:154–60. doi: 10.1001/archneurol.2009.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grigorev VV, Dranyi OA, Bachurin SO. Comparative study of action mechanisms of dimebon and memantine on AMPA- and NMDA-subtypes glutamate receptors in rat cerebral neurons. Bull Exp Biol Med. 2003;136:474–7. doi: 10.1023/b:bebm.0000017097.75818.14. [DOI] [PubMed] [Google Scholar]
- 10.Lermontova NN, Redkozubov AE, Shevtsova EF, Serkova TP, Kireeva EG, Bachurin SO. Dimebon and tacrine inhibit neurotoxic action of beta-amyloid in culture and block L-type Ca(2+) channels. Bull Exp Biol Med. 2001;132:1079–83. doi: 10.1023/a:1017972709652. [DOI] [PubMed] [Google Scholar]
- 11.Bachurin SO, Shevtsova EP, Kireeva EG, Oxenkrug GF, Sablin SO. Mitochondria as a target for neurotoxins and neuroprotective agents. Ann N Y Acad Sci. 2003;993:334–44. doi: 10.1111/j.1749-6632.2003.tb07541.x. discussion 345–9. [DOI] [PubMed] [Google Scholar]
- 12.Doody RS. Dimebon as a potential therapy for Alzheimer’s disease. CNS Spectr. 2009;14:14–6. doi: 10.1017/s1092852900024913. discussion 16–8. [DOI] [PubMed] [Google Scholar]
- 13.Wu J, Li Q, Bezprozvanny I. Evaluation of Dimebon in cellular model of Huntington’s disease. Mol Neurodegener. 2008;3:15. doi: 10.1186/1750-1326-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okun I, Tkachenko ES, Khvat A, Mitkin O, Kazey V, Ivachtchenko VA. From Anti-allergic to Anti-Alzheimer’s: Molecular Pharmacology of Dimebon. Curr Alzheimer Res. 2009 doi: 10.2174/156720510790691100. [DOI] [PubMed] [Google Scholar]
- 15.Upton N, Chuang TT, Hunter AJ, Virley DJ. 5-HT6 receptor antagonists as novel cognitive enhancing agents for Alzheimer’s disease. Neurotherapeutics. 2008;5:458–69. doi: 10.1016/j.nurt.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schaffhauser H, Mathiasen JR, Dicamillo A, Huffman MJ, Lu LD, McKenna BA, Qian J, Marino MJ. Dimebolin is a 5-HT6 antagonist with acute cognition enhancing activities. Biochem Pharmacol. 2009;78:1035–42. doi: 10.1016/j.bcp.2009.06.021. [DOI] [PubMed] [Google Scholar]
- 17.Steele JW, Kim SH, Cirrito JR, Verges DK, Restivo JL, Westaway D, Fraser P, Hyslop PS, Sano M, Bezprozvanny I, Ehrlich ME, Holtzman DM, Gandy S. Acute dosing of latrepirdine (Dimebon), a possible Alzheimer therapeutic, elevates extracellular amyloid-beta levels in vitro and in vivo. Mol Neurodegener. 2009;4:51. doi: 10.1186/1750-1326-4-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamashita M, Nonaka T, Arai T, Kametani F, Buchman VL, Ninkina N, Bachurin SO, Akiyama H, Goedert M, Hasegawa M. Methylene blue and dimebon inhibit aggregation of TDP-43 in cellular models. FEBS Lett. 2009;583:2419–24. doi: 10.1016/j.febslet.2009.06.042. [DOI] [PubMed] [Google Scholar]