

The authors present the results of three independent mass spectrometry–based approaches to assess protein composition of precatalytic and catalytic spliceosomes. Of the 157 proteins identified, only a small subset remains unchanged during the B-to-C transition. Results are consistent across the three analytical approaches, including the least expensive one.
Keywords: spliceosome, quantitative proteomics, stable isotope labeling with amino acids in cell culture (SILAC), isobaric tags for relative and absolute quantification (iTRAQ), spectral count
Abstract
The spliceosome undergoes major changes in protein and RNA composition during pre-mRNA splicing. Knowing the proteins—and their respective quantities—at each spliceosomal assembly stage is critical for understanding the molecular mechanisms and regulation of splicing. Here, we applied three independent mass spectrometry (MS)–based approaches for quantification of these proteins: (1) metabolic labeling by SILAC, (2) chemical labeling by iTRAQ, and (3) label-free spectral count for quantification of the protein composition of the human spliceosomal precatalytic B and catalytic C complexes. In total we were able to quantify 157 proteins by at least two of the three approaches. Our quantification shows that only a very small subset of spliceosomal proteins (the U5 and U2 Sm proteins, a subset of U5 snRNP-specific proteins, and the U2 snRNP-specific proteins U2A′ and U2B′′) remains unaltered upon transition from the B to the C complex. The MS-based quantification approaches classify the majority of proteins as dynamically associated specifically with the B or the C complex. In terms of experimental procedure and the methodical aspect of this work, we show that metabolically labeled spliceosomes are functionally active in terms of their assembly and splicing kinetics and can be utilized for quantitative studies. Moreover, we obtain consistent quantification results from all three methods, including the relatively straightforward and inexpensive label-free spectral count technique.
INTRODUCTION
Proteomic analysis provides essential information about the function and regulation of protein complexes. Numerous mass spectrometry (MS) techniques have been developed to determine protein composition and their quantities (Aebersold and Mann 2003; Steen and Mann 2004; Ong and Mann 2005; Domon and Aebersold 2006; Yates et al. 2009; Bantscheff et al. 2012; Nikolov et al. 2012). To date, the relative quantification technique of stable-isotope labeling followed by MS has frequently been used to quantify proteins from tissues, from different functional states of a cell, from compartments or from pull-down experiments involving protein(–ligand) complexes. However, they have rarely been used to compare directly isolated native protein complexes that represent different functional states of a single molecular machine. Here, we examine the proteomes of isolated human precatalytic B and catalytically active C spliceosomal complexes by quantitative MS.
The spliceosome catalyzes eukaryotic pre-mRNA splicing to generate mature mRNAs and is a highly dynamic and complex macromolecular machine. Spliceosomes consist of one or more of the five uridine-rich small nuclear ribonucleoprotein particles (snRNPs; U1, U2, U5, the U4/U6 di-snRNP, and the U4/U6.U5 tri-snRNP), each of which contains U snRNA(s), a core set of seven Sm or LSm proteins, and numerous U snRNP-specific proteins. In vitro, several different functional states of the spliceosome can be distinguished; these are termed the E, A, B, Bact, and C complexes (Wahl et al. 2009). Transitions between these different states are often accompanied by dramatic rearrangements in the spliceosomal interactions. For instance, the precatalytic B complex, which contains the U4/U6.U5 tri-snRNP, undergoes structural rearrangements that destabilize the U1 and U4 snRNPs to form the activated Bact complex; this then catalyzes the first step of splicing to generate the catalytically active C complex, which in turn catalyzes the second step of splicing to produce the mature mRNA (Wahl et al. 2009). Determining the protein composition of each of the spliceosomal states has been a major goal in the past decade. Initial studies using active spliceosomes assembled in vitro revealed that in addition to the U snRNP-specific proteins, more than 100 proteins are involved in the splicing cycle (Jurica et al. 2002; Rappsilber et al. 2002; Zhou et al. 2002; Jurica and Moore 2003). More detailed studies focused on analyzing the protein composition of the isolated functional states of the A, B, Bact, and C complexes and the mRNPs in humans (Deckert et al. 2006; Behzadnia et al. 2007; Bessonov et al. 2008, 2010), yeast (B, Bact, and C complexes) (Fabrizio et al. 2009), and Drosophila (B and C complexes) (Herold et al. 2009). MS analyses have revealed that the human B and C complexes contain approximately 130 and 150 proteins, respectively. Of these, 105 proteins are stably associated with both complexes (Bessonov et al. 2008). The substantial change in composition from one functional spliceosomal state to another has been clearly shown by the determination of the protein compositions of isolated A, B, Bact, and C complexes, each of which differs remarkably from the others even when isolated under native conditions that allow close comparison (Deckert et al. 2006; Behzadnia et al. 2007; Bessonov et al. 2008, 2010; Fabrizio et al. 2009; Herold et al. 2009). By using a novel 2D gel electrophoresis system to quantify spliceosomal complexes, it has recently been shown that only 60–70 protein factors are moderately or highly abundant in the various spliceosomal complexes (Agafonov et al. 2011). In these previous studies, the protein components of isolated spliceosomes were analyzed in a so-called “semi-quantitative” manner by determining the number of sequenced peptides by MS (peptide count) or by measuring the intensity of stained proteins after 2D gel electrophoresis to determine the relative and absolute abundances, respectively, of the proteins (Deckert et al. 2006; Behzadnia et al. 2007; Merz et al. 2007; Bessonov et al. 2008, 2010; Fabrizio et al. 2009; Agafonov et al. 2011). We applied now here—for the first time—three independent MS-based quantification methods (SILAC metabolic labeling [Ong et al. 2002], iTRAQ chemical labeling [Ross et al. 2004], and the label-free spectral count [Liu et al. 2004]) in determining the protein quantities in purified spliceosomal B and C complexes.
We identified and quantified approximately 200 proteins in both the B and the C complex preparations; this represents >95% of the previously published proteomes of the B and C complexes (Deckert et al. 2006; Bessonov et al. 2008). Strikingly, only a few of the proteins identified are “core” components of both complexes, the quantities of which do not change upon transition from B-to-C complex. These were a subset of U5 snRNP proteins, the U2-specific proteins U2A′ and U2B′′, and the evolutionarily conserved Sm proteins of U5 and U2. Most proteins, in contrast, either joined or left the spliceosome during its transition from the precatalytic B complex to the catalytically active C complex. This study expands our previous investigation of the proteomes of spliceosomal B and C complexes and may further be extended to study the assembly kinetics of spliceosomal complexes in nuclear extract (NE).
RESULTS
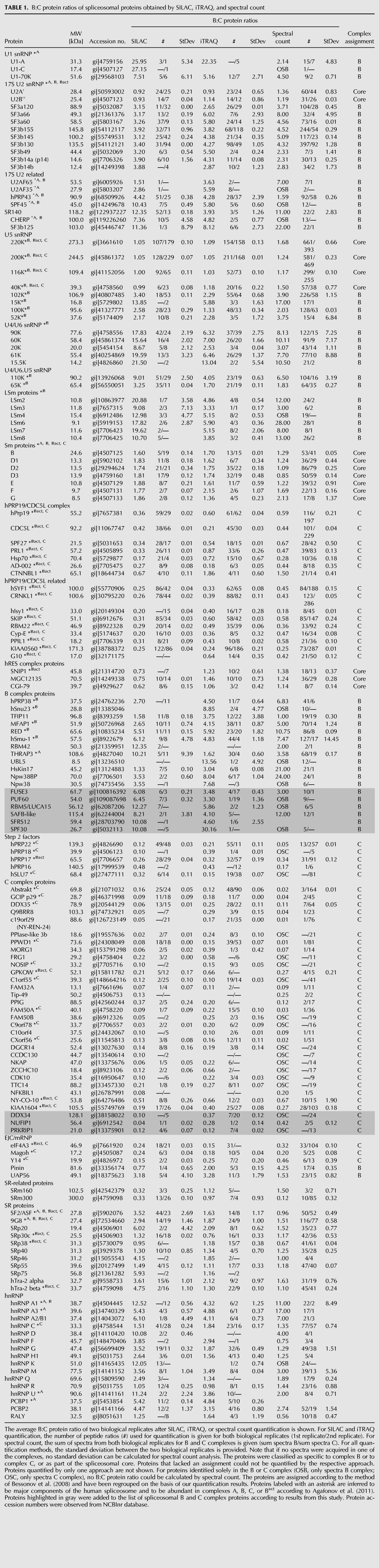
For our quantitative MS studies, we used the same experimental approach as Bessonov et al. (2008), namely, separation of the protein components of glycerol-gradient-purified spliceosomal complexes by 1D gel electrophoresis, and thus reduced the sample complexity to a similar extent (Bessonov et al. 2008). Importantly, as a prerequisite of such an MS-based quantitative analysis, namely, comparing the exact amount of natively purified assemblies, we benefit from the measurement of the amount of 32P-labeled pre-mRNA that is bound to the respective complexes. All quantitative MS-based analyses were performed with two biological replicates. We resumed the protein assignment previously introduced by Deckert et al. (2006), Bessonov et al. (2008), and Agafonov et al. (2011) and adjusted the protein classification of B and C complexes according to our quantification results (Table 1). In addition, we list some proteins that could not unambiguously be assigned to either of the two complexes, owing to their fluctuating quantification results (Supplemental Table 1). Finally, we identified and quantified several proteins that clearly show association with either the B or the C complex by all three approaches and were not classified as B- or C-specific proteins previously by Bessonov et al. (2008) or Agafonov et al. (2011); Table 1.
TABLE 1.
B:C protein ratios of spliceosomal proteins obtained by SILAC, iTRAQ, and spectral count
MS analysis of metabolically stable isotope-labeled (SILAC) spliceosomes
Metabolic labeling with stable isotopes (Ong et al. 2002) is considered to be the gold standard in protein quantification; as proteins are fully labeled, samples can be pooled at an early stage of sample preparation and cleavage of proteins with endoproteinase trypsin always leads to coeluting labeled peptide pairs that are analyzed in the MS. We prepared spliceosomes from HeLa NEs labeled with Lys+6, and Arg+10 (“heavy” NE) and spliceosomes prepared from NEs containing the nonlabeled amino acids Lys+0 and Arg+0 (“light” NE) (Fig. 1A). We had previously verified that the different SILAC NE preparations did not display significant differences in their overall protein abundance, as shown by LC-MS/MS analyses of peptides obtained from equivalent concentrations of the “light” or “heavy” NE proteins hydrolyzed in gel (Nikolov et al. 2011).
FIGURE 1.

Purification of the spliceosomal B and C complexes for proteomic analysis and iTRAQ and SILAC quantification. (A) B and C complexes were purified from “heavy” or “light” SILAC NEs, respectively. The complexes were allowed to assemble onto MS2-tagged pre-mRNA for 6 or 180 min, respectively. The complexes were then isolated by gradient centrifugation and affinity purification; isolated complexes were pooled in equal amounts; the proteins were separated by gel electrophoresis; and the peptides generated were analyzed by LC-MS/MS. (B) B and C complexes were purified from “normal” (light) NE. The proteins were separated by gel electrophoresis, and after in-gel digestion, the peptides generated were analyzed by LC-MS/MS for proteomic analysis, or the peptides generated from the B complex were labeled with iTRAQ reagent 115 and those generated from the C complex were labeled with iTRAQ reagent 116. After pooling, the samples were analyzed by LC-MS/MS (iTRAQ quantification).
In order to ensure that spliceosomal complexes isolated from “light” and “heavy” NEs do not exhibit differences in their activity, we monitored the splicing kinetics (Fig. 2A) and spliceosomal complex formation (Fig. 2B) in “light” and “heavy” SILAC NEs, as well as the RNA compositions of the purified B and C complexes (Fig. 2C). The analyses revealed that catalytically active spliceosomes form in exactly the same manner from “light” and “heavy” SILAC NEs, with no significant quantitative differences in the overall protein composition or functional pre-mRNA splicing activities (Fig. 2). We then isolated spliceosomal B complexes from “light” SILAC NEs and C complexes from “heavy” SILAC NEs (Fig. 1A), as described elsewhere (Bessonov et al. 2008). Proteins from the B and C complex mixtures were then separated by 1D SDS-PAGE and analyzed. We identified and quantified 266 proteins (see Supplemental Fig. 1); this included all of the previously published spliceosomal proteins from the B and C complexes, with the exception of the G10, UBL5, RACK1, TOE1, RBM7, THOC3, and SC35 proteins (Supplemental Table 2). Additionally, we identified and quantified 91 proteins that in previous studies had not been found to be components of the spliceosome and that in most cases represent contaminating proteins commonly detected in large-scale proteomic studies (Supplemental Table 3).
FIGURE 2.

Metabolically labeled NEs retained full catalytic activity, as shown by analyzing the B and C complexes purified from “heavy” or “light” SILAC NEs, respectively. (A) The splicing kinetics were determined from aliquots of splicing reactions taken from 0–180 min and analyzed by denaturing gel electrophoresis. Pre-mRNA and splicing products were visualized by autoradiography. Splicing products first appeared after 10 min. (B) The spliceosomal complex formation was assayed by native agarose gel electrophoresis and visualized by autoradiography. The A and B complexes were first observed after 2 and 4 min, respectively, while the C complex first appeared after 10–15 min. (C) The RNA compositions of purified B (“light” SILAC NE) and C (“heavy” SILAC NEs) complexes were analyzed by denaturing gel electrophoresis and visualized by silver staining (lanes 1,3) or autoradiography (lanes 2,4). B complexes contained U1, U2, U4, U5, and U6 snRNA (lane 1) and a large amount of pre-mRNA (lane 2). C complexes contained U2, U5, and U6 snRNA (lane 3) and splicing products and reduced amounts of pre-mRNA (lane 4).
Chemical labeling of spliceosomal proteins with iTRAQ reagents
We also applied chemical labeling with stable isotopes on purified spliceosomal B and C complexes using iTRAQ reagents (Ross et al. 2004). Chemical labeling is widely accepted as a quantitative MS-based method to analyze proteins derived from different samples. iTRAQ labeling of peptides from purified spliceosomal B and C complexes was performed with a method most recently established after gel separation of proteins as described previously (Schmidt and Urlaub 2009; Schmidt et al. 2013). Extracted peptides from the B complex were labeled with iTRAQ-115 and from the C complex with iTRAQ-116. Our iTRAQ analysis allowed us to quantify almost all previously published spliceosomal proteins from B and C complex preparations (Bessonov et al. 2008), except for the proteins U1-C, RACK1, Tip-49, CCDC130, NFKBIL1, THOC3, DBPA, RBM42, and SC35 (Supplemental Table 4). Similarly to the SILAC results, we found a total of 265 proteins; among these were 87 proteins that had not been listed in the previous analysis by counting peptides; these mainly represented commonly encountered contaminants (Supplemental Table 5).
Proteome analysis: spectral count
Previous analyses of protein components of spliceosomal complexes used the numbers of sequenced peptides in each sample to estimate the quantities of the proteins in their respective complexes (Deckert et al. 2006; Behzadnia et al. 2007; Bessonov et al. 2008, 2010; Fabrizio et al. 2009; Herold et al. 2009; Agafonov et al. 2011). As in these studies spliceosomal complexes were purified to the highest standards in biochemical terms (i.e., kinetic splicing assays, density gradient centrifugation to obtain homogenous populations), the number of peptides sequenced in the subsequent MS analysis was considered to be a valid approach for the estimation of amounts of proteins in various complexes. Nonetheless, a certain ambiguity remained in these studies, namely, whether the peptide numbers and the respective comparison with numbers obtained from other complexes meet the requirements of quantitative protein analysis. As we have now obtained quantitative values for the proteins present in the spliceosomal B and C complex, we set out to compare these values with MS data obtained from a previous data set (Bessonov et al. 2008). Instead of peptide count, we used the spectral count from the same data set of Bessonov et al. (2008) with the software Scaffold 2 (Supplemental Table 5). The ratios of the spectra or peptides of B-complex proteins to C-complex proteins are listed in Supplemental Table 6 (note that ratios could not be assigned to the proteins that were exclusively present in either the B or the C complex; these were labeled as “OSB” [only spectra B complex] or “OSC” [only spectra C complex]). This comparison revealed that assigning a protein abundance on the basis of peptide counting gives results consistent with those of spectral counting, with differences observed for only a few proteins (such as RACK1 and Pinin) (Supplemental Table 6). Moreover, we observed, when compared with the values obtained from the labeling experiments, good agreement in the quantification of the various proteins in the different complexes, so that it can be concluded—with a few exceptions that are discussed below—that spectral count (or even peptide count) is an appropriate method to quantify proteins in various spliceosomal complexes.
Validation of the quantification results
The investigated spliceosomal B and C complexes provide an ideal system for validation of the obtained quantitative results. The expected theoretical B:C protein ratio for the cap-binding proteins (which interact with the 5′ cap structure of the pre-mRNA) is 1:1; for the Sm proteins (which are core subunits for all of the U snRNPs except U6), 2:1 (since the C complex contains the U2 and U5 but lacks the U1 and U4). Indeed, we observe a 1:1 ratio for the CBP20 and CBP80 cap-binding proteins in the B:C complexes by all three methods (SILAC, iTRAQ, and label-free spectral count), with the exception of the spectral count for CPB20, which gave a ratio of 0.67 (Supplemental Tables 2, 4, 6). For the B:C protein ratios for the Sm proteins, both iTRAQ and SILAC yielded an average protein ratio of 1.75, which is close to the expected value of 2 (Fig. 3A). However, spectral count gave the expected value only for the SmF and SmG proteins and showed clearly lower ratios for the other Sm proteins (Fig. 3A). Overall, the quantification of these proteins provided an internal validation of the different quantification methods.
FIGURE 3.

Relative protein abundances of the Sm proteins and the U2 and U5 snRNP-specific proteins obtained by spectral count, SILAC, and iTRAQ. The B:C ratios are shown for the Sm proteins (A), the U2 snRNP-specific proteins (B), and the U5 snRNP specific proteins (C). Ratios of proteins in B versus C complex are plotted on a logarithmic scale; error bars, SD between the two biological replicates. “1” indicates that a protein is present in these complexes in a 1:1 ratio. The different shading of the bars represents the ratios of proteins in B and C complexes derived from the values obtained by SILAC, iTRAQ, and spectral count, respectively (see Table 1; Supplemental Tables).
A stable “core” of U snRNP-specific proteins
The snRNAs of U2 and the U5 snRNPs remain stably associated with the B and C complexes; however, only a subset of the U2- and U5-specific proteins appears in equal amounts within the two complexes (Fig. 3B,C). SILAC, iTRAQ, and spectral count showed that the U2-A′ and U2-B′′ proteins had B:C ratios of approximately 1, whereas the U2 snRNP-associated splicing factors SF3a and SF3b were more abundant in the B complex (Table 1; Fig. 3B). Likewise, only four of the eight U5 snRNP proteins were found to be present in a 1:1 ratio between the two complexes (220K, 200K, 116K, and 40K) (Fig. 3C); we therefore consider the proteins U2A′, U2B′′, U5-220K, 200K, 116K, and 40K together with the Sm proteins of U2 and U5 snRNPs, which are also stably associated with both complexes (see above, Fig. 3A), to be the spliceosomal “core” proteins in the B and C complexes.
Specific proteins of the B and C complexes
Once we had validated our quantification results as described above, we next used the quantitative values of the protein abundances to investigate the correlation of their association with the B and C complexes. To do this, we consider proteins that show a B:C value above 2.0 for at least two of the three methods to be specifically associated with the B complex and those showing a value below 0.5 for at least two of the three methods to be specifically associated with the C complex. We also take the previous ordering and grouping of proteins into account (Bessonov et al. 2008, 2010; Agafonov et al. 2011). Of note, to draw conclusion from the absolute number of the values, that means, whether, for example, an extremely high or low value (e.g., U1-A, 25.95 [SILAC]; PPIase-like 3b, 0.02 [SILAC]) compared with a moderate value (e.g., hPrp19, 0.36 [SILAC]; MFAP, 2.65 [SILAC]) reflects the complete presence or absence in B or C complex, respectively, cannot be unambiguously addressed.
Proteins predominantly associated with spliceosomal B complexes
During the transition from the B to the C complex, the U1 and U4 snRNPs with their associated proteins are destabilized and dissociate; this is clearly reflected by the high B:C ratios observed for these proteins (Table 1; Fig. 4A,B). The U4/U6 snRNP-specific proteins showed very high B:C ratios (average B:C 10.54), as determined by all three approaches (Fig. 4B), showing that they, together with the U4 snRNA, also dissociate from the spliceosome during the transition from the B to the C complex. All three methods revealed high B:C ratios (>3) for the U6 snRNP LSm proteins, showing that, although the U6 snRNA remains associated with the C complex, the LSm proteins dissociate from U6 during the B-to-C transition (Table 1; Fig. 4C). Contrary to the U2A′ and U2-B′′ proteins (see above), the U2 snRNP-specific SF3a and SF3b splicing factors were found to be more abundant in the B complex (Fig. 3B) and thus, however, do not seem to belong in the category of U snRNP “core” proteins of the B and C spliceosomes. Likewise, U5 snRNP-specific proteins 15K, 52K, 100K, and 102K show high B:C ratios as obtained by all three methods and thus represent B-specific proteins (Fig. 3C). In addition, all proteins specific to the tri-snRNP (U4/U6.U5) showed high B:C ratios (Fig. 4B; Table 1); this is consistent with previous studies that have shown dissociation of some tri-snRNP–specific proteins from the spliceosome during its activation (Makarov et al. 2002).
FIGURE 4.

Relative protein abundances in the B and C complexes for the U1, U4/U6, and U4/U6.U5 snRNP-specific proteins and the LSm proteins, as obtained by SILAC, iTRAQ, and spectral count. (A) B:C ratios for U1 snRNP-specific proteins. No protein ratios were obtained for U1-C from the spectral count since this protein was completely absent from the C complex. (B) B:C ratios for the U4/U6- and U4/U6.U5-specific proteins. (C) B:C ratios for the LSm proteins. No protein ratio was obtained from the spectral count for LSm4 since this protein was absent from the C complex. LSm5 was not identified or quantified. For details, see legend to Figure 3.
For several non-snRNP proteins high B:C ratios were observed, showing that they are more abundant in B-complex preparations. Some examples are RED (average B:C 7.39), h-Smu-1 (average B:C 6.12), and UBL5 (B:C 13.56 [iTRAQ]). By using our quantification methods, we classify these proteins as specific for the B complex (hPrp38, hSnu23, TFIP11, MFAP1, RED, hSmu-1, RBM42, TRAP3, UBL5, HsKin17, Npw38, Npw38BP) (Table 1; Fig. 5A). In addition to these proteins, we thus extend the list of B-specific proteins by six proteins (FUSE3, PUF60, RBM5/LUCA15, SAFB-like, SFRS12, and SPF30) (Table 1), which were not present in the protein lists of previous studies (Bessonov et al. 2008; Agafonov et al. 2011).
FIGURE 5.

Relative protein abundances of the B- and C- specific proteins. (A) B:C ratios for B-specific proteins. (B) B:C ratios for C-specific proteins. For several proteins, no values for spectral count were obtained as these proteins had spectra only in the B (hSnu23, UBL5, Npw38, SPF30) or the C (PPIL3b, FRG1, NOSIP, C1orf55, FAM50B, C9orf78, DGCR14, CCDC130, NKAP, ZCCHC10, CDK10, TTC14, DDX34, PRKRIP1) complex. For details, see legend to Figure 3.
Proteins predominantly associated with C complexes
The so-called step 2 splicing factors are required for the second step of pre-mRNA splicing, which occurs in the C complex (for review, see Umen and Guthrie 1995; Smith et al. 2008), and accordingly, those proteins should be more abundant in this complex. Indeed, all of these proteins (hPrp17, hPrp18, hPrp22, hPrp16, and hSlu7) had B:C ratios between 0.1 and 0.5 in iTRAQ and SILAC and close to 0 by spectral count, showing that they were clearly associated with the catalytically active spliceosomal C complex (Fig. 6A; Table 1).
FIGURE 6.

Relative protein abundances of the step 2 factors, the EJC and the hPrp19/CDC5L complex–specific proteins obtained by SILAC, iTRAQ, and spectral count. (A) B:C ratios for step 2 factors and the EJC. No protein ratio was obtained from the spectral count for hPrp18 since this protein was absent from the B complex. (B) B:C ratios for the hPrp19/CDC5L complex–specific proteins. For CTNNBL1, no clear association to either the B or the C complex could be determined. For details, see legend to Figure 3.
Additional proteins have previously been classified as “proteins recruited to the C complex” and “potential C complex–specific proteins” (Bessonov et al. 2008). All of these proteins revealed low B:C ratios by all three quantification methods (Table 1; Fig. 5B; Supplemental Tables 2, 4, 6).
On the basis of these quantification results, we extended the list of C-specific proteins by three proteins (DDX34, NUFIP1, and PRKRIP1) (Table 1), all of which showed high abundance in the C complex, with B:C ratios between 0.04 and 0.37. The DDX34 protein, which was not previously identified within any spliceosomal complex, was clearly identified as being more strongly represented in the C complex by both iTRAQ and SILAC. Spectral-count analysis identified this protein solely in the C complex but not in the B complex.
The hPrp19/CDC5L complex
The hPrp19/CDC5L (NTC in yeast, Chan et al. 2003) complex is essential for pre-mRNA splicing and associates with the spliceosome before the first catalytic step of splicing (Ajuh et al. 2000). Together with U5 snRNP, it forms the remodeled 35S U5 complex in humans (Makarov et al. 2002), and recent studies in yeast found it in the 35S ILS intron-lariat spliceosome (Fourmann et al. 2013). Here, we found that all of these proteins (hPrp19, CDC5L, SPF27, PRL1, Hsp70, AD-002)—with the exception of CTNNBL1, which showed an average B:C ratio of 1.34 (Table 1)—were clearly more abundant in the C complex than the B complex (Table 1; Fig. 6B). The Npw38 and Npw38BP proteins were previously found to comigrate at the top of the gradient during hPrp19/CDC5L complex purification, suggesting that they were co-isolated and/or not stably associated with the hPrp19/CDC5L complex (Makarova et al. 2004). These two proteins were found here to be highly abundant in the B complex compared with the C complex (Table 1; Fig. 5A), demonstrating their predominant association with the B but not with C complex.
The RES complex
The RES (retention and splicing) complex consists of SNIP1, MGC12135, and CGI-79. It binds to the spliceosome before the first step of splicing and is required for efficient intron removal and nuclear pre-mRNA retention (Dziembowski et al. 2004). We found by all three quantification methods that the hRES proteins associate with both the B and C complexes, showing a 1:1 B:C ratio (Table 1). Indeed, this is the only non-snRNP protein complex among the quantified non-snRNP splicing factors that remains constantly associated in the B and C complex.
The exon junction complex
The exon junction complex (EJC) protein complex binds spliced mRNAs in a sequence-independent manner close to site of exon–exon ligation. It is a highly dynamic complex with stably and more weakly associated protein components (Le Hir et al. 2000; Lau et al. 2003; Merz et al. 2007; Singh et al. 2012). We found that the EJC did not show the same B:C ratios for all its proteins. Only for three proteins (eIF4A3, Magoh, and Y14), we observed low B:C ratios (∼0.25) (Table 1; Fig. 6A), showing that they are much more abundant in the C complex, while proteins UAP56 and Pinin are much more abundant in the B complex (B:C ratios of ∼2.5) (Table 1). Other proteins previously assigned to the EJC showed fluctuating protein ratios (Supplemental Table 1), which did not allow us to classify them unambiguously.
DISCUSSION
In this study, we utilize three independent MS quantification techniques to compare the proteomes of stages of the spliceosomal complex during its transition from a precatalytic to a catalytically active state. The various functional states of the spliceosome provide an ideal system to investigate quantitative MS approaches on native and functionally active molecular assemblies. First, the amount of complexes to be compared can be precisely controlled by measuring the amount of radioactively labeled pre-mRNA in each complex; second, the functionality of the complexes can be monitored by corresponding assays.
We found good overall agreement between results from the label-free spectral count and those from the two methods that use stable isotope labeling (SILAC and iTRAQ). The fact that our results are consistent among the different quantification approaches, in spite of the fact that we used different analytical conditions during the quantitative MS analysis (i.e., different MS instruments and liquid chromatography conditions; see Materials and Methods) underlines the reproducibility of the methods employed here. Moreover, as previously generated data sets (Bessonov et al. 2008) were used to explore spectral counting as a label-free MS approach—and, importantly, gave results consistent with those of both the labeling approaches—this demonstrates that the evaluation of the protein abundance among various spliceosomal complexes as performed for A, B, Bact, and C in human spliceosomes and B, Bact, B*, and C yeast spliceosomes is valid and indeed reflects the association of a distinct protein with its corresponding complexes (Deckert et al. 2006; Behzadnia et al. 2007; Bessonov et al. 2008, 2010; Fabrizio et al. 2009; Agafonov et al. 2011).
In agreement with previous comparative studies on MS-based quantification (Hendrickson et al. 2006; Collier et al. 2010; Li et al. 2012), discrepancies between the isotope labeling and the spectral count techniques were seen, especially for small proteins (<20 kDa, e.g., Sm proteins) (Table 1), for which accurate quantification was not achieved by spectral count owing to the limited number of peptides that were generated. The spectral count was also a little less accurate than labeling in quantifying proteins present in equal amounts within the B and C complexes, such as U5-220K (B:C 1.68), U5-40K (B:C 1.50), and CBP20 (0.79) (see Supplemental Tables 2, 4, 6). Although the two methods based on stable isotope labeling yielded similar results, iTRAQ showed overall lower values for proteins strongly represented in the B complex and showed slightly higher values for proteins that are strongly represented in the C complex, than SILAC did. This may be due to the fact that precursor selection for MS/MS is not 100% selective, allowing reporter ions of co-eluting peptides to contribute to the iTRAQ reporter ion intensity of the peptide being analyzed (Bantscheff et al. 2007).
We compared our results with those from the previously published semi-quantitative analyses by Agafonov et al. (2011) and Bessonov et al. (2008) and found that all the studies show the same results to a large extent. Remarkably, we found that most of the U snRNP-specific proteins are abundant in the B complex but not in the C complex. Here, we show that not only are U1 and U4 snRNP-specific proteins destabilized during the transition from complex B to complex C, but also U5 and U2 snRNP-specific proteins and LSm proteins. This leaves a U snRNP-specific protein “core” in the B and the C complex consisting of only the Sm proteins, four U5 snRNP-specific proteins (220K, 200K, 116K, and 40K), and two U2 snRNP-specific proteins (U2A′ and U2B′′).
Evaluation of our quantitative data also allowed us to classify non-snRNP proteins as being specific for the B complex (Table 1). We extended the list of B-specific proteins by six proteins (FUSE3, PUF60, RBM5/LUCA15, SAFB-like, SFRS12, and SPF30) (Table 1), which showed high B:C ratios by at least two of the three methods. However, we have to take into account the fact that these proteins might be residual proteins belonging to the A complex, which was not included in our study. They might associate with the spliceosome at a very early point during pre-mRNA splicing and dissociate during formation of the catalytically active C complex. One example of this is the protein THRAP3, which is classified as B-specific by our results but was found to be specifically associated with the A complex by Agafonov et al. (2011). Along the same lines, we cannot make a clear statement regarding the hnRNP proteins. Although these are more abundant in the B complex, we have no clear evidence as to whether they are B-specific or whether they are residual proteins from the H/A complex.
More precise conclusions can be drawn for C complex proteins. The hPrp19/CDC5L complex (Ajuh et al. 2000; Makarova et al. 2004) was found to be associated with the B complex but was shown to be more abundant in the C complex; we show here that the quantitative values are approximately twice as great in the C as in the B complex, suggesting that this complex interacts loosely with the B complex and then becomes more stably associated with the C complex. Similarly, Agafonov et al. (2011) found the hPrp19/CDC5L complex to be less abundant in the B complex but highly abundant in the Bact and C complexes. We did not analyze the intermediate complex Bact in our study, and the slightly higher B:C ratio of the hPrp19/CDC5L proteins might reflect the association of these proteins during the B-to-C transition. In contrast, the second-step factors (hPrp16, hPrp17, hPrp18, hPrp 22, and hSLU7) clearly show high abundance in the C complex with very low B:C ratios (∼0.25), showing their association with the activated C complex only. Only three EJC proteins (eIF4A3, Magoh, and Y14) were also found to be clearly associated with the C complex. Accordingly, we define other proteins showing very low B:C ratios as “C-specific proteins” (Table 1). Some of these proteins have already been found to be abundant in the C complex by Agafonov et al. (2011); however, we are now able to extend the number of C-complex proteins (e.g., c19orf29, FAM32A, c10orf4, DDX34, NUFIP1) (Table 1). Importantly, DDX34 has never been described in the context of spliceosomal complexes. This protein is a probable ATP-dependent RNA helicase (gi|38158022), but its function has not yet been described, either in yeast or in humans. DDX34 thus represents a potential target for future studies.
Alongside the proteins that were clearly classifiable into B and C complex proteins, we identified some proteins that could not clearly be assigned owing to their inconsistent quantification values (Supplemental Table 1). For these proteins, different B:C ratios were obtained by the different quantification approaches (e.g., WDR70, SKIV2L2, DDX3) (Supplemental Table 1). There are several possible reasons why these proteins do not show a clear quantitative association. First, they may represent transiently bound proteins that easily dissociate from the complexes during purification and are thus present to different extents in the different complex preparations. Examples are the pre-mRNA/mRNA-binding proteins (Supplemental Table 1), which showed very inconsistent quantitative trends and at least two of which (YB-1 and ASR2B) were found to be present in all spliceosomal complexes (A, B, Bact, and C) detected in each case at a different abundance (Agafonov et al. 2011). Second, they may be components of the Bact complex, the intermediate complex in the B-to-C transition. We did not quantify the proteome of this complex, and the B:C ratios of proteins abundant in the Bact complex fluctuate between different preparations and quantification approaches. Indeed, some of these proteins were found by Agafonov et al. (2011) to be specific for the Bact complex, and a recent study in which the human Bact complex was analyzed confirms this assumption, as several of these proteins were shown to be abundant in the Bact complex only (Bessonov et al. 2010); examples are hPrp2, PPIL2, RNF113A, MGC20398, and MGC23918 (Supplemental Table 1).
CONCLUSIONS
We applied three independent MS-based quantification techniques to compare the proteomes of the precatalytic and the catalytically active spliceosomes (i.e., B and C complexes). Overall, we confirm, but also extend, results from previous studies that addressed the relative abundances of proteins in the respective complexes by semi-quantitative approaches. We have found that the label-free spectral count technique provided a suitable method for quantifying highly purified samples (such as the spliceosome or other RNPs). However, it has its limits when quantifying proteins of low molecular weight or small fold changes. This is the first report of SILAC used to label and subsequently purify a molecular machine that was functionally as active as its nonlabeled counterpart (as shown here for the spliceosome in terms of splicing efficiency and kinetics). Thus, it may be applicable to monitoring the assembly kinetics of the spliceosome in short time-frames in order to address the dynamic incorporation and release of proteins in its various functional states or to gain insight into the changes of protein modification during the splicing cycle. Overall, iTRAQ (or a similar approach using isotope-labeled reagents) is easier to apply to the quantitative investigation of spliceosomal proteins, in particular as a relatively large amount of NE is required for the assembly of spliceosomes. Thus, a reliable quantification technique based on chemical labeling with stable isotopes would be of benefit for in-depth quantitative analyses of spliceosomes from sources with low quantities.
MATERIALS AND METHODS
Preparation of metabolically labeled “light” and “heavy” NEs
Dulbecco’s modified Eagle medium (DMEM; PAA Laboratories) lacking L-arginine and L-lysine was supplemented with 10% (v/v) dialyzed fetal bovine serum (PAA Laboratories), 1 × penicillin/streptomycin, and 50 mg/L of normal (“light”) or stable-isotope-labeled (“heavy”) L-arginine and L-lysine. HeLa S3 cells were grown in custom-made DMEM containing either “light” or “heavy” L-arginine and L-lysine. Cells were grown for at least six passages at 100 mL (∼0.5 × 108 to 1.0 × 108 cells/mL) in 200 mL spinner flasks. The cells were then expanded to higher volumes and transferred to a 2.5 L fermenter. The cells were grown under standard conditions (50 mg/L L-arginine and L-lysine) with continuous perfusion of medium (0.5–1.0 v/24 h). A total of 1.5 L (5 × 106 cells/mL) of cells was harvested, and NEs were prepared according to the method of Dignam et al. (1983) from “light”- or “heavy”-labeled cells. L-arginine and L-lysine, and stable isotope-labeled L-arginine (13C615N4; Arg+10) and L-lysine (13C6; Lys+6), were purchased from Sigma-Aldrich.
Generation of aptamer-tagged pre-mRNA and in vitro splicing
MS2-tagged PM5 pre-mRNA was generated as previously described (Deckert et al. 2006; Bessonov et al. 2008). In vitro splicing was performed in either “light” or “heavy” HeLa NE, using 32P-labeled, m7G(5′)ppp(5′)G-capped, and MS2-tagged PM5 pre-mRNA. RNA was separated on a 8.0 M urea–10% (v/v) polyacrylamide gel. Spliceosomal complex assembly was analyzed by native gel electrophoresis on a 2% (m/v) agarose gel.
Affinity selection of spliceosomal B and C complexes
Spliceosomal B and C complexes for proteomic analysis and SILAC and iTRAQ quantification were isolated as previously described (Bessonov et al. 2008). Briefly, 32P-labeled, m7G(5′)ppp(5′)G-capped, and MS2-tagged PM5 pre-mRNA was incubated with MS2-MBP fusion protein. Spliceosomal complexes were allowed to assemble from “light” or “heavy” NEs for 6 min (B complex) or 180 min (C complex). For SILAC quantification, B complexes were assembled from “heavy” NEs, and C complexes were assembled from “light” NEs. Assembled complexes were separated on 10%–30% (v/v) glycerol gradients, and 40–45S gradient fractions were subjected to affinity selection on amylose beads.
Sample preparation and MS for SILAC quantification
Three pmoles of affinity-purified B and C complexes was mixed in equal amounts according to the 32P-labeled pre-mRNA. Proteins were separated by gel electrophoresis on a 4%–12% Bis-Tris precast gel (NuPAGE, Invitrogen) and stained with colloidal Coomassie blue. The entire gel lane was cut into 25 pieces, and proteins were digested in-gel as described previously (Shevchenko et al. 1996). Samples were redissolved in 10% (v/v) acetonitrile/0.15% (v/v) formic acid (FA) and analyzed on a CAP-LC system coupled to a Q-ToF Ultima mass spectrometer (Waters) or on an Agilent HP 1100 series system coupled to a LTQ-Orbitrap XL (Thermo Fisher Scientific).
Sample preparation and MS for iTRAQ or spectral count quantification
Four pmoles each of affinity-purified B and C complexes were separated by 8%/14% (v/v) SDS-PAGE, respectively, and stained with Coomassie blue. Entire gel lanes were cut into 60–70 slices. Proteins were digested in-gel as described previously (Shevchenko et al. 1996) except that 50 mM triethylammonium bicarbonate buffer (TEAB, Sigma-Aldrich) instead of ammonium bicarbonate buffer was used for the iTRAQ preparation. For iTRAQ labeling, the extracted peptides were dissolved in 20 μL 100 mM TEAB buffer. Internal standards were prepared by mixing 5 μL TEAB buffer with 5 μL aliquots of samples generated from gel slices cut at the same height from both gel lanes. iTRAQ reagents were reconstituted at room temperature in 70 μL ethanol per vial. iTRAQ reagent (5 μL) was added to each sample, and samples were incubated at room temperature for 1 h with gentle mixing. Internal standards were labeled with iTRAQ-114, and samples from the B and C complexes were labeled with iTRAQ-115 and iTRAQ-116, respectively. The remaining iTRAQ reagent was quenched by adding 5 μL of 50 mM glycine and incubating at room temperature for 30 min with gentle mixing. Samples to be compared, such as those containing the peptides generated from B and C complexes, were pooled with their relevant internal standards and then dried in a vacuum centrifuge (Schmidt and Urlaub 2009). For the MS analysis, iTRAQ or spectral count samples were dissolved in 10% (v/v) acetonitrile with 0.15% (v/v) FA and subsequently analyzed on a Waters Q-TOF Ultima coupled to a Waters CAP-LC system.
LC-coupled ESI MS–MS/MS on a Q-ToF mass spectrometer
To analyze samples over the CAP-LC system coupled to the Q-ToF Ultima mass spectrometer (Waters), peptides were separated online by reversed-phase chromatography using 0.1% (v/v) FA as mobile phase A and 80% (v/v) acetonitrile/0.15% (v/v) FA as mobile phase B. The peptides were loaded onto a trap column (μ-Precolumn Cartridge, Acclaim PepMap100 C18, 300 μm i.d. × 5 mm, LC Packings) and separated at a flow rate of 200 nL/min on an analytical column packed in-house (C18, Reprosil, Maisch) with a gradient of 7%–40% mobile phase B over 50 min. Eluted peptides were analyzed directly in the Q-ToF mass spectrometer in a data-dependent manner. MS scans were acquired for 1 sec followed by three MS/MS spectra for 3 sec, each with an ion-mass window set to 2.5 Da. The MS-to-MS/MS switch was set to 15 counts/sec, and the MS/MS-to-MS was set to an intensity below a threshold of 2 counts/sec. Charge state recognition was used to estimate the collision energy for the selected precursors. Scan time and interscan time were set to 0.9 sec and 0.1 sec, respectively. Peak lists were generated from raw data by using MassLynx v4.0 software with the following settings: smooth window 4.00, number of smooth 2, smooth mode Savitzky-Golay, percentage of peak height to calculate centroid spectra at 80% with no baseline subtraction.
LC-coupled ESI MS–MS/MS on a LTQ-Orbitrap mass spectrometer
Samples were analyzed on an HP 1100 series system (Agilent) coupled to a hybrid Linear Ion Trap–Orbitrap mass spectrometer (LTQ-Orbitrap XL, Thermo Fisher Scientific). Peptides were separated by online reversed-phase nanoflow chromatography, using 0.1% (v/v) FA as the mobile phase A and 95% (v/v) acetonitrile/0.1% (v/v) FA as the mobile phase B. Peptides were loaded onto a trap column packed in-house (1.5 cm, 360 μm o.d., 150 μm i.d., ReproSil-Pur C18-AQ, 5 μm, Dr. Maisch) and separated at a flow rate of 300 nL/min on an analytical C18 capillary column (30 cm, 360 μm o.d., 75 μm i.d., ReproSil-Pur C18-AQ, 5 μm) with a gradient of 0%–38% mobile phase B over 35 min. Eluted peptides were analyzed directly in the mass spectrometer (LTQ-Orbitrap XL; Thermo Fisher Scientific). The LTQ-Orbitrap was operated in a data-dependent mode. Survey full-scan MS spectra were acquired in the LTQ-Orbitrap (m/z 350–1400) with a resolution of 30 000 at m/z 400, and an automatic gain control target of 5 × 105. The five most intense ions were selected for CID (collision-induced dissociation) MS/MS fragmentation and detection in the linear ion trap, with previously selected ions dynamically excluded for 60 sec. Singly charged ions and ions with unrecognized charge states were also excluded. Internal calibration of the Orbitrap was performed using the lock mass option (lock mass: m/z 445.120025) (Olsen et al. 2005). Mascot generic format (mgf) files were generated from raw data using Mascot Daemon v2.2.2 (Matrix Science).
Data analysis and quantification
Peak lists generated were searched against NCBI nonredundant database (October 8, 2007; 5539442 sequences), by using Mascot v.2.2.04 as search engine. The mass accuracy filter used was 0.2 Da for the parent and fragment ions for the Q-ToF mass spectrometer. For the Orbitrap mass spectrometer, this was 5 ppm for precursor and 0.5 Da for product ions. Peptides with no or at most two missed cleavage sites were defined as tryptic peptides. Carbamidomethylation of cysteines and oxidation of methionine residues were allowed as variable modifications. For SILAC and iTRAQ quantification, “heavy” arginine (Arg+10) and “heavy” lysine (Lys+6) and iTRAQ modifications, respectively, were allowed as fixed modifications.
SILAC quantification
SILAC quantification was carried out by using unique peptides with the MSQuant software v1.2. Data normalization was performed on proteins known to be present in a 1:1 ratio (e.g., the 5′ pre-mRNA cap-binding proteins CBP20 and CBP80, the U5-220K and U5-200K proteins).
iTRAQ quantification
Non-normalized peptide ratios for iTRAQ quantification were obtained from Mascot v2.2.04 for unique peptides with a minimum peptide score of 20. Proteins were quantified from the main bands by calculating the mean ratio after manual removal of outliers. Data normalization was performed on proteins known to be present in a 1:1 ratio, as above. Protein ratios obtained were further validated by three independent procedures: (1) calculating the labeling efficiency for each protein in each band; (2) using the same amounts of nonmodified trypsin (Roche), resulting in a 1:1 ratio for autoproteolytic trypsin peptides; and (3) analyzing peak intensities of the reporter ions for the internal standards (iTRAQ-114) of low-scoring peptides. As the internal standard was prepared by pooling aliquots from iTRAQ-115– and iTRAQ-116–labeled samples the following equation represents the intensity ratios: intensity[iTRAQ-114] = ⅓ intensity[iTRAQ-115] + ⅓ intensity[iTRAQ-116] whereby [iTRAQ−114] = (5 µL/(20 µL − 5 µL)) [iTRAQ−115] + (5 µL/(20 µL − 5 µL)) [iTRAQ-116] (see also Schmidt and Urlaub 2009).
Spectral count
Unweighted spectral count for proteins identified in B and C complexes was obtained by using the software Scaffold 2. B:C protein ratios were calculated manually from the obtained number of spectra for each protein.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank P. Kemkes, H. Kohansal, M. Raabe, and U. Plessmann for excellent technical assistance; I. Lemm for help with SILAC cell culture; and V.A. Raker for help in manuscript preparation. M.G. was supported by the Danish Agency for Science, Technology and Innovation (DASTI). This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB860) to R.L. and H.U.
Footnotes
Freely available online through the RNA Open Access option.
REFERENCES
- Aebersold R, Mann M 2003. Mass spectrometry-based proteomics. Nature 422: 198–207 [DOI] [PubMed] [Google Scholar]
- Agafonov DE, Deckert J, Wolf E, Odenwalder P, Bessonov S, Will CL, Urlaub H, Luhrmann R 2011. Semiquantitative proteomic analysis of the human spliceosome via a novel two-dimensional gel electrophoresis method. Mol Cell Biol 31: 2667–2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajuh P, Kuster B, Panov K, Zomerdijk JC, Mann M, Lamond AI 2000. Functional analysis of the human CDC5L complex and identification of its components by mass spectrometry. EMBO J 19: 6569–6581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B 2007. Quantitative mass spectrometry in proteomics: A critical review. Anal Bioanal Chem 389: 1017–1031 [DOI] [PubMed] [Google Scholar]
- Bantscheff M, Lemeer S, Savitski MM, Kuster B 2012. Quantitative mass spectrometry in proteomics: Critical review update from 2007 to the present. Anal Bioanal Chem 404: 939–965 [DOI] [PubMed] [Google Scholar]
- Behzadnia N, Golas MM, Hartmuth K, Sander B, Kastner B, Deckert J, Dube P, Will CL, Urlaub H, Stark H, et al. 2007. Composition and three-dimensional EM structure of double affinity-purified, human prespliceosomal A complexes. EMBO J 26: 1737–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessonov S, Anokhina M, Will CL, Urlaub H, Luhrmann R 2008. Isolation of an active step I spliceosome and composition of its RNP core. Nature 452: 846–850 [DOI] [PubMed] [Google Scholar]
- Bessonov S, Anokhina M, Krasauskas A, Golas MM, Sander B, Will CL, Urlaub H, Stark H, Luhrmann R 2010. Characterization of purified human Bact spliceosomal complexes reveals compositional and morphological changes during spliceosome activation and first step catalysis. RNA 16: 2384–2403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SP, Kao DI, Tsai WY, Cheng SC 2003. The Prp19p-associated complex in spliceosome activation. Science 302: 279–282 [DOI] [PubMed] [Google Scholar]
- Collier TS, Sarkar P, Franck WL, Rao BM, Dean RA, Muddiman DC 2010. Direct comparison of stable isotope labeling by amino acids in cell culture and spectral counting for quantitative proteomics. Anal Chem 82: 8696–8702 [DOI] [PubMed] [Google Scholar]
- Deckert J, Hartmuth K, Boehringer D, Behzadnia N, Will CL, Kastner B, Stark H, Urlaub H, Luhrmann R 2006. Protein composition and electron microscopy structure of affinity-purified human spliceosomal B complexes isolated under physiological conditions. Mol Cell Biol 26: 5528–5543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res 11: 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domon B, Aebersold R 2006. Mass spectrometry and protein analysis. Science 312: 212–217 [DOI] [PubMed] [Google Scholar]
- Dziembowski A, Ventura AP, Rutz B, Caspary F, Faux C, Halgand F, Laprevote O, Seraphin B 2004. Proteomic analysis identifies a new complex required for nuclear pre-mRNA retention and splicing. EMBO J 23: 4847–4856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Dannenberg J, Dube P, Kastner B, Stark H, Urlaub H, Luhrmann R 2009. The evolutionarily conserved core design of the catalytic activation step of the yeast spliceosome. Mol Cell 36: 593–608 [DOI] [PubMed] [Google Scholar]
- Fourmann JB, Schmitzova J, Christian H, Urlaub H, Ficner R, Boon KL, Fabrizio P, Luhrmann R 2013. Dissection of the factor requirements for spliceosome disassembly and the elucidation of its dissociation products using a purified splicing system. Genes Dev 27: 413–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson EL, Xia Q, Wang T, Leigh JA, Hackett M 2006. Comparison of spectral counting and metabolic stable isotope labeling for use with quantitative microbial proteomics. Analyst 131: 1335–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold N, Will CL, Wolf E, Kastner B, Urlaub H, Luhrmann R 2009. Conservation of the protein composition and electron microscopy structure of Drosophila melanogaster and human spliceosomal complexes. Mol Cell Biol 29: 281–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurica MS, Moore MJ 2003. Pre-mRNA splicing: Awash in a sea of proteins. Mol Cell 12: 5–14 [DOI] [PubMed] [Google Scholar]
- Jurica MS, Licklider LJ, Gygi SR, Grigorieff N, Moore MJ 2002. Purification and characterization of native spliceosomes suitable for three-dimensional structural analysis. RNA 8: 426–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CK, Diem MD, Dreyfuss G, Van Duyne G 2003. Structure of the Y14-Magoh core of the exon junction complex. Curr Biol 13: 933–941 [DOI] [PubMed] [Google Scholar]
- Le Hir H, Izaurralde E, Maquat LE, Moore MJ 2000. The spliceosome deposits multiple proteins 20–24 nucleotides upstream of mRNA exon–exon junctions. EMBO J 19: 6860–6869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Adams RM, Chourey K, Hurst GB, Hettich RL, Pan C 2012. Systematic comparison of label-free, metabolic labeling, and isobaric chemical labeling for quantitative proteomics on LTQ Orbitrap Velos. J Proteome Res 11: 1582–1590 [DOI] [PubMed] [Google Scholar]
- Liu H, Sadygov RG, Yates JR III 2004. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem 76: 4193–4201 [DOI] [PubMed] [Google Scholar]
- Makarov EM, Makarova OV, Urlaub H, Gentzel M, Will CL, Wilm M, Luhrmann R 2002. Small nuclear ribonucleoprotein remodeling during catalytic activation of the spliceosome. Science 298: 2205–2208 [DOI] [PubMed] [Google Scholar]
- Makarova OV, Makarov EM, Urlaub H, Will CL, Gentzel M, Wilm M, Luhrmann R 2004. A subset of human 35S U5 proteins, including Prp19, function prior to catalytic step 1 of splicing. EMBO J 23: 2381–2391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz C, Urlaub H, Will CL, Luhrmann R 2007. Protein composition of human mRNPs spliced in vitro and differential requirements for mRNP protein recruitment. RNA 13: 116–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolov M, Stutzer A, Mosch K, Krasauskas A, Soeroes S, Stark H, Urlaub H, Fischle W 2011. Chromatin affinity purification and quantitative mass spectrometry defining the interactome of histone modification patterns. Mol Cell Proteomics 10: M110.005371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolov M, Schmidt C, Urlaub H 2012. Quantitative mass spectrometry-based proteomics: An overview. Methods Mol Biol 893: 85–100 [DOI] [PubMed] [Google Scholar]
- Olsen JV, de Godoy LM, Li G, Macek B, Mortensen P, Pesch R, Makarov A, Lange O, Horning S, Mann M 2005. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol Cell Proteomics 4: 2010–2021 [DOI] [PubMed] [Google Scholar]
- Ong SE, Mann M 2005. Mass spectrometry-based proteomics turns quantitative. Nat Chem Biol 1: 252–262 [DOI] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M 2002. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1: 376–386 [DOI] [PubMed] [Google Scholar]
- Rappsilber J, Ryder U, Lamond AI, Mann M 2002. Large-scale proteomic analysis of the human spliceosome. Genome Res 12: 1231–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, et al. 2004. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics 3: 1154–1169 [DOI] [PubMed] [Google Scholar]
- Schmidt C, Urlaub H 2009. iTRAQ-labeling of in-gel digested proteins for relative quantification. Methods Mol Biol 564: 207–226 [DOI] [PubMed] [Google Scholar]
- Schmidt C, Hesse D, Raabe M, Urlaub H, Jahn O 2013. An automated in-gel digestion/iTRAQ-labeling workflow for robust quantification of gel-separated proteins. Proteomics 13: 1417–1422 [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68: 850–858 [DOI] [PubMed] [Google Scholar]
- Singh G, Kucukural A, Cenik C, Leszyk JD, Shaffer SA, Weng Z, Moore M 2012. The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell 151: 750–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ, Query CC, Konarska MM 2008. “Nought may endure but mutability”: Spliceosome dynamics and the regulation of splicing. Mol Cell 30: 657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen H, Mann M 2004. The ABC’s (and XYZ’s) of peptide sequencing. Nat Rev Mol Cell Biol 5: 699–711 [DOI] [PubMed] [Google Scholar]
- Umen JG, Guthrie C 1995. The second catalytic step of pre-mRNA splicing. RNA 1: 869–885 [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Luhrmann R 2009. The spliceosome: Design principles of a dynamic RNP machine. Cell 136: 701–718 [DOI] [PubMed] [Google Scholar]
- Yates JR, Ruse CI, Nakorchevsky A 2009. Proteomics by mass spectrometry: Approaches, advances, and applications. Annu Rev Biomed Eng 11: 49–79 [DOI] [PubMed] [Google Scholar]
- Zhou Z, Licklider LJ, Gygi SP, Reed R 2002. Comprehensive proteomic analysis of the human spliceosome. Nature 419: 182–185 [DOI] [PubMed] [Google Scholar]