

In Gram-negative bacteria, acquired 16S rRNA methyltransferases ArmA and NpmA confer high-level resistance to all clinically useful aminoglycosides by modifying, respectively, G1405 and A1408 in the A-site. These enzymes must coexist with several endogenous methyltransferases that are essential for fine-tuning of the decoding center, such as RsmH and RsmI in Escherichia coli, which methylate C1402 and RsmF C1407. The authors show that resistance methyltransferases impair endogenous methylation with various consequences on cell fitness.
Keywords: aminoglycoside resistance, 16S rRNA methyltransferase, fitness, translation
Abstract
In Gram-negative bacteria, acquired 16S rRNA methyltransferases ArmA and NpmA confer high-level resistance to all clinically useful aminoglycosides by modifying, respectively, G1405 and A1408 in the A-site. These enzymes must coexist with several endogenous methyltransferases that are essential for fine-tuning of the decoding center, such as RsmH and RsmI in Escherichia coli, which methylate C1402 and RsmF C1407. The resistance methyltransferases have a contrasting distribution—ArmA has spread worldwide, whereas a single clinical isolate producing NpmA has been reported. The rate of dissemination of resistance depends on the fitness cost associated with its expression. We have compared ArmA and NpmA in isogenic Escherichia coli harboring the corresponding structural genes and their inactive point mutants cloned under the control of their native constitutive promoter in the stable plasmid pGB2. Growth rate determination and competition experiments showed that ArmA had a fitness cost due to methylation of G1405, whereas NpmA conferred only a slight disadvantage to the host due to production of the enzyme. MALDI MS indicated that ArmA impeded one of the methylations at C1402 by RsmI, and not at C1407 as previously proposed, whereas NpmA blocked the activity of RsmF at C1407. A dual luciferase assay showed that methylation at G1405 and A1408 and lack of methylation at C1407 affect translation accuracy. These results indicate that resistance methyltransferases impair endogenous methylation with different consequences on cell fitness.
INTRODUCTION
Aminoglycosides that are used to treat severe infections caused by Gram-positive and -negative bacteria cause translational errors and inhibit translocation. These bactericidal antibiotics bind to the highly conserved A-site of 16S rRNA in the 30S small subunit and interfere with the decoding of mRNA. Aminoglycoside resistance is mediated by a variety of mechanisms (Magnet and Blanchard 2005). Among them, methylation or substitution of the bases involved in drug binding at the A-site lead to decrease or loss of affinity of the antibiotic for its target (Pfister et al. 2003; Liou et al. 2006). Resistance to aminoglycosides by ribosomal methylation has been observed in a wide range of Gram-negative pathogens and confers high-level resistance to all aminoglycosides available to treat systemic infections, except streptomycin (Wachino and Arakawa 2012).
In Escherichia coli, the 16S ribosomal RNA is modified by 10 endogenous methyltransferases and a pseudouridine synthase (Basturea et al. 2012). These modifications are required for proper maturation of the 30S subunit, fine-tuning of local rRNA structure, and RNA–RNA interactions (Roy-Chaudhuri et al. 2010). In particular, in the decoding center (nucleotides 1400–1500) where aminoglycosides bind, endogenous methyltransferases RsmI and RsmH methylate C1402 (Kimura and Suzuki 2010), RsmE U1498 (Basturea et al. 2006), and RsmF C1407 (Fig. 1; Andersen and Douthwaite 2006). Thus, endogenous methyltransferases must have evolved to be sterically compatible and/or temporally displaced such that each of the enzymes is able to modify its own nucleotide.
FIGURE 1.
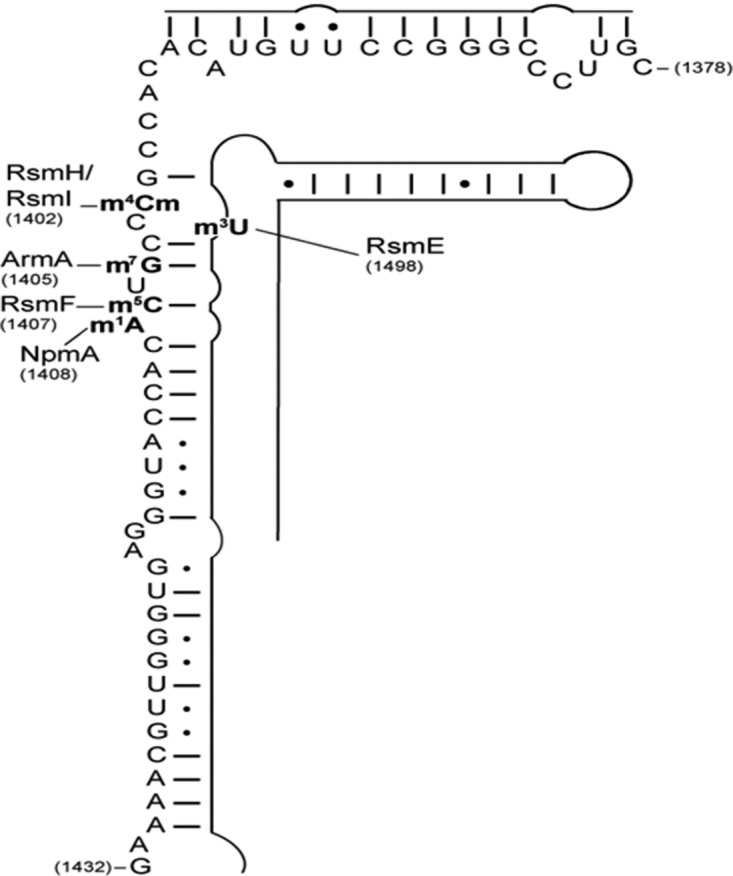
Schematic representation of 16S rRNA helix 44 (positions 1378–1432) secondary structure isolated for mass spectrometry analysis. The sites of post-transcriptional endogenous (RsmH, RsmI, RsmF, and RsmE) and exogenous (ArmA and NpmA) methylation are indicated.
The most prevalent type of resistance methyltransferases, ArmA (aminoglycoside resistance methyltransferase A), confers high-level resistance to 4,6-disubstituted 2-deoxystreptamines (2-DOS) (kanamycin and gentamicin groups) by methylation of the N7 position of guanine 1405 (m7G1405) (Galimand et al. 2003; Liou et al. 2006). Since the description of ArmA, eight other m7G1405 methyltransferases, RmtA to RmtH, sharing 25%–46% identity have been reported in Gram-negative bacilli. The second type is composed of a unique member, NpmA, which methylates the N1 position of A1408 (m1A1408) and confers high-level resistance to both 4,6- and 4,5-disubstituted 2-DOS (neomycin group) (Wachino et al. 2007). Since G1405 and A1408 are located within the A-site (Fig. 1) of the decoding region, acquisition of these methyltransferases could have a biological cost.
Methylation by ArmA and NpmA, like RsmI, RsmH, and RsmF, takes place after the assembly of the 30S subunits (Andersen and Douthwaite 2006; Liou et al. 2006; Wachino et al. 2007; Kimura and Suzuki 2010). This raises the question as to how well the resistance methyltransferases modifying G1405 and A1408 can be accommodated in this functionally crowded region of the ribosome. Mutations at the decoding site, in particular, at G1405 and A1408 (O'Connor et al. 1997), and lack of endogenous methylation (Roy-Chaudhuri et al. 2010; Qin et al. 2012) can alter translation fidelity. However, the impact of exogenous methylation at these positions has not been studied.
The two types of resistance methyltransferases have a contrasting distribution: ArmA, and to a lesser extent the Rmt enzymes, are spread worldwide, whereas a single Escherichia coli isolate producing NpmA has been reported to date (Wachino and Arakawa 2012). The fitness cost associated with most resistance mechanisms is an important biological parameter that influences emergence and dissemination of resistance and its persistence in the absence of selective pressure (Andersson and Hughes 2010).
We have quantified the biological cost due to production of ArmA and NpmA and studied the interference between these exogenous and the endogenous methyltransferases and the consequences of their production on translation accuracy. To allow comparison between the two methyltransferases, all experiments were performed in isogenic E. coli strains harboring the structural genes for the enzymes cloned under the control of their native promoter on a stable low-copy-number plasmid. We show that both resistance enzymes interfere with endogenous methyltransferases with different consequences on translation efficacy and host fitness.
RESULTS AND DISCUSSION
Resistance methylation at the A-site confers various degrees of biological cost to the host
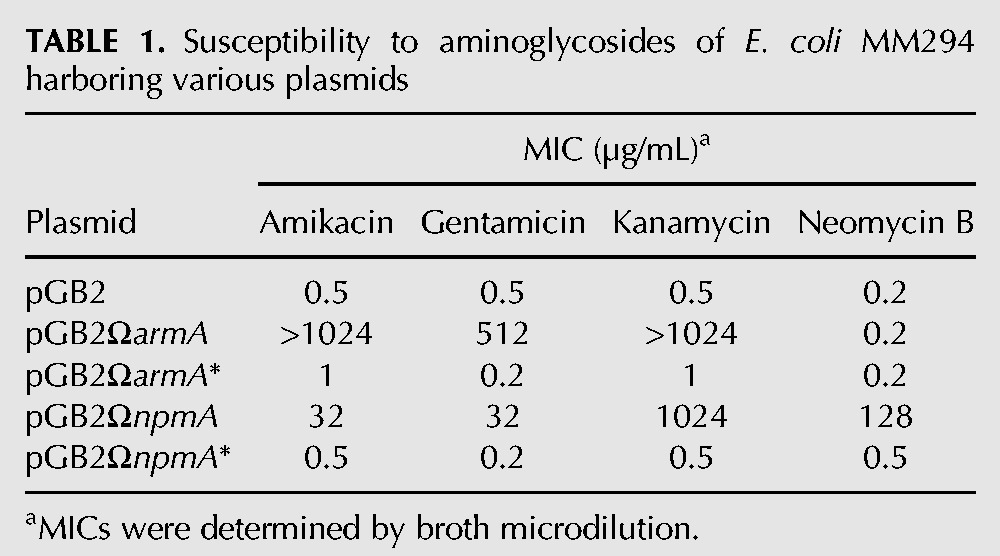
The armA and npmA genes and their inactive point mutants armA* and npmA* unable to bind the S-adenosyl-L-methionine cofactor were cloned into E. coli MM294 (Bachmann 1972) in the stable low-copy-number non-self-transferable plasmid pGB2 (Churchward et al. 1984) under the control of their native promoter (Supplemental Table S1). Expression of functional enzymes was confirmed by determination of the MICs of aminoglycosides for the hosts. E. coli MM294 harboring armA or npmA was resistant to high levels of 4,6-disubstituted 2-DOS or to 4,5- and 4,6-disubstituted 2-DOS, respectively, whereas strains containing pGB2, pGB2ΩarmA*, or pGB2ΩnpmA* remained susceptible to the drugs (Table 1).
TABLE 1.
Susceptibility to aminoglycosides of E. coli MM294 harboring various plasmids
The strength of the promoters for the structural genes was evaluated by comparing the mean fluorescence intensity (MFI) of MM294/pGB2ΩParmAgfpmut1 (MFI of 13) and pGB2ΩPnpmAgfpmut1 (MFI of 22) to that of MM294/pGB2 (MFI of 2.5). These results indicate that the genes were expressed constitutively and that Npma was produced at higher levels than ArmA.
Growth rates of the strains were determined in monocultures at the beginning of the exponential phase in the absence of antibiotics. The growth rate of E. coli MM294/pGB2ΩarmA was significantly decreased compared with that of E. coli MM294/pGB2 (relative growth rate of 0.87), whereas the growth rates of E. coli MM294/pGB2ΩarmA* and E. coli MM294/pGB2 were similar (Table 2). These results indicate that methylation at G1405 reduces the fitness of the host. In contrast, E. coli MM294/pGB2ΩnpmA or /pGB2ΩnpmA* had the same growth rate as MM294/pGB2 (relative growth rates of 1) (Table 2).
TABLE 2.
Relative growth rates of E. coli with various plasmids

For a more sensitive fitness evaluation, in vitro competition experiments were performed between strains differing only by the presence or absence of the methyltransferase gene in its active or inactive form. A marked competitive disadvantage was observed for cells expressing ArmA that were outcompeted by those transformed with pGB2 or pGB2ΩarmA* with a loss of 7.3% or 8.2% per generation, respectively (Fig. 2A), confirming that G1405 methylation was responsible for the burden to the cells. In contrast, competition between MM294/pGB2 and MM294/pGB2ΩnpmA revealed only a small disadvantage of the latter cells (loss of 2.7% per generation) (Fig. 2B). In addition, there was no competitive disadvantage between MM294/pGB2ΩnpmA and MM294/pGB2ΩnpmA* (0.4% loss per generation), suggesting that methylation at A1408 was not costly per se but rather that expression of NpmA, produced at high levels in MM294, could account for the slight fitness decrease.
FIGURE 2.

Competition experiments between susceptible and resistant strains. Competition was carried out in LB broth at 37°C. MM294 harboring (A) pGB2 and pGB2ΩarmA or pGB2ΩarmA* and (B) pGB2ΩnpmA were mixed at a 1:1 ratio at an initial inoculum of 103 CFU and transferred every 12 h (corresponding to about 20 generations) in fresh medium for up to six passages. The competition index (CI) was calculated as the CFU ratio of the resistant and susceptible strains (R/S) at time (t1) divided by the same R/S at time (t0), and the selection coefficient s was then calculated as the slope of the following linear regression model s= ln(CI)/[t × ln(2)], where t is the number of generations. Values are the mean ± SE of at least three independent experiments.
As opposed to our findings, it was reported that acquisition of rmtC, which like ArmA methylates G4015, does not entail a fitness cost for the host (Gutierrez et al. 2012). In this study, RmtC was shown to block endogenous methylation at C1407 by RsmF in E. coli, but unexpectedly, an rsmF knockout was found to display slower growth and reduced fitness in competition with the parental strain (Gutierrez et al. 2012). In our experimental conditions, there was no reduction in growth rate of MM294ΔrsmF relative to E. coli MM294 (Table 2) and no fitness decrease in competition experiments performed between MM294▵araB▵rsmF and MM294▵araB (relative fitness of 1). Only ArmA expression significantly reduced E. coli MM294ΔrsmF growth rate and to the same extent as in MM294 (Table 2). Different experimental conditions could account for these contradictory results. In Gutierrez et al. (2012), growth rates were measured every hour over a 12-h period as opposed to every 3 min at the beginning of the exponential phase (Foucault et al. 2009). Competition experiments should explore the entire growth cycle, i.e., the lag phase, the exponential, and the stationary phases on several cycles. As reported in Gutierrez et al. (2012), 20 generations were reached after the fourth transfer performed every 24 h; this would correspond to an estimated, rather unusual, 4-h doubling time for E. coli. In addition, the proportion of resistant to susceptible strains was determined on 100 colonies by replica plating or PCR (Gutierrez et al. 2012), whereas we surveyed 500–1000 colonies by replica plating on selective media.
Our data indicate that ArmA expression is costly for E. coli MM294, whereas deletion of rsmF is not deleterious for the host, suggesting that there is no link, as opposed to that proposed in Andersen and Douthwaite (2006) and Gutierrez et al. (2012), between the fitness cost associated with expression of ArmA and lack of methylation by RsmF.
The slight cost observed with NpmA was probably due to constitutive expression of the methyltransferase, a finding already reported for methyltransferase Cfr, which modifies the conserved 23S rRNA residue A2503 (LaMarre et al. 2011).
ArmA interferes with endogenous methylation at position C1402
In an attempt to elucidate the molecular mechanism responsible for the fitness cost due to ArmA production, the pattern of methylation at the decoding site was studied by MALDI MS. This technique can accurately measure the masses of small RNAs to within 0.2 Da and thus detect the presence or absence of a methyl group. Digestion of the 16S rRNA fragment by RNase T1, which specifically cleaves at the 3′ end of unmethylated guanosines, and by RNase A, which cleaves after unmethylated cytosines and uracils, gives rise to mass spectra from which methylation at C1402, C1407, and G1405 can be detected.
We first studied the 16S rRNA sequence from C1378 to G1432 (Fig. 1) containing the endogenous methylation sites C1402 (RsmH and RsmI) and C1407 (RsmF) from E. coli MM294 and that containing only the endogenous di-methylation at C1402 from E. coli MM294ΔrsmF, both digested with RNase T1. In MM294/pGB2, the rRNA fragment produced the expected MS spectrum with peaks at m/z 1305 and 3195 (Fig. 3A). These correspond to tetranucleotide m4CmCCGp from C1402 to G1405 with two methyl groups at nucleotide C1402, one at the N4 position (RsmH) and the other at the 2′-O-ribose (RsmI) (Kimura and Suzuki 2010) and to the decanucleotide Um5CACACCAUGp from U1406 to G1415 with one methyl group at nucleotide C1407 (Andersen and Douthwaite 2006), respectively.
FIGURE 3.

MALDI MS analysis of ArmA methylation in 16S rRNA fragment C1378–G1432. (A) Spectrum of RNase T1 rRNA from E. coli MM294/pGB2. The empirical m/z values are given above the peaks and match the theoretical masses (see below) to within 0.1 Da. (B) Spectrum from E. coli MM294/pGB2ΩarmA. Fragments at m/z 1305 and 3195 are missing and fused in the longer fragment at m/z 4483. (Inset) Enlargement of the spectrum around m/z 4480. Spectrum of RNase T1 rRNA from (C) E. coli MM294▵rsmF/pGB2 and (D) E. coli MM294▵rsmF/pGB2ΩarmA. Fragments at m/z 1305 and 3181 are missing and are fused in the longer fragment at m/z 4469. (Inset) Enlargement of the spectrum around m/z 4460. (E) MALDI tandem MS of RNase A rRNA from E. coli MM294/pGB2ΩarmA. The fragment at m/z 986 corresponding to GCCp fragment (positions 1401–1403) was selected and subjected to further fragmentation. Peaks corresponding to mCCp (Z2), Cp (W1), and GmC (c2) are indicated. These ions confirm that C1402 was monomethylated. (F) Theoretical masses of the monoisotopic E. coli RNase T1 fragments with 3′-linear phosphate (p). Only fragments that are tetranucleotides and larger are shown.
The rRNA isolated from MM294/pGB2ΩarmA could no longer be cleaved at G1405 by RNase T1, as indicated by the disappearance of the m/z 1305 (containing m4Cm1402) and 3195 (containing m5C1407) peaks and the appearance of a new peak at m/z 4483 (Fig. 3B). The latter corresponds to the C1402–G1415 fragment resulting from the fusion of the two smaller fragments but with only three methyl groups instead of four as expected (no peak at m/z 4497) (Fig. 3B). Resistance to RNase T1 cleavage at G1405 and the complete loss of the m/z 1305 and 3195 peaks indicated that ArmA methylation at G1405 is stoichiometric. In MM294/pGB2ΩarmA*, the rRNA fragment produced a spectrum pattern identical to that of MM294/pGB2. As already mentioned, in E. coli expression of m7G1405 resistance methyltransferases such as Sgm or RmtC was proposed to reduce endogenous RsmF methylation at C1407 (Cubrilo et al. 2009; Gutierrez et al. 2012). The MS spectrum of the fragment purified from MM294ΔrsmF showed peaks at m/z 1305 (containing m4Cm1402) and m/z 3181 (fragment U1406–G1415), the latter being 14 Da less than that from the rsmF+ strain due to loss of C1407 methylation (Fig. 3C). In MALDI MS analysis of the 16S rRNA fragment from MM294ΔrsmF/pGB2ΩarmA (Fig. 3D), peaks at m/z 1305 and 3181 were missing and replaced by a longer fragment at m/z 4469 corresponding to the C1402–G1415 sequence but with only two methyl groups. The m7G1405 modification must be present in the peak at m/z 4469 to prevent its cleavage by RNase T1; taken together, these data indicate that it is one of the two rRNA methylations at C1402 that is missing rather than that at C1407 as previously suggested (Cubrilo et al. 2009; Gutierrez et al. 2012).
To further confirm that ArmA blocks C1402 and not C1407 methylation, the rRNA fragment isolated from E. coli MM294/pGB2ΩarmA was partially digested with RNase A. The peak at m/z 986 with the sequence GCCp (positions 1401–1403) was selected and subjected to MALDI tandem MS fragmentation. Presence of peaks corresponding to mCCp (Z2), Cp (W1), and GmC (c2) (Fig. 3E) confirmed that C1402 was monomethylated, suggesting that the resistance methyltransferase interferes with one of the endogenous methyltransferases at this position. Fragmentation of the m/z 986 ions from MM294/pGB2ΩarmA and MM294ΔrsmF/pGB2ΩarmA provided identical spectra. Two endogenous methylations occur at C1402: N4-methylation (RsmH) and 2′-O-methylation (RsmI), but their sequential order is not known (Wei et al. 2012).
Recently, in Pseudomonas aeruginosa, it was shown that within the decoding center of 16S rRNA only endogenous methylation due to orthologs of RsmH, RsmI (m4Cm1402), and RsmE (m3U1498) is occurring and that expression of resistance methyltransferase RmtD, modifying m7G1405, impedes one of the methylations at C1402 without specifying which of RsmH or RsmI was blocked (Gutierrez et al. 2013).
To determine which activity, RsmH or RsmI, was blocked by ArmA, we performed MALDI MS of 16S rRNA isolated from E. coli BW25113, a strain from which single knockouts BW25113▵rsmH and BW25113▵rsmI (Baba et al. 2006) and the double knockout BW25113▵rsmH▵rsmI (Kimura and Suzuki 2010) are available. Plasmids pGB2 and pGB2ΩarmA were introduced in each strain. In BW25113 the fragment produced the expected MS spectrum with peaks at m/z 1305 (containing m4Cm1402) and 3195 (containing m5C1407) (Supplemental Fig. S1A). In the MS spectrum from E. coli BW25113/pGB2ΩarmA, peaks at m/z 1305 and 3195 were missing and combined in the longer sequence at m/z 4483 containing only three methyl groups (Supplemental Fig. S1B). The spectrum from E. coli BW25113▵rsmH/pGB2ΩarmA showed the disappearance of peaks at m/z 1291 (containing Cm1402) and 3195 (containing m5C1407), which were replaced by a longer fragment at m/z 4469 corresponding to the C1402–G1415 sequence with only two methyl groups instead of three (RsmI, RsmF, and ArmA) (Supplemental Fig. S1C,D). In the spectrum BW25113▵rsmH▵rsmI/pGB2ΩarmA, peaks at m/z 1277 (containing unmethylated C1402) and 3195 were missing and fused in a longer fragment at m/z 4469 corresponding to the C1402–G1415 sequence with two methyl groups due to RsmF and ArmA, as expected (Supplemental Fig. S1E,F). In contrast, the spectrum from BW25113▵rsmI/pGB2ΩarmA showed that the peaks at m/z 1291 (containing m4C1402) and 3195 were missing and replaced by a longer fragment at m/z 4483 corresponding to the C1402–G1415 sequence with, as expected, three methyl groups due to RsmH, RsmF, and ArmA (Supplemental Figs. S1G, S2). Taken together, these data indicate that ArmA interferes with RsmI without affecting the activity of RsmH and RsmF.
It is possible that ArmA, which operates late in the 30S ribosomal biogenesis pathway, recognizes as a substrate the same 30S intermediary as RsmI impeding methylation at C1402. Site-directed hydroxyl radical probing failed to define the regions of 16S rRNA involved in the interaction with Sgm and ArmA (Husain et al. 2010; Zarubica et al. 2011). Nucleotides found to be protected or deprotected in the presence of the methyltransferases are scattered over a wide area, suggesting that ArmA and Sgm induce global conformational changes in the 30S ribosomal subunit that allow both enzymes to access the buried nucleotide G1405. (Husain et al. 2010). Zarubica et al. (2011) hypothesize that the ArmA family of enzymes exploits a labile property of the mature 30S ribosomal subunit formation, rather than an intermediate on the pathway to subunit formation, to gain access to G1405. This disorganization may impede, somehow, access of RsmI to its target. In addition, lack of methylation at C1402 was also demonstrated to be disadvantageous for E. coli, imposing a delay in cell growth and altering ribosome functioning (Kimura and Suzuki. 2010). However, we cannot conclude if the high fitness cost imposed by ArmA to E. coli MM294 is a direct consequence of methylation at G1405, an indirect effect of interference with RsmI methylation at C1402, or a mix of both.
NpmA interferes with endogenous methylation at C1407
To study if the resistance methyltransferase NpmA also interferes with endogenous methylation at C1402 or C1407, we performed MALDI MS of 16S rRNA digested with RNase T1 isolated from MM294/pGB2ΩnpmA (Fig. 4). The peak observed at m/z 1305 (containing m4Cm1402) indicates that NpmA did not interfere with RsmI and RsmH. In contrast, the 16S rRNA sequence UCACACCAUGp from U1406–G1415 containing the methylation sites of RsmF (m5C1407) and NpmA (m1A1408) was monomethylated with an m/z 3195 (Fig. 4A). Putative interference between NpmA and RsmF at their respective adjacent targets was studied by MALDI MS of the 16S rRNA fragment isolated from MM294ΔrsmF/pGB2ΩnpmA. Again, a peak at m/z 3195 indicating a singly methylated C1406–G1415 fragment was observed (Supplemental Fig. S3A). To confirm interference, rRNA isolated from MM294/pGB2ΩnpmA was partially digested with RNase A and the peak corresponding to m/z 970 with the monomethylated sequence CACp (1407–1409) was analyzed by MALDI tandem MS fragmentation. The presence of fragments corresponding to mACp (Y2), CmA (a2), and C (d1), together with that corresponding to the methylated adenine (mA at m/z 148), confirmed that A1408 was methylated but not C1407 (Fig. 4B). MALDI MS performed on the 16S rRNA fragment from MM294/pGB2ΩnpmA* showed a peak at m/z 3195 due to RsmF modification (Supplemental Fig. S3B), confirming that methylation at A1408 blocks methylation at adjacent C1407.
FIGURE 4.

MALDI MS analysis of NpmA methylation in 16S rRNA fragment C1378–G1432. (A) Spectrum of RNase T1 rRNA from E. coli MM294/pGB2ΩnpmA. The empirical m/z values are given above the peaks and match the theoretical masses (see below) to within 0.1 Da. (Inset) Higher mass region of rRNA showing the monomethylated C1406–G1415 fragment. (B) MALDI tandem MS of RNase A nucleotides from E. coli MM294/pGB2ΩnpmA. The CACp fragment (positions 1407–1409) at m/z 970 was selected and subjected to further fragmentation. Peaks corresponding to C (d1), CmA (a2), and mACp (Y2) are indicated. (C) Theoretical masses of the monoisotopic E. coli RNase T1 fragments with 3′-linear phosphate (p). Only fragments that are tetranucleotides and larger are shown.
As opposed to ArmA, the interaction of NpmA with the 30S subunit does not induce any global structural rearrangement but rather only local changes (Husain et al. 2011). It has been proposed that nucleotide A1408 may be flipped out and more solvent-accessible in complex with NpmA than in the 30S subunit before modification (Husain et al. 2011). Hence, it is likely that interference between NpmA and RsmF, which methylate adjacent nucleotides, could be due to a clash between exogenous and endogenous methyltransferases that affects access to their target bases A1408 and C1407. Production of NpmA and lack of RsmF does not entail a fitness cost to the host. This is consistent with the observation that NpmA, which impedes the activity of RsmF, has no consequences on host fitness.
Alteration of the methylation pattern at the A-site affects translation accuracy
The functional role of the decoding site in rRNA has been generally analyzed by examining the effect of mutations on defined ribosomal functions. It was shown that mutations in the decoding center promote stop codon readthrough and frameshifting during elongation (O'Connor et al. 1997). Lack of post-transcriptional modification also affects translation initiation, frameshifting, and codon readthrough (Kimura and Suzuki 2010; Husain et al. 2011). However, to the best of our knowledge, the effect of resistance methylation at G1405 and A1408 on ribosome function has not yet been studied. To evaluate fidelity of initiation, reading frame maintenance, and stop readthrough in the presence of methylation at G1405 or A1408, the Renilla/Firefly luciferases reporter system (Kimura and Suzuki 2010) was used. Plasmids pQE-Luc (Supplemental Table S1) were introduced into E. coli MM294 and MM294▵rsmF, and the consequence of the production of ArmA, ArmA*, and NpmA was measured by dividing the chemiluminescence of Firefly luciferase (FLuc) by that of Renilla luciferase (RLuc) and compared with MM294/pGB2 (relative F/R value) (Fig. 5). No differences were observed for translation initiation at AUG and AUU codons for both ArmA and NpmA. A 40% increase (P < 0.01) in UGA readthrough efficiency was observed in E. coli MM294/pGB2ΩarmA as opposed to MM294/pGB2ΩarmA*. A nearly identical increase in UGA readthrough has been reported in E. coli in the absence of RsmI, which is responsible for 2′-O-methylation at C1402 (Kimura and Suzuki 2010). Lack of RsmI is also associated with a 30% increase in +1 frameshift (Kimura and Suzuki 2010) that was not observed in the presence of ArmA (Fig. 5). We do not know if the ArmA-induced increase in UGA readthrough is due to the absence of methylation at C1402 by RsmI, to the extra methylation at G1405, or to the combination of both.
FIGURE 5.

Translational accuracy in E. coli MM294 in the presence of ArmA, ArmA*, or NpmA or in the absence of RsmF. Strain MM294 harboring pGB2 (white), pGB2ΩarmA (strips), pGB2ΩarmA* (gray), or pGB2ΩnpmA (dotted) was transformed with plasmids encoding Renilla luciferase (RLuc) starting with the AUG codon and Firefly luciferase (FLuc) starting with codon AUG or AUU on a single transcript. Translation initiation efficiency was measured by dividing the chemiluminescence of FLuc by that of RLuc and normalizing the ratio to that of MM294/pGB2 (relative F/R value). To measure the efficiency of +1 reading frame maintenance and UGA readthrough in the presence of ArmA or NpmA, fusion constructs containing the RLuc and FLuc open reading frames separated by short windows containing a stop codon (UGA) or a frameshift site (+1) were used. The efficiency of translation initiation at AUG and the accuracy of UGA readthrough in the absence of RsmF (black) were also determined. In this case, the ratio FLuc/RLuc was normalized to the ratio FLuc/RLuc obtained for E. coli MM294. The values are the means of three independent experiments carried out in quintuplates with error deviations. (*) Significant difference (P< 0.01) of the mean value.
In the presence of NpmA, a decrease in the +1 reading frame maintenance (16%) and in UGA readthrough (20%) was observed (Fig. 5). We further evaluated the efficiency of UGA readthrough in the MM294ΔrsmF strain and found a diminution of 24% (Fig. 5). These data indicate that both C1407 and A1408 are important for translation accuracy, and it is likely that the role played by NpmA results from impeachment of RsmF activity.
Plasmid pIP1204-mediated ArmA has an impact on the host
The armA gene was first detected in 2003 in a multiresistant Klebsiella pneumoniae clinical isolate (Galimand et al. 2003). It is located on the self-transferable IncL/M plasmid pIP1204 of ∼90 kb and part of functional composite transposon Tn1548 (Galimand et al. 2005). Since then it has disseminated worldwide in Enterobacteriaceae, Pseudomonas aeruginosa, and Acinetobacter baumannii, where it is borne by large (57–196 kb) transferable plasmids belonging to diverse incompatibility groups such as IncA/C, IncF, IncL/M, and IncN (for review, see Wachino and Arakawa 2012). Spread of armA linked with genes for resistance to β-lactams and fluoroquinolones results thus from the combinatorial genetics of Tn1548-like elements’ transposition and plasmid conjugation.
We therefore studied the biological cost of ArmA when mediated by plasmid pIP1204 by comparing the growth rate of E. coli MM294/pIP1204 or MM294/pIP1204▵armA in which armA was deleted by allele replacement. The growth rate of E. coli MM294/pIP1204 was significantly decreased compared with that of MM294 (relative growth rate of 0.89 ± 0.09; P < 0.01), but deletion of armA resulted in normal growth (relative growth rate of 0.98 ± 0.04). Compared with MM294/pIP1204, the relative growth rate of MM294/pIP1204▵armA was 1.10 ± 0.04 (P < 0.01), indicating that it is expression of ArmA that is responsible for the fitness cost. Similar results were obtained in MM294▵rsmF (data not shown).
MALDI MS analysis of the 16S rRNA C1378–G1432 sequence from MM294/pIP1204 digested by RNase T1 showed the presence of two peaks corresponding to the C1402–G1415 sequence (Supplemental Fig. S4A)—one at m/z 4497 with four methyl groups: two at C1402 (RsmH and RsmI), one at C1407 (RsmF), and another one at G1405 (ArmA), and the second peak at m/z 4483 with three methylations as in MM294/pGB2ΩarmA (Fig. 3B). The presence of the C1402–G1415 fragments resistant to cleavage by RNase T1 indicated that ArmA methylation is complete but that one of these endogenous modifications is no longer stoichiometric, as already shown for other G1405 methyltransferases (Cubrilo et al. 2009; Gutierrez et al. 2012). To discriminate between partial interference at position C1402 or C1407, pIP1204 was introduced into MM294ΔrsmF. MALDI MS of 16S rRNA isolated from MM294ΔrsmF/pIP1204 showed peaks at m/z 4483 (with three methyl groups) and at m/z 4469 (containing two methyl groups) corresponding to the C1402–G1415 sequence (Supplemental Fig. S4B). There was full inhibition of RsmI methylation at position C1402 in MM294/pGB2ΩarmA (Fig. 3B), whereas in MM294/pIP1204, there was only a partial block of methylation at C1402 (Supplemental Fig. S4A). These differences could be accounted for by a gene dosage effect due to the different copy number per genome of the two plasmids. The level of expression of the armA gene was measured by qRT-PCR, and mRNA of the constitutively expressed single-copy housekeeping gene rpoB was used as a control. Expression of armA in MM294/pGB2ΩarmA was 7.8 ± 1.3-fold (P< 0.01) higher than that in MM294/pIP1204.
CONCLUSION
Aminoglycoside resistance methyltransferases modify the functionally critical A-site in 16S rRNA, a region already heavily methylated by endogenous enzymes (Basturea et al. 2012). It was previously shown that methyltransferases Sgm and RmtC, which confer resistance by modification of G1405, impede RsmF methylation at C1407 (Cubrilo et al. 2009; Gutierrez et al. 2012).
Our study further documents examples of clashes between exogenous resistance methyltransferases and endogenous enzymes. We provide evidence that acquired A1408 methyltransferase NpmA impedes housekeeping RsmF, which methylates adjacent C1407 (Fig. 4), and that the G1405 methyltransferase ArmA interferes with endogenous C1402 RsmI (Supplemental Figs. S1, S2). Interestingly, the functional consequences for the cell of the defect in endogenous methylation differ: In our experimental conditions, lack of C1407 methylation did not alter cell growth (Table 2), whereas that at C1402 resulted, as previously shown, in growth impairment (Kimura and Suzuki 2010).
Surprisingly, ArmA has disseminated into numerous bacterial genera worldwide (Galimand et al. 2005; Wachino and Arakawa 2012), whereas a single E. coli clinical isolate producing NpmA has been reported (Wachino et al. 2007). This contrasting epidemiology could be accounted for by the fact that the armA gene, which is part of the functional transposon Tn1548 (Galimand et al. 2005) carried by conjugative plasmid pIP1204 (Galimand et al. 2003), is physically linked to other resistance genes, in particular, to those for resistance to β-lactams and fluoroquinolones (Périchon et al. 2007; Wachino and Arakawa 2012), the two drug classes the most extensively used. The fitness cost resulting from constitutive expression of ArmA could diminish its persistence under nonselective conditions, but its dissemination is favored under selective pressure by various classes of antibiotics, and its location on two superposed mobile genetic elements could explain its worldwide distribution. On the contrary, NpmA was found to be located on a plasmid that does not encode other resistances (Wachino et al. 2007). In this genetic context, lack of coresistance results in the absence of coselection, thus limiting its dissemination. However, since expression of npmA does not entail a cost to the host, the gene can persist in the absence of selection.
Extensive analysis of 16S rRNA from isogenic E. coli strains by MALDI MS and MALDI tandem MS fragmentation led to the demonstration that it was not RsmF, as has been proposed (Cubrilo et al. 2009; Gutierrez et al. 2012), but one of the two C1402 methylations that was blocked by ArmA (Fig. 3E; Supplemental Fig. S2). Dimethylation at C1402 has a critical role in the function of the P-site, and, using a dual luciferase reporter assay, Kimura and Suzuki (2010) showed that deletion of rsmH and rsmI affects efficiency of non-AUG initiation and the fidelity of translation. ArmA, which modifies G1405, impedes methylation at C1402 by RsmI (Fig. 3E), increases UGA readthrough (Fig. 5) and has a fitness cost for the host (Fig. 2). In contrast, NpmA, which methylates A1408 and blocks the activity of RsmF at adjacent C1407 (Fig. 4), decreases translation accuracy and UGA readthrough, as observed for the ▵rsmF strain (Fig. 5), but confers only a slight growth disadvantage to the host (Fig. 2).
In numerous instances, the study of antibiotic resistance mechanisms has contributed to our understanding of bacterial physiology. This work on the interference between resistance and endogenous 16S rRNA methyltransferases has contributed to a better characterization of the role of these housekeeping enzymes in the fine-tuning of the ribosomal decoding center with consequences on translation accuracy and cell fitness.
MATERIALS AND METHODS
Bacterial strains and plasmids
The origin and properties of the E. coli strains and plasmids are described in Supplemental Table S1. Bacteria were grown in Luria-Bertani (LB) broth at 37°C. To study the putative biological cost associated with expression of resistance, the armA and npmA genes and their inactive point mutants armA* and npmA* were cloned with their own promoters in the stable low-copy-number plasmid pGB2 (Churchward et al. 1984; Supplemental Table S1) into E. coli. Methyltransferase mutants unable to bind the S-adenosyl-L-methionine cofactor were constructed by site-directed mutagenesis; for ArmA*, Lys180 was changed to Ala (Schmitt et al. 2009), and for NpmA, Asp30 and Asp55 were both changed to Ala (Husain et al. 2011).
E. coli MM294▵rsmF was constructed by allele replacement using plasmid pKOBEGA (Chaveroche et al. 2000). Briefly, an FRT-flanked tetracycline-resistance gene was amplified using plasmid p1500 DNA as a template with the 70-nucleotide (nt) 5′-primer (▵rsmF P1) composed of 50 nt of the region upstream of rsmF and 20 nt of p1500 and the 70-nt 3′-primer (▵rsmF P2) consisting of 50 nt downstream from rsmF (including the stop codon) and 20 nt from p1500 (Supplemental Table S2). The resulting PCR fragment was used to delete the rsmF gene with selection on tetracycline (Chaveroche et al. 2000). Plasmid pCP20 encoding the FLP recombinase was then introduced to eliminate the FRT-flanked tetracycline gene, as described (Cherepanov and Wackernagel 1995). Deletion was confirmed by PCR using external primers pair ▵rsmF up and ▵rsmF down (Supplemental Table S2).
E. coli MM294/pIP1204▵armA and E. coli MM294▵rsmF/pIP1204▵armA were constructed similarly by the allele replacement using plasmid pKOBEG (Chaveroche et al. 2000). The PCR fragment obtained with the 70-nt 5′-primer (▵armA P1) and 3′-primer (▵armA P2) (Supplemental Table S2) was used to delete the armA gene with selection on tetracycline. The FRT-flanked tetracycline gene was next eliminated as described (Cherepanov and Wackernagel 1995). Deletion was confirmed by PCR using external primers pair ΔarmA up and ΔarmA down (Supplemental Table 2) and by the loss of resistance of 4,6-disubstituted deoxystreptamines.
Inactivation of the araB gene in E. coli MM294 and MM294ΔrsmF was carried out as described (Marcusson et al. 2009). In the absence of araB, cells grown in MacConkey agar supplemented with 1% arabinose are white, while araB+ cells are pink.
Plasmid pGB2ΩParmA and pGB2ΩPnpmA were constructed by successive PCR amplifications. In a first PCR, ParmA and PnpmA were amplified from pGB2ΩarmA and pGB2ΩnpmA, respectively. A second PCR enabled a gfpmut1 fragment to be obtained using plasmid pAT505 (Grillot-Courvalin et al. 1998) as a template. In a second step, the resulting PCR products were then linked by overlapping PCR to obtain ParmAgfpmut1 and PnpmAgfpmut1 fragments that were cloned in pGB2 (Supplemental Table S1).
Growth competitions and growth rate measurements
Growth rates were determined in microplates coupled to a spectrophotometer Multiscan (Thermoscientific). Strains were grown overnight at 37°C, and the cultures were diluted to inoculate ∼1 × 103 bacteria into 200 µL of LB in a 96-well microplate that was incubated at 37°C with shaking. Absorbance was measured at 600 nm every 3 min. Each culture was replicated three times in the same microplate. Growth rates, measured in five independent experiments, were determined at the beginning of the exponential phase, and relative growth rates were calculated as the ratio of the growth rate of the strains versus that of the E. coli parental strain.
Competition experiments were performed in cocultures with ∼103 CFU of the susceptible and the resistant strains mixed in 10 mL of LB at an initial ratio of 1:1 and grown for 12 h (about 20 generations). The mixed culture was transferred to fresh LB by 106-fold serial dilution every 12 h for up to six passages. The total number of viable cells was determined at the end of every transfer on nonselective plates, and the proportion of resistant strains was determined by replica-plating between 500 and 1000 colonies on LB and LB supplemented with 50 µg/mL kanamycin or on LB and McConkey agar supplemented with 1% arabinose, depending on the pair of strains studied. The competition index (CI) was calculated as the CFU ratio of the resistant and susceptible strains (R/S) at time (t1) divided by the same ratio at time (t0), and the selection coefficient s was then calculated as the slope of the following linear regression model: s = ln(CI)/[t × ln(2)], where t is the number of generations. The fitness of the susceptible strain was set to 1, and the relative fitness of the resistant strains was determined as 1 + s (Foucault et al. 2009).
Luciferase reporter assays
E. coli MM294 and MM294ΔrsmF bearing pGB2, pGB2ΩarmA, pGB2ΩarmA*, or pGB2ΩnpmA were transformed with plasmids pQE-Luc (AUG), pQE-Luc (AUU), pQE-Luc (+1), and pQE-Luc (UGA), and translation accuracy was determined as described previously (Kimura and Suzuki 2010). To evaluate the efficiency of translation initiation at non-AUG codons, the reporter genes Rluc (Renilla), starting with AUG codon, and Fluc (Firefly) starting with codons AUG or AUU on a single transcript were used. To measure the efficiency of stop codon readthrough and reading frame maintenance, the reporter constructs carry the Rluc and Fluc genes separated by short windows containing a stop codon (UGA) or a frameshift site (+1). Bacterial lysates were analyzed in a GLOMAX96 Microplate Luminometer using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. The efficiency of AUG and AUU initiation, UGA readthrough, and +1 reading frame maintenance was measured by dividing the chemiluminescence of Firefly luciferase by that of Renilla luciferase (F/R value) and referring to the F/R value of the indicated control.
Matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) MS analysis of 16S rRNA
Total rRNA was extracted from ribosomal particles isolated from E. coli MM294, MM294▵rsmF, BW25113, BW25113▵rsmH, BW25113▵rsmI, or BW25113▵rsmH▵rsmI expressing ArmA, ArmA*, NpmA, or NpmA* grown in the absence of aminoglycoside. The 16S rRNA sequence from C1378 to G1432 was isolated by hybridization to a complementary 55-mer deoxyoligonucleotide as described (Fig. 1; Andersen et al. 2004). Fifty picomoles of total rRNA was incubated with 500 pmol of the deoxyoligonucleotide for 5 min at 70°C in 100 µL of hybridization buffer (250 mM HEPES, 500 mM KCl at pH 7.0), followed by slow cooling over 2 h to 45°C. Unhybridized rRNA was digested with 20 units of Mung bean nuclease and 0.5 µg of RNase A, and the rRNA fragment protected by the deoxyoligonucleotide was isolated by gel electrophoresis as described (Andersen et al. 2004). Finally, 1 µg of the rRNA fragment was digested with 20 units of RNase T1 for 4 h at 37°C in 10 µL and 5 µL of 0.5 M 3-hydroxypicolinic acid (3-HPA) or with 2 µg of RNase A for 4 h at 37°C in 10 µL and 5 µL of 100 mM NH4OAc. Cyclic phosphates were hydrolyzed with 2.5 µL of 0.5 M HCl for 30 min at room temperature. Samples were dried and rehydrated in 10 µL of H2O. One microliter of the digest was mixed with 9 µL of MALDI matrix (3-HPA), and 1 µL of the mixture was spotted on the MALDI plate and air-dried (“dried droplet method”). MALDI-TOF MS and MALDI-TOF/TOF MS/MS analyses were performed directly on the digestion products using a Voyager DE-STR MALDI-TOF mass spectrometer and a 5800 MALDI-TOF/TOF Analyzer mass spectrometer (both AB Sciex), respectively. Acquisitions were performed in negative ion mode. For MS/MS experiments, precursor ions were accelerated at 8 kV, and the MS/MS spectra were acquired using 1 keV of collision energy with CID gas (argon) at a pressure of 3.5 × 10−6 Torr. MS data were processed using DataExplorer 4.4 (AB Sciex). To identify the methylation sites in the trinucleotides GCCp (positions 1401–1403) and CACp (positions 1407–1409), the scheme of McLuckey (McLuckey et al. 1992) was followed. For calculation of fragment masses, the NUKE software was used (Institute for Medical Physics and Biophysics, University of Münster, Germany).
RNA isolation and quantitative reverse transcriptase PCR (qRT-PCR)
Total RNA from E. coli strains MM294/pGB2ΩarmA, MM294/pGB2ΩarmA*, or MM294/pIP1204 was extracted from exponentially grown bacterial cells in antibiotic-free medium (OD600 nm = 0.8–0.9) using TRIzol Reagent (Invitrogen). RNA samples were treated with the TURBO DNA-free Kit (Applied Biosystems) to remove any genomic DNA carryover. cDNA synthesis and qRT-PCR were performed with a LightCycler RNA amplification kit SYBR Green I (Roche Diagnostic) and 0.5 µM gene-specific primers for armA (armA For/armA Rev) and rpoB (rpoB For/rpoB Rev) (Supplemental Table S2) according to the manufacturer's instructions. Amplification and detection of specific products were performed using the LightCycler sequence detection system (Roche) with the following cycle profile: 1 cycle at 95°C for 30 sec, followed by 45 cycles at 95°C for 5 sec, 55°C for 10 sec, and 72°C for 20 sec. Each experiment was performed in duplicate at least twice independently.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported in part by the European Union FP7-PAR grant that included a fellowship in support to V.S.L. and by an unrestricted grant from Reckitt Benckiser. We thank T. Suzuki for providing the Dual Luciferase System and strains BW25113▵rsmI and BW25113▵rsmIΔrsmH, the NBRP-E. coli at NIG for providing strain BW25113▵rsmH, and Peter E. Reynolds for reading of the manuscript.
REFERENCES
- Andersen NM, Douthwaite S 2006. YebU is a m5C methyltransferase specific for 16 S rRNA nucleotide 1407. J Mol Biol 359: 777–786 [DOI] [PubMed] [Google Scholar]
- Andersen TE, Porse BT, Kirpekar F 2004. A novel partial modification at C2501 in Escherichia coli 23S ribosomal RNA. RNA 10: 907–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DI, Hughes D 2010. Antibiotic resistance and its cost: Is it possible to reverse resistance? Nat Rev Microbiol 8: 260–271 [DOI] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol Syst Biol 2: 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann BJ 1972. Pedigrees of some mutant strains of Escherichia coli K-12. Bacteriol Rev 36: 525–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basturea GN, Rudd KE, Deutscher MP 2006. Identification and characterization of RsmE, the founding member of a new RNA base methyltransferase family. RNA 12: 426–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basturea GN, Dague DR, Deutscher MP, Rudd KE 2012. YhiQ is RsmJ, the methyltransferase responsible for methylation of G1516 in 16S rRNA of E. coli. J Mol Biol 415: 16–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaveroche MK, Ghigo JM, d'Enfert C 2000. A rapid method for efficient gene replacement in the filamentous fungus Aspergillus nidulans. Nucleic Acids Res 28: e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov PP, Wackernagel W 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158: 9–14 [DOI] [PubMed] [Google Scholar]
- Churchward G, Belin D, Nagamine Y 1984. A pSC101-derived plasmid which shows no sequence homology to other commonly used cloning vectors. Gene 31: 165–171 [DOI] [PubMed] [Google Scholar]
- Cubrilo S, Babic F, Douthwaite S, Maravic Vlahovicek G 2009. The aminoglycoside resistance methyltransferase Sgm impedes RsmF methylation at an adjacent rRNA nucleotide in the ribosomal A site. RNA 15: 1492–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foucault ML, Courvalin P, Grillot-Courvalin C 2009. Fitness cost of VanA-type vancomycin resistance in methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 53: 2354–2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galimand M, Courvalin P, Lambert T 2003. Plasmid-mediated high-level resistance to aminoglycosides in Enterobacteriaceae due to 16S rRNA methylation. Antimicrob Agents Chemother 47: 2565–2571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galimand M, Sabtcheva S, Courvalin P, Lambert T 2005. Worldwide disseminated armA aminoglycoside resistance methylase gene is borne by composite transposon Tn1548. Antimicrob Agents Chemother 49: 2949–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillot-Courvalin C, Goussard S, Huetz F, Ojcius DM, Courvalin P 1998. Functional gene transfer from intracellular bacteria to mammalian cells. Nat Biotechnol 16: 862–866 [DOI] [PubMed] [Google Scholar]
- Gutierrez B, Escudero JA, San Millan A, Hidalgo L, Carrilero L, Ovejero CM, Santos-Lopez A, Thomas-Lopez D, Gonzalez-Zorn B 2012. Fitness cost and interference of Arm/Rmt aminoglycoside resistance with the RsmF housekeeping methyltransferases. Antimicrob Agents Chemother 56: 2335–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez B, Douthwaite S, Gonzalez-Zorn B 2013. Indigenous and acquired modifications in the aminoglycoside binding sites of Pseudomonas aeruginosa rRNAs. RNA Biol 10: 1324–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain N, Tkaczuk KL, Tulsidas SR, Kaminska KH, Cubrilo S, Maravic-Vlahovicek G, Bujnicki JM, Sivaraman J 2010. Structural basis for the methylation of G1405 in 16S rRNA by aminoglycoside resistance methyltransferase Sgm from an antibiotic producer: A diversity of active sites in m7G methyltransferases. Nucleic Acids Res 38: 4120–4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain N, Obranic S, Koscinski L, Seetharaman J, Babic F, Bujnicki JM, Maravic-Vlahovicek G, Sivaraman J 2011. Structural basis for the methylation of A1408 in 16S rRNA by a panaminoglycoside resistance methyltransferase NpmA from a clinical isolate and analysis of the NpmA interactions with the 30S ribosomal subunit. Nucleic Acids Res 39: 1903–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Suzuki T 2010. Fine-tuning of the ribosomal decoding center by conserved methyl-modifications in the Escherichia coli 16S rRNA. Nucleic Acids Res 38: 1341–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMarre JM, Locke JB, Shaw KJ, Mankin AS 2011. Low fitness cost of the multidrug resistance gene cfr. Antimicrob Agents Chemother 55: 3714–3719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou GF, Yoshizawa S, Courvalin P, Galimand M 2006. Aminoglycoside resistance by ArmA-mediated ribosomal 16S methylation in human bacterial pathogens. J Mol Biol 359: 358–364 [DOI] [PubMed] [Google Scholar]
- Magnet S, Blanchard JS 2005. Molecular insights into aminoglycoside action and resistance. Chem Rev 105: 477–498 [DOI] [PubMed] [Google Scholar]
- Marcusson LL, Frimodt-Moller N, Hughes D 2009. Interplay in the selection of fluoroquinolone resistance and bacterial fitness. PLoS Pathog 5: e1000541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLuckey SA, Van Berkel GJ, Glish GL 1992. Tandem mass spectrometry of small multiply charged oligonucleotides. J Am Soc Mass Spectrom 3: 60–70 [DOI] [PubMed] [Google Scholar]
- O'Connor M, Thomas CL, Zimmermann RA, Dahlberg AE 1997. Decoding fidelity at the ribosomal A and P sites: Influence of mutations in three different regions of the decoding domain in 16S rRNA. Nucleic Acids Res 25: 1185–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Périchon B, Courvalin P, Galimand M 2007. Transferable resistance to aminoglycosides by methylation of G1405 in 16S rRNA and to hydrophilic fluoroquinolones by QepA-mediated efflux in Escherichia coli. Antimicrob Agents Chemother 51: 2464–2469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister P, Hobbie S, Vicens Q, Bottger EC, Westhof E 2003. The molecular basis for A-site mutations conferring aminoglycoside resistance: Relationship between ribosomal susceptibility and X-ray crystal structures. Chembiochem 4: 1078–1088 [DOI] [PubMed] [Google Scholar]
- Qin D, Liu Q, Devaraj A, Fredrick K 2012. Role of helix 44 of 16S rRNA in the fidelity of translation initiation. RNA 18: 485–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy-Chaudhuri B, Kirthi N, Culver GM 2010. Appropriate maturation and folding of 16S rRNA during 30S subunit biogenesis are critical for translational fidelity. Proc Natl Acad Sci 107: 4567–4572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt E, Galimand M, Panvert M, Courvalin P, Mechulam Y 2009. Structural bases for 16 S rRNA methylation catalyzed by ArmA and RmtB methyltransferases. J Mol Biol 388: 570–582 [DOI] [PubMed] [Google Scholar]
- Wachino J, Arakawa Y 2012. Exogenously acquired 16S rRNA methyltransferases found in aminoglycoside-resistant pathogenic Gram-negative bacteria: An update. Drug Resist Updat 15: 133–148 [DOI] [PubMed] [Google Scholar]
- Wachino J, Shibayama K, Kurokawa H, Kimura K, Yamane K, Suzuki S, Shibata N, Ike Y, Arakawa Y 2007. Novel plasmid-mediated 16S rRNA m1A1408 methyltransferase, NpmA, found in a clinically isolated Escherichia coli strain resistant to structurally diverse aminoglycosides. Antimicrob Agents Chemother 51: 4401–4409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Zhang H, Gao ZQ, Wang WJ, Shtykova EV, Xu JH, Liu QS, Dong YH 2012. Crystal and solution structures of methyltransferase RsmH provide basis for methylation of C1402 in 16S rRNA. J Struct Biol 179: 29–40 [DOI] [PubMed] [Google Scholar]
- Zarubica T, Baker MR, Wright HT, Rife JP 2011. The aminoglycoside resistance methyltransferases from the ArmA/Rmt family operate late in the 30S ribosomal biogenesis pathway. RNA 17: 346–355 [DOI] [PMC free article] [PubMed] [Google Scholar]