

Gene-deleted mice show that serpinB1 serves as an endogenous restraint to limit untoward expansion of lymphocytes with a Th17 phenotype.
Keywords: lymphocytes, population size, cell proliferation, inflammation, cytokine regulation
Abstract
SerpinB1 is an endogenous inhibitor of serine proteases recognized for its anti-inflammatory and host-protective properties. Although loss of serpinB1 in mice does not result in gross immune deregulation, serpinb1a−/− mice display increased mortality and inflammation-associated morbidity upon challenge with influenza virus. Here, we show that IL-17A+ γδ and CD4+ Th17 cells are already expanded in the lungs of serpinb1a−/− mice at steady-state. Both γδ and αβ+ CD4+ CCR6+ T cells isolated from the lungs of naive serpinb1a−/− mice displayed a skewed transcriptional profile relative to WT cells, including increased Th17 signature transcripts [Il17a, l17f, and Rorc (RORγt)] and decreased Th1 signature transcripts [Ifng, Cxcr3, and Tbx21 (T-bet)] in γδ T cells. In addition to the lung, IL-17A+ γδ and CD4+ Th17 cells were increased in the spleen of naive serpinb1a−/− mice, despite normal αβ and γδ T cell development in the thymus. Within the γδ T cell compartment, loss of serpinb1a prompted selective expansion of Vγ4+ and Vγ6/Vδ1+ cells, which also displayed elevated expression of the proliferating cell nuclear antigen, Ki-67, and IL-17A. Given that serpinb1a is preferentially expressed in WT IL-17A+ γδ and CD4+ Th17 cell subsets vis-à-vis other T cell lineages, our findings reveal a novel function of serpinB1 in limiting untoward expansion of lymphocytes with a Th17 phenotype.
Introduction
SerpinB1 is an ancient protein that functions as an effective inhibitor of serine proteases; it is broadly expressed in immune cells at levels that vary by several logs. Studies with rat models and gene-deleted mice identified serpinB1 as a protective immunomodulator that preserves host antimicrobial defense, prevents tissue injury, and restricts proinflammatory responses. In influenza virus respiratory infection, for example, we have shown previously that mortality and morbidity are elevated in serpinb1a−/− mice compared with WT animals. In this model, high-dose infection with influenza A/Philadelphia/82 causes rapid infiltration of macrophages and neutrophils that produces a storm of injurious cytokines and enhances lung histopathology—features that are characteristic of human infection with highly pathogenic influenza strains. Despite normal virus clearance in serpinb1a−/− mice, pulmonary tissue injury and immune cell death are increased, as is the production of inflammatory cytokines by infiltrating monocytes/macrophages (IL-6, TNF-α) and T cells (IL-17A). Both the number and proportion of IL-17+ γδ T cells, which contribute to influenza pathogenicity but not to viral clearance [1], are increased in the lungs of infected serpinb1a−/− mice relative to WT mice [2].
Among hematopoietic cells, serpinb1a expression is highest in neutrophils, and its role in these cells is understood to include protection of the bone marrow reserve of postmitotic neutrophils, as well as restriction of the neutrophil extracellular traps (NETs)-generating death pathway of mature, activated tissue neutrophils [3, 4]. The function of serpinB1 in lymphocytes is less clear, although its expression has been described previously in subsets of T lineage cells, including αβ and γδ T cells [5]. In CD4+ αβ+ Th cells, which differentiate from naive precursor cells into a variety of specialized effector cells upon stimulation through the TCR, costimulatory molecules (e.g., CD28), and cytokines, serpinb1a has been shown to be highly induced upon Th17 differentiation in a Stat3-dependent manner [6].
γδ T cells are the first T cells to appear in the fetal thymus; they fulfill innate-like and adaptive-immune functions. Although more limited than αβ cells in terms of antigen receptor diversity, γδ T cells nonetheless include moderately diverse subsets, as well as subsets with invariant (monoclonal) and nearly invariant TCRs [7–9]. Unlike αβ T cells, which largely home to the LN and spleen, extrathymic γδ T cells are found principally in peripheral tissues and organs. γδ T cell subsets are defined by the expression of particular γ- and/or δ-V genes, where specific subsets are confined to limited anatomical sites, and in the extreme case, the invariant Vγ5/Vδ1+ subset is localized to a single location and microenvironment—the skin epidermal layer. The exclusiveness of location reflects the fact that γδ T subsets expressing the same V gene carry out similar functions. Also, γδ T cells, unlike αβ T cells, are largely preprogrammed prior to emigration from the thymus [10, 11]. It is the combination of preprogramming during development in the thymus together with retention of plasticity in response to environmental cues that exquisitely equip γδ T cells for their role as sentinel tissue cells capable of rapid responses upon sensing invading microbes. Interestingly, recent work from the Immunological Genome Consortium Project described that similar to its Th17-linked expression in CD4+ αβ+ T cells, serpinb1a expression is elevated in the Vγ4+ γδ T cell subset, which also preferentially expresses a host of Th17-associated transcripts, such as II17a, Il22, and Rorc, relative to other γδ subsets [5].
Here, we sought to identify the underlying factors that predispose serpinb1a−/− mice to overly robust and detrimental responses in the influenza infection model, focusing on the increased numbers and proportions of IL-17-expressing Th17 cells and γδ T cells. Even before infection, we show that loss of serpinb1a leads to homeostatic expansion of Th17 and IL-17+ γδ T cell subsets, without altering T cell development in the thymus. These results lend novel insight into the link between serpinB1 and IL-17-associated inflammation.
MATERIALS AND METHODS
Mice
SerpinB1−/− (serpinb1a−/−) mice were generated in the 129S6/SvEv/Tac (129S6) background [12]. Unchallenged serpinb1a−/− mice are healthy with normal growth, reproduction, and tissue morphology. WT 129S6 mice from Taconic Labs (Hudson, NY, USA) were maintained with serpinb1a−/− mice in the animal facility of the Immune Disease Institute or Boston Children's Hospital for at least several weeks or were bred in the latter facility. Animal studies were approved by the Institutional Animal Care and Use Committees of the Immune Disease Institute and/or Boston Children's Hospital.
Influenza A virus and infection model
Influenza A virus strain Philadelphia 82 [13] was kindly provided by Dr. Kevan Hartshorn (Boston University School of Medicine, Boston, MA, USA). The virus was harvested from 10-day-old embryonated hen's eggs, purified on sucrose gradients, dialyzed against PBS [14], titered as ffc on Madin-Darby canine kidney cells, and stored in aliquots at −80°C [2]. Groups of weight- and age-matched male mice were sedated with 100 mg/kg ketamine and 10 mg/kg xylazine and were inoculated intranasally by applying 15 μl inoculum onto each nare containing a sublethal dose, 28 × 106 ffc/mouse [2]. The mice were killed on Day 2 for harvest of lung tissue.
Harvest of lung cells
Groups of infected or naive male mice, ∼10 weeks old, were killed, and the lungs were aseptically removed into 2.0 ml DMEM, 5% FCS, 400 U/ml (4–5 mg/ml) collagenase type 1 (Worthington, Lakewood, NJ, USA), and 62 U/ml rDNase (Roche, Indianapolis, IN, USA). The lungs were minced with scissors to 1–2 mm and incubated with agitation at 37°C for 30 min. FCS (0.5 ml) was added, and the digested lungs were further disrupted by gently pushing the suspended tissue through a 70-μm nylon cell sieve (Fisher Scientific, Pittsburgh, PA, USA) using a syringe plunger and 10 ml DMEM with 10% FCS to optimize cell yield. The lung cells were pelleted, resuspended in red blood cell lysis buffer (PharmLyse; BD Biosciences, San Jose, CA, USA), incubated at ambient temperature for 5 min, combined with equal volume PBS with 2% FCS, and pelleted.
Flow cytometry and cell staining
Single-cell preparations of lung, spleen, or thymus were suspended in PBS, stained with viability dye (ViD; Invitrogen, Carlsbad, CA, USA), pelleted, washed, and resuspended in PBS with 2% FCS. Cell staining was done with fluorochrome- or biotin-labeled antibodies to CD45 (Clone 30-FM), CD3 (17A2), CD4 (GK1.5), CD8α (53-6.7), CD8β (YTS156.7.7), CD11b (M1/70), CD19 (GD5), Ly6G (1A8), F4-80 (BM8), CD27 (LG.3A10), γδ TCR (GL3), glycophorin (TER-119), IL-17A (hereafter IL-17; TC11-18H10.1), and IFN-γ (XMG-1.2; BioLegend, San Diego, CA, USA) and CCR6 (Clone 140,706) from R&D Systems (Minneapolis, MN, USA). Intracellular staining of FoxP3 (Clone JK-16S) and Ki-67 (Clone SOLA12) was done with antibody, reagents, and protocols from eBioscience (San Diego, CA, USA).
γδ TCR-V regions were detected with fluorochrome-labeled antibodies to Vγ1-TCR (Clone 2.1), Vγ4-TCR (UC3-10A6), and Vγ5-TCR (536) from BioLegend. The nomenclature of Heilig and Tonegawa [15] is used throughout. To detect Vγ6-TCR, cells were stained sequentially with GL3 antibody, followed by 17D1 [16]; the latter antibody was kindly provided by Drs. Robert Tigelaar and Julie Lewis. When used in combination with the anti-γδ TCR antibody, GL3, 17D1 detects Vγ6/Vδ1, as well as Vγ5Vδ1 [17].
For cytokine synthesis assays, single-cell preparations were suspended in DMEM with 10% FCS, and aliquots of 106 cells/ml were cultured with PMA (50 ng/ml), ionomycin (500 ng/ml), and 1 μg brefeldin A (1 μl GolgiPlug; BD Biosciences) for 4 h or with rIL-1β (20 ng/ml; BioLegend), rIL-23 (40 ng/ml; BioLegend), and brefeldin A for 6 h. The cells were pelleted and resuspended in PBS with 2% FCS, stained with surface antibodies, fixed and permeabilized with BD Cytofix/Cytoperm, and stained intracellularly for IL-17 or IFN-γ. Data were acquired on a Canto II cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
Cell sorting
To isolate immune-cell lineages, lung cells from five mice/group were pooled prior to the red blood cell lysis step and were enriched by negative selection using biotinylated antibodies to CD11b, CD8β, and CD19 and glycophorin from BioLegend, together with EasySep immunomagnetic reagents (Stemcell Technologies, Vancouver, BC, Canada), according to the manufacturer's protocol. The enriched cells were stained and sorted on a FACSAria II cytometer, and the target cell populations were collected directly into RNeasy lysis buffer (Qiagen, Valencia, CA, USA).
Microarrays and data analysis
Total RNA was isolated using RNeasy isolation kits (Qiagen), according to the manufacturer's protocol. Total RNA, from 50,000 to 100,000 cells, was processed and hybridized by the Boston University Microarray Resource Facility (Boston, MA, USA). All procedures were performed as described in GeneChip Whole Transcript Sense Target Labeling Assay Manual (Affymetrix, Santa Clara, CA, USA; current version available at www.affymetrix.com) and Ambion Whole Transcript Expression Kit protocol (Life Technologies, Carlsbad, CA, USA; current version available at http://tools.lifetechnologies.com/content/sfs/manuals/cms_064619.pdf). RNA was hybridized to Mouse Gene 1.0 ST chips (Affymetrix). Data were analyzed using GenePattern software (Broad Institute, Cambridge, MA, USA; http://www.broadinstitute.org/cancer/software/genepattern/); PCA was performed using population PCA software (http://cbdm.hms.harvard.edu/LabMembersPges/SD.html). For array normalization, samples were robust multiarray average-normalized using Affymetrix Power Tools (gcbg background correction, quantile normalization, median polish) on the annotated set of probe sets. Unannotated probe sets were removed prior to normalization. The sequences presented in this article have been submitted to the National Center for Biotechnology Information Gene Expression Omnibus website (http://www.ncbi.nim.nih.gov/geo) under Accession Number GSE46297.
Statistical analysis
All analyses were performed using GraphPad Prism software. Data were expressed as means ± sem and were analyzed by the unpaired Student's t-test. P < 0.05 was considered statistically significant.
Online Supplemental material
Five supplemental figures (described in Results) are available online.
RESULTS
IL-17+ γδ T cells and CD4+ Th17 cells are expanded in the lungs of naive serpinb1a−/− mice
To test for altered programming of pulmonary immune defense in the absence of serpinb1a−/−, we examined myeloid and lymphoid cell subsets present in the lungs of naive serpinb1a−/− mice, comparing them with cells from WT control animals. The animals were studied at 10 weeks of age when lung cell maturation is complete. For further comparison, some WT and serpinb1a−/− animals were sublethally infected with influenza virus and killed on Day 2, as described previously [2]. As anticipated, monocyte and neutrophil counts in the lungs of uninfected (naive) mice were low compared with Day 2 influenza-infected mice, and no genotype-dependent differences were found (Supplemental Fig. 1). In contrast, IL-17+ γδ T cells (hereafter, IL-17+ γδ T cells) were increased significantly in lungs of naive serpinb1a−/− mice compared with WT mice (Fig. 1A). The nearly threefold increase reflects higher total γδ cell number (Fig. 1A) and increased percentage of IL-17 producers among γδ cells (39.0±2.7% for serpinb1a−/− vs. 23.7±3.3% for WT). As noted previously for influenza-infected mice, no differences were found for IFN-γ producing γδ T cells between naive WT and serpinb1a−/− mice (Fig. 1A, right), indicating that the skewing of serpinb1a−/− γδ T cells toward an IL-17 phenotype is not accompanied by a broader deregulation of γδ T cell function. CD4+ Th cells expressing IL-17 were also increased in lungs of naive serpinb1a−/− mice compared with WT mice, whereas total CD4+ T cells, IFN-γ+ CD4 cells, and Tregs were not different between the genotypes (Fig. 1B). Moreover, the absolute counts of IL-17+ CD4 cells for naive mice of each genotype were comparable with the counts on Day 2 of infection (Fig. 1B). By contrast, numbers of total lung CD4+ T cells following influenza virus infection decreased in both genotypes. Reduced CD4+ T cell numbers in the lung in highly pathogenic influenza has been described previously in these and other mice [2, 18] and similarly occurs in humans [19]. Of note, the absolute counts of IL-17+ γδ T cells in the lungs of naive mice of each genotype were also quantitatively comparable with Day 2 infected animals (Fig. 1), strongly suggesting that IL-17 skewing of pulmonary γδ and CD4 T cells pre-exists in naive serpinb1a−/− mice.
Figure 1. Increase of IL-17+ γδ and CD4 T cells in lungs of naive serpinb1a−/− mice.

Matched groups of WT and serpinb1a−/− mice were killed without infection or as a control on Day 2 of sublethal infection with influenza virus. Suspensions of lung cells were cultured with PMA and ionomyin (and brefeldin A) and stained with surface antibodies and intracellularly for IL-17 and IFN-γ. Cells were gated on lymphocytes (low side-scatter, CD45+CD11bneg) and then on γδ TCR or CD4. (A) γδ T cells. Shown left to right are representative contour plots, followed by quantitation of total, IL-17+, and IFN-γ+ γδ T cells. (B) CD4 T cells. Shown are contour plots followed by quantitation of total, IL-17+, and IFN-γ+ CD4 cells and Tregs (CD4+FoxP3+). Numbers in quadrants indicate percentage in each. Means ± sem for eight to 12 mice/group from two to three experiments. *P < 0.05; ***P < 0.001. sb1−/−, serpinb1a−/−. Counts of monocytes, macrophages, and neutrophils in naive lungs are shown in Supplemental Fig. 1.
Th17 skewing of αβ+ and γδ+ pulmonary T cell lineages in the absence of serpinB1
To characterize more broadly the Th17 skewing in γδ and CD4+ T cells upon loss of serpinb1a, we isolated γδ and CD4+ T cell subsets from the lungs of uninfected serpinb1a−/− and WT mice and determined their transcriptional profiles via microarray experiments. PCA of pulmonary immune-cell lineages indicated that serpinb1a−/− did not fundamentally change the transcriptional identity of CD4+ CCR6+ αβ T cells, Tregs, or γδ T cells (Fig. 2B). However, serpinb1a−/− did lead to noticeable changes in gene expression, including increased Il17a expression in CD4+ CCR6+ αβ T cells and γδ T cells (Fig. 2). In addition to Il17a, serpinb1a−/− CD4+ CCR6+ αβ T cells and γδ T cells expressed elevated levels of other Th17 signature mRNAs, including Il17f, Rorc, Ccr6 itself, and Ccr4. Lower levels of Th1-associated transcripts, namely Ifng, Cxcr3, and Tbx21, were noted in γδ T cells but not in CD4+ CCR6+ αβ T cells (Fig. 2D). In contrast, only minor differences in gene expression were observed between serpinb1a−/− and WT Tregs, consistent with their low-level expression of serpinb1a (Fig. 2A). Thus, loss of serpinb1a skews pulmonary CD4+ αβ and γδ T cell compartments toward a Th17 phenotype in the absence of overt infection.
Figure 2. Transcriptome analysis of T cell lineages in lungs of naive WT and serpinb1a−/− mice.

(A) Transcription levels of serpinb1a in three populations of WT T cells expressed as arbitrary units (A.U.). (B) Principal components (PC) analysis of the six analyzed populations. PC1 accounts for 85.4% of the genotype variation, PC2 for 9.9%, and PC4 for <1%. PC3, which is not displayed, accounts for 3.3%, but no genotype-dependent differences were seen. (C) Heat map of all 2131 genes. The data were analyzed using hierarchical clustering. Mean normalized values from two independent analyses were used for cluster analysis. (D) Transcriptional levels of signature genes differentially expressed between serpinb1a−/− [knockout (KO)] and WT γδ T cells (upper) and CCR6+ CD4 cells (lower). (E) Increase of CCR6+ γδ and CD4 T cells and (F) CD27neg CCR6+ γδ T cells in lungs of WT and serpinb1a−/− mice. (A–D; mean of duplicates) Data represent evaluations of RNA from two isolates of lung cells, each from five or more mice/cell type. (E and F) Means ± sem or representative data for eight mice/group from two experiments. *P < 0.05; ***P < 0.001.
Skewing of T cell lineage genes, as observed by microarray experiments performed on bulk cell populations, may be a result of cell-intrinsic responses or differences in subset composition. Based on the limited number of altered transcripts and their specificity, primarily Th17 and Th1 signature genes, we reasoned that the microarray differences were primarily a result of differences at the level of subset composition. This interpretation means that the IL-17 bias in serpinb1a−/− mice reflects increased numbers of otherwise “normal” T cell subsets, as opposed to spurious activation of IL-17 expression in non-Th17 lineage αβ and γδ T cells. Consistent with this notion, we found that CCR6+ γδ and CD4 T cells were increased in the lungs of serpinb1a−/− mice compared with WT mice, as judged by surface-antibody staining and FACS analyses (Fig. 2E). As expected for Th17 cells of naive WT mice, IL-17 expression by pulmonary γδ T cells was almost entirely restricted to the CCR6+ subsets for both genotypes (not shown), and the expanded CCR6+ γδ cells were exclusively CD27neg (Fig. 2F), indicating that characteristic features of Th17 phenotype cells were maintained in serpinb1a−/− γδ T cells.
Systemic Th17 skewing of γδ and αβ CD4 T cells in serpinb1a−/− mice
We next asked whether Th17 skewing of the γδ and αβ CD4 T cell compartments in serpinb1a−/− mice was restricted to the lung or if similar changes could be seen in other lymphoid organs. We found that spleens of naive serpinb1a−/− mice contain increased numbers and proportions of Th17-skewed γδ cells compared with spleens of WT mice, as judged by the three- to fourfold increase of CCR6+ and IL-17-producing γδ cells (Fig. 3A), with no differences of IFN-γ+ γδ T cells compared with spleens of WT mice (Supplemental Fig. 2A). Increases in CCR6+ and IL-17+ γδ T cells within the spleens of serpinb1a−/− animals were detected as early as 4 weeks of age and were maintained in 10-week-old animals (Fig. 3A). IL-17+ and CCR6+ CD4 αβ cells were also increased modestly in serpinb1a−/− mice at 4 and 10 weeks, although the differences were not significant in all cases (Fig. 3B). To test for the specificity of Th17 skewing of CD4+ spleen cells, an enlarged group of 10-week-old mice was studied. Whereas IL-17+ CD4 cells were increased in spleens of serpinb1a−/− mice compared with WT, there were no genotype-dependent differences of IFN-γ+ CD4 cells (Supplemental Fig. 2B). These findings indicate that serpinb1a−/− has systemic and Th17-specific effects on skewing of the γδ and CD4+ T cell compartments, which are present early in life.
Figure 3. Increase of IL-17+ and CCR6+ γδ T cells in spleens of serpinb1a−/− mice.

Splenocytes of 4- and 10-week-old, naive WT and serpinb1a−/− mice were evaluated for surface antigens or IL-17 expression as in Fig. 1. Cells were gated on lymphocytes (CD45+CD11bneg) and then on γδ TCR or CD4. (A) γδ Cells. Shown are total, IL-17+, and CCR6+ γδ T cells. (B) CD4 cells. Shown are total, IL-17+, and CCR6+ CD4 cells. Means ± sem for eight mice/group at 4 weeks and six mice/group at 10 weeks, each from two experiments. *P < 0.05; **P < 0.01; ***P < 0.001. Related findings are in Supplemental Fig. 2.
Given that systemic changes in mature T cell homeostasis may be a secondary effect of altered T cell lymphopoiesis, we examined the thymic development of αβ and γδ T cells in serpinb1a−/− mice, we did this in part because, as γδ T cells, like other innate lymphocytes, emerge from the thymus as preprogrammed, memory-like cells [10, 11]. Among αβ thymocytes, no genotype differences were observed in counts of immature CD4+CD8+ or mature CD4+ or CD8+ cells (Supplemental Fig. 3A). Among γδ thymocytes, no differences were found between serpinb1a−/− and WT mice in total cells or in the very low numbers of γδ cells expressing IL-17 (Supplemental Fig. 3B). We also assessed CD27 expression on γδ thymocytes as a more accurate measure of IL-17 programming; immature γδ thymocytes preprogrammed as IFN-γ, and IL-4 producers have been shown to express CD27, whereas γδ thymocytes that give rise to IL-17 producers are CD27neg [20]. The percentages of CD27+ and CD27neg γδ thymocytes were also not different between serpinb1a−/− and WT mice (Supplemental Fig. 3B). These data collectively suggest that loss of serpinb1a results in peripheral expansion of CD4+ Th17 cells and IL-17+ γδ cells and that this phenotype is not a result of altered thymic development.
Serpinb1a selectively regulates expansion of IL-17-producing subsets of γδ T cells
As our microarray studies indicated that γδ T cells display elevated serpinb1a gene expression relative to other T cell lineages (e.g., Th1, Tregs), and γδ T cells have the most pronounced Th17 skewing in the absence of serpinb1a, we focused on this compartment for further analysis. Specific γδ subsets sharing location and function are defined by the expression of particular γ- and/or δ-V genes, and each subset can thus be marked by costaining with a common and a V-region-specific TCR-γδ antibody. We used this approach coupled with flow cytometric analysis to quantify γδ subsets in naive mice. Three γδ T cell subsets, namely Vγ1+, Vγ4+, and Vγ6/Vδ1+ cells, were detected in the lung and spleen of WT and serpinb1a−/− mice (Fig. 4); no Vγ5+ cells were detected (see Fig. 4 legend). Together, these three subsets account for most of the γδ T cells in naive lung (estimated at 85–95%) but only ∼75% of γδ T cells in naive spleen. Upon comparing the two genotypes, we found that the increase of total γδ T cells in the lung and spleen of serpinb1a−/− mice could be accounted for completely by the increase of Vγ4+ and Vγ6/Vδ1+ subset cells; there were no genotype-dependent differences in the number of Vγ1+ γδ cells (Fig. 4).
Figure 4. Selective increase of Vγ4+ and Vγ6/Vδ1+ γδ T cells in lung and spleen of serpinb1a−/− mice.
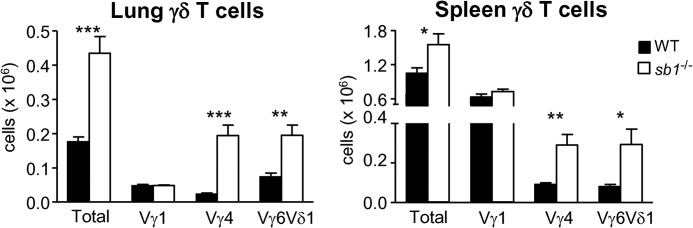
Lung (left) and spleen (right) cells of WT and serpinb1a−/− mice were stained with GL-3, followed by antibodies for Vγ1, Vγ4, Vγ5, and 17D1, which detect Vγ5/Vδ1 and Vγ6/Vδ1. Shown are absolute counts of total, Vγ1+, Vγ4+, and Vγ6/Vδ1+ γδ T cells. No Vγ5+ cells were detected (data not shown), and thus, 17D1 antibody staining identified Vγ6/Vδ1+ cells. The data are means ± sem for eight mice/group in three experiments. *P < 0.05; **P < 0.01; ***P < 0.001. Similar results were obtained for Vγ1+, Vγ4+, and Vγ6/Vδ1+ spleen cells of 4-week-old mice (data not shown).
Intracellular staining experiments revealed that IL-17 production by γδ T cells of lung and spleen was confined to Vγ4+ and Vγ6/Vδ1+ cells; no IL-17+ Vγ1 cells were detected. Furthermore, the percentages of Vγ4+ and Vγ6/Vδ1+ subsets producing IL-17 were increased for serpinb1a−/− mice compared with WT mice (Fig. 5A and B). In contrast to Vγ4+ cells, which preferentially produced IL-17, stimulated Vγ1+ γδ T cells produced IFN-γ but not IL-17 (Supplemental Fig. 4). IFN-γ production by Vγ1+ and Vγ4+ cells did not differ between the genotypes (Supplemental Fig. 4), consistent with the absence of genotype differences for total IFN-γ+ γδ+ T cells (Figs. 1A and 3A).
Figure 5. Increased percentage of IL-17+-producing cells within the serpinb1a−/− Vγ4+ and Vγ6/Vδ1+ subsets.

Lung and spleen cells of WT and serpinb1a−/− mice were cultured with PMA and ionomycin. The cells were surface-stained as in Fig. 4 and intracellularly for IL-17. Events were gated on lymphocytes and then on specific γδ subsets. (A) IL-17+ cells quantified as percentage of the Vγ1+, Vγ4+, and Vγ6/Vδ1+ subsets. (B) Representative contour plots of IL-17-stained Vγ4+ and Vγ6/Vδ1+ cells; there were no IL-17+ Vγ1 cells. (C) Enumeration of IL-17+ Vγ1+, Vγ4+, and Vγ6/Vδ1+ cells. Means ± sem for four mice/genotype. **P < 0.01; ***P < 0.001. Related findings are in Supplemental Figs. 4 and 5.
Whereas the above assays for IL-17 used PMA and ionomycin stimulation, it is thought that inflammatory cytokines, such as IL-1β and IL-23, are the principal drivers of IL-17 in γδ T cells in vivo [21]. Stimulation of WT and serpinb1a−/− γδ+ T cells with these cytokines showed that as with PMA and ionomycin stimulation, IL-17 production was increased in total γδ T cells and Vγ4+ cells from serpinb1a−/− versus WT mice (Supplemental Fig. 5A). IL-1β plus IL-23 stimulation only induced IL-17 in γδ+ splenocytes as anticipated (Supplemental Fig. 5A), and the most robust IL-17 response was observed within Vγ4+ cells (Supplemental Fig. 5B). By factoring in the increased percentages and the increased IL-17 expression, our data indicate a five- to tenfold overall increase of IL-17+ Vγ4+ and Vγ6/Vδ1+ cells in lung and spleen of serpinb1a−/− mice (Fig. 5C).
Serpinb1a regulates proliferation of Vγ4 and Vγ6δ1 but not Vγ1 γδ T cells
The increased frequency of Vγ4+ and Vγ6/Vδ1+ subset cells in serpinb1a−/− mice could be a result of preferential expansion of these cells or relative loss of competing subsets. To address the mechanism underlying the increased size of the Vγ4+ and Vγ6/Vδ1+ compartments in serpinb1a−/− mice, lung and spleen cells were stained for Ki-67, a nuclear antigen expressed only in cells undergoing active proliferation [22]. Cycling of WT γδ T cells, as measured by the proportion of Ki-67+ cells, varied, depending on subset and anatomical site. In WT mice, for example, a larger percentage of Vγ6/Vδ1+ cells was Ki-67+ relative to Vγ4+ or Vγ1+ cells, and Ki-67 staining/expression was generally higher in cells harvested from the lung compared with those from spleen (Fig. 6A and B). However, the percentage of cycling (Ki-67+) Vγ4+ cells was further increased substantially (fourfold) in serpinb1a−/− mice relative to WT animals (Fig. 6B). Ki-67 expression was also increased consistently, although not “significantly”, in Vγ6/Vδ1+ cells in the lung and spleen of serpinb1a−/− mice (Fig. 6B). Importantly, the frequency of cycling Vγ1+ cells was not different in serpinb1a−/− mice compared with WT mice, again indicating that the function of serpinB1 in regulating proliferation of γδ T cells is subset-specific (Fig. 6B). To link these findings with microarray-based gene-expression differences (Fig. 2D), we examined one of these—Rorc—in γδ subset cells. Whereas Vγ1+ cells of both genotypes were RORγtneg, Vγ4+ cells included RORγtneg and RORγt+ populations, and the frequency of RORγt+ Vγ4+ cells was increased among serpinb1a−/− Vγ4+ cells compared with WT Vγ4+ cells (Fig. 6C). Moreover, the serpinb1a−/−-proliferating (Ki-67+) Vγ4+ cells were primarily RORγt+ cells (Fig. 6D), indicating that SerpinB1 selectively regulates expansion of the RORγt+ Vγ4+ γδ T cells.
Figure 6. Selectively increased proliferation of serpinb1a−/− Vγ4+ and Vγ6/Vδ1+ γδ T cells.

Freshly isolated lung (A and B) and spleen (A–D) cells of naive WT and serpinb1a−/− mice were surface-stained with γδ subset antibodies, as in Fig. 4, and intracellularly for (A, B, and D) the proliferation marker Ki-67 and (C and D) RORγt. (A) Dot plots showing Ki-67 staining of Vγ1+, Vγ4+, and Vγ6/Vδ1+ subsets. (B) Ki-67+ cells quantified as percentage within each subset. (C) RORγt staining of Vγ1+ and Vγ4+ cells. (Left) Flow cytometry plots. (Right) RORγ+ cells quantified within the Vγ4+ subset. (D) Vγ4 cells costained for Ki-67 and RORγt. (Left) Contour plots. (Right) Quantitation of Ki-67+ RORγneg and Ki-67+ RORγ+ cells within the Vγ4 subsets. Means ± sem or representative data for four mice/group. **P < 0.01; ***P < 0.001.
DISCUSSION
We have shown that serpinB1 regulates the homeostasis of multiple proinflammatory T cell lineages. In particular, loss of serpinb1a leads to spontaneous and subset-specific expansion of CD4+ Th17 and IL-17+ γδ T cells. Th17 skewing of γδ and CD4 T cells in serpinb1a−/− mice is specific, as neither IFN-γ+ γδ cells nor other CD4+ T cell subsets (Th1, Tregs) are altered in the absence of serpinb1a. Moreover, these alterations in sentinel T cell populations at steady-state are likely responsible for the predisposition of serpinb1a−/− mice to increased pathology upon infection; serpinb1a−/− mice challenged with high-dose influenza virus display increased morbidity and mortality in association with enhanced tissue injury, increased death of infiltrating leukocytes, and enhanced and prolonged inflammatory cytokine production by infiltrating monocytes (IL-6, TNF-α) and T cells (IL-17), primarily γδ T cells [2].
The fundamental characteristics of cells with a Th17/IL-17 phenotype, including absence of CD27 and expression of CCR6 and Rorc, were maintained in the absence of serpinb1a. This result suggested that the Th17/IL-17 bias in serpinb1a−/− mice is primarily a result of increased numbers of otherwise normal T cell subsets, as opposed to spurious activation of IL-17 expression in non-Th17 lineage αβ and γδ T cells. Consistent with this notion, global transcriptional profiling of T cells from the lung of naive serpinb1a−/− mice revealed not only increased expression of Il17a but also other Th17 signature genes (Il17f, Ccr4, Ccr6 itself, and Rorc) in γδ+ and αβ+ CD4+ CCR6+ T cells and decreased expression of Th1-associated transcripts (Ifng, Cxcr3, and Tbx21) in γδ+ T cells relative to WT counterpart T cells. Few perturbations in gene expression were observed in serpinb1a−/− γδ and αβ+ CD4+ CCR6+ T cells that were unrelated to IL-17 and IFN-γ. Even fewer genotype-specific changes in gene expression were seen in Tregs, consistent with their low-level serpinb1a expression.
Pronounced expansion of Th17/IL-17-biased γδ+ and αβ+ CD4+ T cells was observed in the lung, where disturbances in T cell population dynamics may contribute directly to increased pathology upon pulmonary infection, and the spleen of serpinb1a−/− mice. The fact that IL-17+ γδ and CD4+ Th17 cells are increased at multiple sites within serpinb1a−/− mice suggests that the principal defect is a result of altered homeostasis, not local recruitment to the lung. Furthermore, this function of serpinB1 in γδ and αβ T cells is likely restricted to the mature peripheral subsets, as we observed that serpinb1a−/− had no bearing on αβ or γδ T cell development in the thymus.
Little is known about the function of serpinB1 in peripheral CD4+ T cell subsets, although subset-selective expression of serpinb1a can be seen by microarray analysis of effector/memory T cell subsets. Specifically, serpinb1a gene expression is induced in murine naive T cells upon in vitro activation in the presence of Th17-polarizing cytokines such as TGF-β and IL-6 [6]. The same transcriptional-profiling experiments reveal that Th17-specific serpinb1a up-regulation requires Stat3. In line with these data, we demonstrate that serpinb1a is also expressed in endogenous CD4+ CCR6+ effector/memory T cells, which include all of the naturally occurring Th17 cells in vivo. Indeed, high-level serpinb1a gene expression can be seen by analyses of previously published microarray data evaluating IL-17+ Th17 cells isolated from the CNS of mice with active experimental autoimmune encephalomyelitis or the gut of mice treated with anti-CD3 therapeutic antibodies [23]. These data thus argue that serpinB1 is linked to the development and function of Th17 cells. However, in contrast to other Th17 regulatory molecules (basic leucine zipper transcription factor ATF-like, Irf4, Stat3, RORγt), which are highly expressed in Th17 cells and are required for Th17 cell development and function, our results suggest that serpinB1 functions as an endogenous restraint mechanism by blunting Th17 cell growth and expansion.
Further study of the mechanism by which serpinB1 restricts expansion of Th17 CD4+ and γδ+ T cells will be important. In general, clade B serpins, also called ov-serpins (ovalbumin-related serpins) are nucleocytoplasmic proteins best known for their cytoprotective functions [24–26]. Cytoprotection can result from neutralization by the serpin of its cognate protease, usually a serine protease released from granules or a cysteine protease, such as a cathepsin (called cross-class inhibition). In this way, endogenous human and murine serpinB9 (PI-9, Spi6) protects activated cytolytic T cells and NK cells from their own granzyme B [27] and is thought to protect bystander and target cells, including tumor cells [28]. Whereas serpins function in these mechanisms to increase population size, the current findings identify a specific effect of serpinB1 to restrict proliferation of CCR6+ CD4 and γδ+ T cells and thus, limit population size at homeostasis. Cases where serpins function to restrict cell proliferation are few but may be informative. The best-characterized is cell-cycle regulation of murine fibroblasts, in which the transcription factor CDP/Cux undergoes proteolytic activation by cathepsin-L to generate the isoform that stably interacts with DNA and allows cells to progress into S-phase [29]. Proteolytic activation of CDP/Cux could be inhibited by small molecule inhibitors of cysteine proteases. Further studies showed that ectopic expression of a serpin—the avian protein serpinB10b/MENT—blocks murine fibroblast proliferation by inhibiting cathepsin-L and repressing cell entry into S-phase [30]. Interestingly, serpinB10b/MENT also blocks the cysteine protease(s) that degrades retinoblastoma protein, another cell-cycle regulator. An analogous proliferation-restricting role has been envisioned for serpinA3g (Spi2a), an intracellular serpin of murine hematopoietic stem cells and memory CD8 cells. SerpinA3g, which inhibits cathepsin-L and cathepsin-B, was proposed to prevent cell entry into S-phase and thus, protect quiescent memory CD8 T cells from replicative senescence [31].
Efforts to identify the serpinB1-inhibited protease that regulates proliferation of CCR6+ CD4+ and γδ+ T cells will need to consider the multiplicity of cognate proteases for this serpin; these include not only neutrophil elastase but also proteinase-3, pancreatic elastase, chymotrypsin, cathepsin G, mast cell chymase, prostate-specific antigen [32], and granzyme H [33], all of which are serine proteases; at least a subset of cysteine cathepsins is also inhibited (unpublished findings). On the other hand, we speculate that the specificity of the proliferative restriction to the most mature CCR6+ CD4+ and γδ+ T cells is a result of the very high serpinB1 expression levels specific to these subsets.
Analysis of TCR-V gene use showed that IL-17+ γδ T cells in lung and spleen of WT and serpinb1a−/− mice were confined to the Vγ4+ and Vγ6/Vδ1+ subsets, and the absolute number of these γδ T cell subsets and the percent expressing IL-17 were increased in serpinb1a−/− mice. Expansion of Vγ6/Vδ1+ and Vγ4+ γδ T cells in lung and spleen of serpinb1a−/− mice relative to WT mice strongly suggests that distorted subset expansion occurs locally. That expansion occurs locally is likely as γδ T cell programming takes place in the thymus, and the number of γδ thymocytes programmed as IL-17 producers was not different in serpinb1a−/− and WT mice. Indeed, for the Vγ6/Vδ1+ γδ T cells, local expansion is the only possibility, as these invariant cells, like Vγ5/Vδ1+ cells in the epidermis, are generated only in the embryonic thymus and then exported to populate peripheral niches [34]. Expansion was verified by the proliferation marker Ki-67, which revealed increased percentages of cycling Vγ4+ and Vγ6/Vδ1+ but not Vγ1+ γδ cells in lung and spleen of serpinb1a−/− mice. Moreover, double staining for Ki-67 and the Th17 transcription factor RORγδ revealed that the proliferating serpinb1a−/− Vγ4+ cells were primarily within the RORγt+ population. This indicates that it is only the most mature Th17 phenotype T cells that are subject to increased proliferation in the absence of serpinb1a. Thus, the extreme selectivity of the serpinb1 effect on proliferation/expansion appears to account also for the increased frequency of IL-17+ T cells within the select subsets.
The Vγ4+ and Vγ6/Vδ1+ subsets of γδ T cells, although rarely studied together, have both been identified as important in the lung and as IL-17 producers. IL-17 γδ T cells share many protective and pathological functions with Th17 cells. However, γδ cells, owing to their preprogramming and localization within tissues, can produce IL-17 within hours of sensing microbial products or stress-induced epithelial antigen or microbe-induced cytokines, such as IL-23 and IL-1β [21, 35, 36]. For better or for worse, the resulting γδ T cell-generated IL-17 initiates early innate responses, most notably, recruitment of neutrophils [35], and also molds adaptive responses directly and indirectly (reviewed in ref. [9]). The Vγ6/Vδ1+ γδ T cells are reportedly relatively rare in adult spleen, liver, and LN and somewhat more numerous in the lung [17]. Lung content of Vγ6/Vδ1+ cells in naive WT mice varies, depending on genetic background, amounting to ∼7% of total γδ cells in C57BL/10 mice, 30% in BALB/c [17], and 50% in the 129S6 mice studied here, strongly suggesting that the homeostatic set point for this population is regulated by modulatory genes.
The Vγ4+ subset of γδ T cells, which are oligoclonal, includes major IL-17+ cells in the dermal layer of the skin; these are locally proliferating cells that provide cutaneous immunosurveillance but also induce skin inflammation in models of psoriasis [37–39]. Vγ4+ γδ T cells also accumulate in draining LNs in collagen-induced arthritis [40]. In lungs infected with Mycobacterium tuberculosis, γδ T cells are the predominant IL-17-producing cells [41], and these consist of the Vγ4+ and Vγ6/Vδ1+ subsets [5, 42].
In a recent study, the Immunological Genome Consortium determined the transcriptomic profiles for γδ T cells resident in adult and fetal thymus; these include most of the CD24hi immature subsets as well as mature Vγ1+ and Vγ4+ cells [5] (the nomenclature has been switched here to the Heilig and Tonegawa system). Comparison of expression profiles showed that immature Vγ4+ thymocytes differ from three other immature subsets (Vγ7+ and Vγ1+ paired with different Vδ genes) [5]. Of the subsets studied, mature Vγ4+ cells had the most distinct gene expression, owing largely to increased expression of a gene cluster related to IL-17 production and responsiveness. Prominent among the gene-expression differences in mature Vγ4+ thymocytes was the >60-fold increase of serpinb1a compared with immature Vγ4+ thymocytes, the second-highest gene expression increase after Il23r and substantially increased compared with mature Vγ1+ thymocytes.
In this context, our findings that show proliferative expansion of the serpinb1ahigh Vγ4+ and the Vγ6/Vδ1+ subset but not the serpinb1alow Vγ1+ subset in lung and spleen of naive serpinb1a−/− mice strongly suggests that serpinB1 negatively regulates the homeostatic set point of mature IL-17+ producer T cell subsets at peripheral sites. A previous study of factors regulating splenic γδ T cell proliferation described a mechanism involving CD28 signals and autocrine IL-2 production [43]. However, the CD28/IL-2-mediated pathway controls IFN-γ-producing and IL-17-producing γδ cells and is thus distinct from the SerpinB1-dependent mechanism identified here, which is highly specific for IL-17-producing cells. Our findings indicate that serpinB1 restricts the size of IL-17+ sentinel T cell compartments at steady-state, which in turn, affects the strength of the immune response to infection and other challenges. Therapeutic strategies based on γδ T cells are important for current and future application in autoimmune disease and cancer. Indeed, further study to delineate the mechanism underlying serpinB1-dependent regulation of IL-17+ cell proliferation might lay the foundation for subset-selective immunomodulation of γδ T cells during autoimmunity and cancer.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a Pilot Award of the GlaxoSmithKline/Immune Disease Institute Research Alliance and by grant HL-066548 from the National Heart, Lung, and Blood Institute, U.S. National Institutes of Health, Bethesda, MD, USA.
We thank Kevan L. Hartshorn and Mitchell White (Boston Medical Center, Boston, MA, USA) for providing virus; Robert Tigelaar and Julie Lewis (Yale University School of Medicine, New Haven, CT, USA) for 17D1 antibodies; Natasha Barteneva PCMM Flow Cytometry Core Facility, Boston, MA, USA] for guidance in cell sorting; and Scott Davis (Tempero Pharmaceuticals, Cambridge, MA, USA) for technical support with microarray data analyses. We thank Jessica Cooley for figure preparation and Fauve Dela Cruz for maintaining the mouse colony.
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- −/−
- deficient
- CDP/Cux
- CCAAT-displacement protein/cut homeobox
- ffc
- fluorescent focal count(s)
- FoxP3
- forkhead box P3
- IL-17
- IL-17A
- MENT
- myeloid and erythroid nuclear termination stage-specific protein
- PCA
- principal component analysis
- ROR
- retinoic acid receptor-related orphan receptor
- SerpinB1
- monocyte neutrophil elastase inhibitor
- Spi2a/6
- serine proteinase inhibitor 2a/6
- Tbx21
- T-box 21
- Treg
- regulatory T cell
- V
- variable
AUTHORSHIP
P.Z., L.H., and K.F. performed research and analyzed and interpreted data. M.S.S. and E.R-O. designed research, interpreted data, and wrote the paper.
DISCLOSURES
The authors declare no conflict of interest.
REFERENCES
- 1. Crowe C. R., Chen K., Pociask D. A., Alcorn J. F., Krivich C., Enelow R. I., Ross T. M., Witztum J. L., Kolls J. K. (2009) Critical role of IL-17RA in immunopathology of influenza infection. J. Immunol. 183, 5301–5310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gong D., Farley K., White M., Hartshorn K. L., Benarafa C., Remold-O'Donnell E. (2011) Critical role of serpinB1 in regulating inflammatory responses in pulmonary influenza infection. J. Infect. Dis. 204, 592–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Benarafa C., LeCuyer T. E., Baumann M., Stolley J. M., Cremona T. P., Remold-O'Donnell E. (2011) SerpinB1 protects the mature neutrophil reserve in the bone marrow. J. Leukoc. Biol. 90, 21–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Farley K., Stolley J. M., Zhao P., Cooley J., Remold-O'Donnell E. (2012) A serpinB1 regulatory mechanism is essential for restricting neutrophil extracellular trap generation. J. Immunol. 189, 4574–4581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Narayan K., Sylvia K. E., Malhotra N., Yin C. C., Martens G., Vallerskog T., Kornfeld H., Xiong N., Cohen N. R., Brenner M. B., Berg L. J., Kang J. (2012) Intrathymic programming of effector fates in three molecularly distinct γδ T cell subtypes. Nat. Immunol. 13, 511–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Durant L., Watford W. T., Ramos H. L., Laurence A., Vahedi G., Wei L., Takahashi H., Sun H. W., Kanno Y., Powrie F., O'Shea J. J. (2010) Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 32, 605–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. O'Brien R. L., Born W. K. (2010) γδ T cell subsets: a link between TCR and function? Sem. Immunol. 22, 193–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bonneville M., O'Brien R. L., Born W. K. (2010) γδ T cell effector functions: a blend of innate programming and acquired plasticity. Nat. Rev. Immunol. 10, 467–478 [DOI] [PubMed] [Google Scholar]
- 9. Vantourout P., Hayday A. (2013) Six-of-the-best: unique contributions of γδ T cells to immunology. Nat. Rev. Immunol. 13, 88–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shibata K., Yamada H., Nakamura R., Sun X., Itsumi M., Yoshikai Y. (2008) Identification of CD25+ γ δ T cells as fetal thymus-derived naturally occurring IL-17 producers. J. Immunol. 181, 5940–5947 [DOI] [PubMed] [Google Scholar]
- 11. Jensen K. D., Su X., Shin S., Li L., Youssef S., Yamasaki S., Steinman L., Saito T., Locksley R. M., Davis M. M., Baumgarth N., Chien Y. H. (2008) Thymic selection determines γδ T cell effector fate: antigen-naive cells make interleukin-17 and antigen-experienced cells make interferon γ. Immunity 29, 90–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Benarafa C., Priebe G. P., Remold-O'Donnell E. (2007) The neutrophil serine protease inhibitor serpinb1 preserves lung defense functions in Pseudomonas aeruginosa infection. J. Exp. Med. 204, 1901–1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hartshorn K. L., White M. R., Voelker D. R., Coburn J., Zaner K., Crouch E. C. (2000) Mechanism of binding of surfactant protein D to influenza A viruses: importance of binding to haemagglutinin to antiviral activity. Biochem. J. 351, 449–458 [PMC free article] [PubMed] [Google Scholar]
- 14. Hartshorn K. L., Collamer M., Auerbach M., Myers J. B., Pavlotsky N., Tauber A. I. (1988) Effects of influenza A virus on human neutrophil calcium metabolism. J. Immunol. 141, 1295–1301 [PubMed] [Google Scholar]
- 15. Heilig J. S., Tonegawa S. (1986) Diversity of murine γ genes and expression in fetal and adult T lymphocytes. Nature 322, 836–840 [DOI] [PubMed] [Google Scholar]
- 16. Mallick-Wood C. A., Lewis J. M., Richie L. I., Owen M. J., Tigelaar R. E., Hayday A. C. (1998) Conservation of T cell receptor conformation in epidermal γδ cells with disrupted primary Vγ gene usage. Science 279, 1729–1733 [DOI] [PubMed] [Google Scholar]
- 17. Roark C. L., Aydintug M. K., Lewis J., Yin X., Lahn M., Hahn Y. S., Born W. K., Tigelaar R. E., O'Brien R. L. (2004) Subset-specific, uniform activation among V γ 6/V δ 1+ γ δ T cells elicited by inflammation. J. Leukoc. Biol. 75, 68–75 [DOI] [PubMed] [Google Scholar]
- 18. Tumpey T. M., Lu X., Morken T., Zaki S. R., Katz J. M. (2000) Depletion of lymphocytes and diminished cytokine production in mice infected with a highly virulent influenza A (H5N1) virus isolated from humans. J. Virol. 74, 6105–6116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cunha B. A. (2008) The clinical diagnosis of severe viral influenza A. Infection 36, 92–93 [DOI] [PubMed] [Google Scholar]
- 20. Ribot J. C., deBarros A., Pang D. J., Neves J. F., Peperzak V., Roberts S. J., Girardi M., Borst J., Hayday A. C., Pennington D. J., Silva-Santos B. (2009) CD27 is a thymic determinant of the balance between interferon-γ- and interleukin 17-producing γδ T cell subsets. Nat. Immunol. 10, 427–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sutton C. E., Lalor S. J., Sweeney C. M., Brereton C. F., Lavelle E. C., Mills K. H. (2009) Interleukin-1 and IL-23 induce innate IL-17 production from γδ T cells, amplifying Th17 responses and autoimmunity. Immunity 31, 331–341 [DOI] [PubMed] [Google Scholar]
- 22. Scholzen T., Gerdes J. (2000) The Ki-67 protein: from the known and the unknown. J. Cell. Physiol. 182, 311–322 [DOI] [PubMed] [Google Scholar]
- 23. Esplugues E., Huber S., Gagliani N., Hauser A. E., Town T., Wan Y. Y., O'Connor W., Jr., Rongvaux A., Van Rooijen N., Haberman A. M., Iwakura Y., Kuchroo V. K., Kolls J. K., Bluestone J. A., Herold K. C., Flavell R. A. (2011) Control of TH17 cells occurs in the small intestine. Nature 475, 514–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Remold-O'Donnell E. (1993) The ovalbumin family of serpin proteins. FEBS Lett. 315, 105–108 [DOI] [PubMed] [Google Scholar]
- 25. Bird P. I. (1999) Regulation of pro-apoptotic leucocyte granule serine proteinases by intracellular serpins. Immunol. Cell Biol. 77, 47–57 [DOI] [PubMed] [Google Scholar]
- 26. Ashton-Rickardt P. G. (2013) An emerging role for serine protease inhibitors in T lymphocyte immunity and beyond. Immunol. Lett. 152, 65–76 [DOI] [PubMed] [Google Scholar]
- 27. Bird C. H., Sutton V. R., Sun J., Hirst C. E., Novak A., Kumar S., Trapani J. A., Bird P. I. (1998) Selective regulation of apoptosis: the cytotoxic lymphocyte serpin proteinase inhibitor 9 protects against granzyme B-mediated apoptosis without perturbing the Fas cell death pathway. Mol. Cell. Biol. 18, 6387–6398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Medema J. P., de Jong J., Peltenburg L. T., Verdegaal E. M., Gorter A., Bres S. A., Franken K. L., Hahne M., Albar J. P., Melief C. J., Offringa R. (2001) Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc. Natl. Acad. Sci. USA 98, 11515–11520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goulet B., Baruch A., Moon N. S., Poirier M., Sansregret L. L., Erickson A., Bogyo M., Nepveu A. (2004) A cathepsin L isoform that is devoid of a signal peptide localizes to the nucleus in S phase and processes the CDP/Cux transcription factor. Mol. Cell. 14, 207–219 [DOI] [PubMed] [Google Scholar]
- 30. Irving J. A., Pike R. N., Dai W., Bromme D., Worrall D. M., Silverman G. A., Coetzer T. H., Dennison C., Bottomley S. P., Whisstock J. C. (2002) Evidence that serpin architecture intrinsically supports papain-like cysteine protease inhibition: engineering α(1)-antitrypsin to inhibit cathepsin proteases. Biochemistry 41, 4998–5004 [DOI] [PubMed] [Google Scholar]
- 31. Ashton-Rickardt P. G. (2010) Serine protease inhibitors and cytotoxic T lymphocytes. Immunol. Rev. 235, 147–158 [DOI] [PubMed] [Google Scholar]
- 32. Cooley J., Takayama T. K., Shapiro S. D., Schechter N. M., Remold-O'Donnell E. (2001) The serpin MNEI inhibits elastase-like and chymotrypsin-like serine proteases through efficient reactions at two active sites. Biochemistry 40, 15762–15770 [DOI] [PubMed] [Google Scholar]
- 33. Wang L., Li Q., Wu L., Liu S., Zhang Y., Yang X., Zhu P., Zhang H., Zhang K., Lou J., Liu P., Tong L., Sun F., Fan Z. (2013) Identification of SERPINB1 as a physiological inhibitor of human granzyme H. J. Immunol. 190, 1319–1330 [DOI] [PubMed] [Google Scholar]
- 34. Havran W. L., Allison J. P. (1988) Developmentally ordered appearance of thymocytes expressing different T-cell antigen receptors. Nature 335, 443–445 [DOI] [PubMed] [Google Scholar]
- 35. Shibata K., Yamada H., Hara H., Kishihara K., Yoshikai Y. (2007) Resident Vδ1+ γδ T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J. Immunol. 178, 4466–4472 [DOI] [PubMed] [Google Scholar]
- 36. Martin B., Hirota K., Cua D. J., Stockinger B., Veldhoen M. (2009) Interleukin-17-producing γδ T cells selectively expand in response to pathogen products and environmental signals. Immunity 31, 321–330 [DOI] [PubMed] [Google Scholar]
- 37. Sumaria N., Roediger B., Ng L. G., Qin J., Pinto R., Cavanagh L. L., Shklovskaya E., Fazekas de St Groth B., Triccas J. A., Weninger W. (2011) Cutaneous immunosurveillance by self-renewing dermal γδ T cells. J. Exp. Med. 208, 505–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gray E. E., Suzuki K., Cyster J. G. (2011) Cutting edge: identification of a motile IL-17-producing γδ T cell population in the dermis. J. Immunol. 186, 6091–6095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cai Y., Shen X., Ding C., Qi C., Li K., Li X., Jala V. R., Zhang H. G., Wang T., Zheng J., Yan J. (2011) Pivotal role of dermal IL-17-producing γδ T cells in skin inflammation. Immunity 35, 596–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roark C. L., French J. D., Taylor M. A., Bendele A. M., Born W. K., O'Brien R. L. (2007) Exacerbation of collagen-induced arthritis by oligoclonal, IL-17-producing γ δ T cells. J. Immunol. 179, 5576–5583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lockhart E., Green A. M., Flynn J. L. (2006) IL-17 production is dominated by γδ T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J. Immunol. 177, 4662–4669 [DOI] [PubMed] [Google Scholar]
- 42. Yoshida Y. O., Umemura M., Yahagi A., O'Brien R. L., Ikuta K., Kishihara K., Hara H., Nakae S., Iwakura Y., Matsuzaki G. (2010) Essential role of IL-17A in the formation of a mycobacterial infection-induced granuloma in the lung. J. Immunol. 184, 4414–4422 [DOI] [PubMed] [Google Scholar]
- 43. Ribot J. C., Debarros A., Mancio-Silva L., Pamplona A., Silva-Santos B. (2012) B7-CD28 costimulatory signals control the survival and proliferation of murine and human γδ T cells via IL-2 production. J. Immunol. 189, 1202–1208 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.