

Abstract
Recent studies indicate high density lipoproteins (HDL) and their major structural protein, apolipoprotein A1 (apoA1), recovered from human atheroma, are dysfunctional and extensively oxidized by myeloperoxidase (MPO), while in vitro oxidation of apoA1/HDL by MPO impairs its cholesterol acceptor function. We developed a high affinity monoclonal antibody (mAb) that specifically recognizes apoA1/HDL modified by the MPO/H2O2/Cl-system using phage display affinity maturation. An oxindolyl alanine (2-OH-Trp) moiety at tryptophan 72 of apoA1 is the immunogenic epitope. Mutagenesis studies confirm a critical role for apoA1 Trp72 in MPO-mediated inhibition of ABCA1-dependent cholesterol acceptor activity of apoA1 in vitro and in vivo. ApoA1 containing a 2-OH-Trp72 group (oxTrp72-apoA1) is in low abundance within the circulation, but accounts for 20% of the apoA1 in atherosclerotic plaque. OxTrp72-apoA1 recovered from human atheroma or plasma was lipid-poor, virtually devoid of cholesterol acceptor activity, and demonstrated both potent pro-inflammatory activities on endothelial cells and impaired HDL biogenesis activity in vivo. Elevated oxTrp72-apoA1 levels in subjects presenting to a cardiology clinic (n=627) were associated with increased cardiovascular disease risk. Circulating oxTrp72-apoA1 levels may serve as a way to monitor a pro-atherogenic process in the artery wall.
Introduction
High density lipoprotein (HDL) is a complex and heterogeneous assembly of proteins and lipids. Defined by its buoyant density isolation characteristics, its compositional heterogeneity mirrors its functional heterogeneity, which includes cholesterol acceptor activity, anti-inflammatory, anti-apoptotic, anti-thrombotic, microRNA delivery, and innate immune functions1–6. Approximately 75% of the protein content of HDL is apolipoprotein A1 (apoA1), which serves as the primary protein scaffolding upon which the lipid cargo-carrying particle is built. The majority of apoA1 within the circulation is associated with HDL, which contains two or more apoA1, depending upon both the degree of particle maturation and the protein complement associated with a given HDL particle. Epidemiology studies show an inverse association between circulating HDL cholesterol (HDLc) or apoA1 levels and coronary artery disease (CAD)7. Animal studies using both genetic and direct infusion models similarly show global anti-atherosclerotic functions of the lipoprotein8–12. Recent clinical studies, however, reveal significant gaps in our knowledge about HDL. For example, while several clinical intervention studies employing either direct infusion of HDL forms or infusion of extracorporeal delipidated HDL/apoA1 have shown evidence of anti-atherosclerotic effects13–15, several distinct classes of HDL cholesterol-elevating drugs have, in contrast, failed to demonstrate significant therapeutic benefit16–18. Moreover, genetic studies demonstrate genetic variants that influence total circulating HDLc levels are not mechanistically linked to CAD development19. Taken as a whole, recent studies underscore the complexity and evolving understanding of the relationship between circulating levels of HDLc and/or apoA1 and CAD pathogenesis.
One possible explanation of the paradox between observed associations between HDLc levels and reduced CAD risks, versus the negative outcomes of recent therapeutic intervention studies targeting HDLc levels, is that a function of the HDL particle, and not the HDLc mass itself, is more clinically relevant diagnostically and therapeutically. For example, measurement of cholesterol efflux activity of HDL, or apolipoprotein-B depleted serum, may serve as a superior independent predictor of prevalent CAD risk compared to HDLc4. Moreover, alternative functional measures of HDL or its associated proteins may provide improved clinical value and prognostic efficacy5,20–24. Furthermore, current measures of HDL mass (either HDLc or apoA1) in the circulation do not appear to adequately reflect the pathobiology of a diseased artery wall. Recent studies of total apoA1 (both HDL-associated and lipid-poor forms) from human aortic tissues revealed the biological function and HDL particle distribution of apoA1 within the artery wall is markedly distinct from that of circulating apoA1 and HDL. Specifically, apoA1 in human aorta was found to be predominantly lipid-poor, not associated with HDL, extensively oxidatively cross-linked, and functionally impaired25. ApoA1 serves as a selective target (100–500 fold) for oxidative modification by MPO-generated and NO-derived oxidants within the artery wall, resulting in site-specific oxidative modifications26–28. Parallel in vitro studies have shown marked reductions in ATP-binding cassette transporter A1 (ABCA1)-dependent cholesterol efflux function, lecithin cholesterol acyl transferase (LCAT) activity, lipid binding activity, and non-cholesterol related activities (e.g. anti-apoptotic, anti-inflammatory) in either HDL or apoA1 oxidized ex vivo by the MPO/H2O2/Cl- system to an extent similar to that observed in apoA1 recovered from human lesions26,28–30. These findings, and studies demonstrating pro-inflammatory activities for HDL recovered from subjects with CAD or chronic inflammatory conditions associated with CAD risk21,31–33, suggest that the molecular processes that impair apoA1/HDL function in the artery wall may be diagnostic and therapeutic targets for CAD.
Herein we report tryptophan 72 of apoA1 serves as a selective target for MPO-dependent oxidative modification forming an oxindolyl alanine (2-hydroxy-L-trytophan, 2-OH-Trp; IUPAC name 2-amino-3-(2-oxo-1,3-dihydroindol-3-yl)propanoic acid) moiety on 20% of apoA1 recovered from human atherosclerotic lesions. Functional characterization studies of lesion and plasma apoA1 harboring this site-specific modification reveals a dysfunctional apolipoprotein with both significantly impaired cholesterol efflux acceptor activity and pro-inflammatory function. Moreover, clinical studies indicate levels of this dysfunctional apoA1 form within the circulation reflect a presumed pathophysiological process within the artery wall.
Results
Development of antibody specific for MPO-modified apoA1
Direct studies of HDL function in the human artery wall to date have been limited. In order to recover and study the chemical modifications and biological activities of HDL modified by MPO-generated oxidants in the artery wall, we sought to develop immunological tools to specifically detect and immunopurify apoA1 modified by the MPO/H2O2/Cl− system. This specific oxidized form of apoA1/HDL was selected since in vitro studies demonstrate this pathway readily inhibits cholesterol acceptor activity of both apoA1 and HDL under pathophysiologically plausible conditions26–27,29. Over 30,000 hybridoma cell lines were generated and screened after immunizing BALB/c mice with native reconstituted HDL particles (apoA1:POPC:cholesterol, 1:100:10, mol:mol:mol) exposed to either MPO-generated chlorinating oxidants (the MPO/H2O2/Cl− system), or MPO-generated nitrating oxidants (the MPO/H2O2/NO2− system) (Supplementary Fig. 1a). To ensure potential epitopes formed on the antigens would be those likely formed under (patho)physiologically plausible conditions, a low molar ratio of H2O2:apoA1 was used (5:1) and the content of protein-bound 3-chlorotyrosine or 3-nitrotyrosine were determined and shown to be within the range previously observed on apoA1 recovered from human atherosclerotic plaque26,29. A monoclonal antibody (mAb) was identified (clone 8B5.2) that specifically recognized both apoA1 and HDL exposed to the MPO/H2O2/Cl− system, but not apoA1 or HDL in their native (unoxidized) forms, or following exposure to alternative oxidant systems including the MPO/H2O2/NO2− oxidant system (Supplementary Fig. 1a).
While mAb 8B5.2 had the desired specificity, its affinity proved to be too low for either efficient immunoprecipitation or use in an ELISA format to detect endogenous circulating levels of oxTrp72-apoA1 (Supplementary Fig. 1b–d). We used phage display technology to affinity mature (as single chain antibody) mAb 8B5.2 and then after sequencing gain of function mutants, we genetically engineered the recombinant IgG form as described under Methods. Briefly, human apoA1 exposed to the MPO/H2O2/Cl− system was used as positive selection bait. Native human apoA1 and human apoA1 exposed to MPO-generated nitrating oxidants (see Methods) were both used as negative selection bait after the first round selection. During the last 2 affinity maturation cycles, multiple gain of function subclones were sequenced and reproducible gain of function mutations identified. All gain of function mutants were then incorporated into a single clone and verified to be functional as a high affinity binder with appropriate specificity. This “combined” gain of function clone was then converted into a chimeric humanized double chain IgG mAb (Fig. 1a–c). We termed this affinity-matured recombinant humanized mAb r8B5.2, which displayed a 1,600-fold enhanced affinity compared to the starting (parental) mAb, attaining a KD of 1 × 10−10 M (Supplementary Fig. 1c). More detailed characterization studies confirmed the recombinant affinity-matured antibody (mAb r8B5.2) retained exclusive specificity for recognition of apoA1 exposed to the MPO/H2O2/Cl− system (Fig. 1d). Further characterization of mAb r8B5.2 revealed it had sufficient affinity to immunoprecipitate MPO-oxidized apoA1, and when used in ELISA format, had adequate sensitivity to detect and quantify apoA1/HDL following oxidation by the MPO/H2O2/Cl− system at levels of oxidation comparable to those observed in plasma or serum (Supplementary Fig. 1d).
Figure 1. Phage display affinity maturation to form a high affinity monoclonal antibody specific for apoA1 oxidized by the MPO-H2O2-halide system.
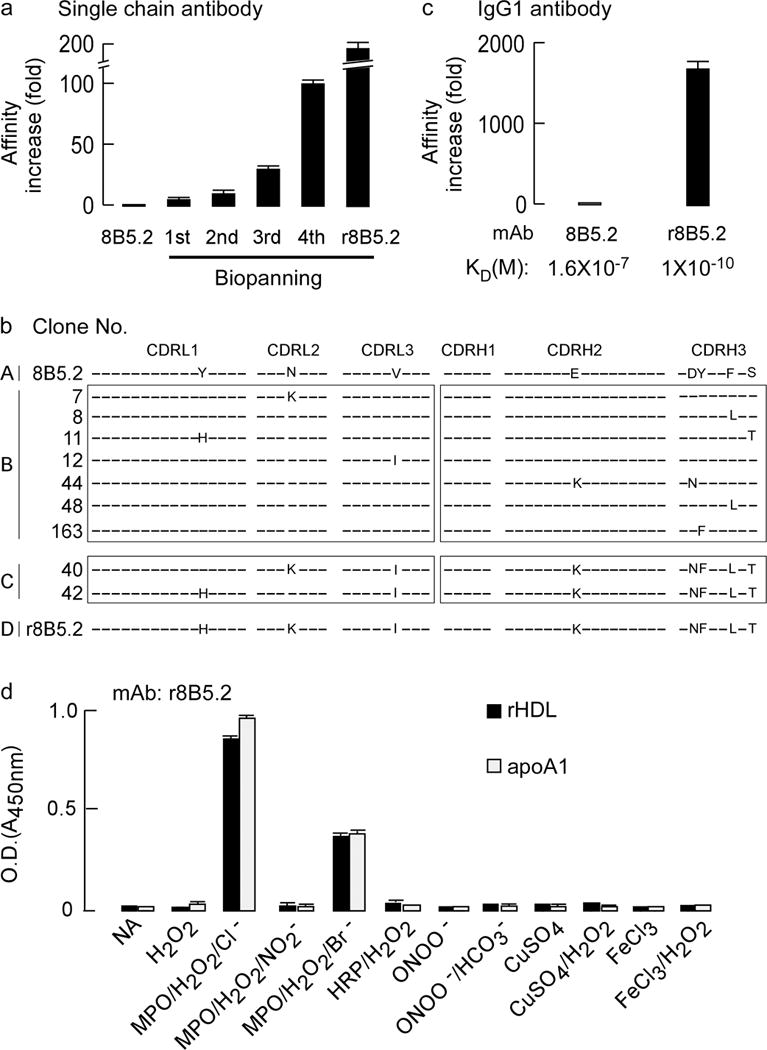
(a) Fold-increase in apparent binding affinity following multiple rounds of biopanning after phage display affinity maturation of parental mouse mAb 8B5.2 single chain antibody (scFv). (b) Complementarity determining region (CDR) DNA sequences of original mAb 8B5.2 and that of nine affinity matured scFv r8B5.2 subclones identified with lowest dissociation rate. CDRL1–3: CDRs of antibody light chain. CDRH1–3: CDRs of antibody heavy chain. The letters indicate identities of the mutated amino acids and differences between wild type and affinity matured clones. (c) The increase in apparent affinity (KD) of parental mAb 8B5.2 and affinity matured scFv r8B5.2 after conversion to full length mouse and human chimeric IgG1, designated as r8B5.2. (d) mAb r8B5.2 recognizes with high specificity only apoA1 or rHDL exposed to the MPO-H2O2-halide system, but not the indicated alternative oxidation systems. ApoA1 or rHDL were coated onto 96 well plates and detected with r8B5.2, as described in Methods. Data are mean± S.D. of triplicate determinations.
MPO oxidation of apoA1 Trp72 inhibits cholesterol acceptor function
No studies to date have directly examined the functional significance of MPO modification of apoA1 or HDL within human atheroma. Rather, the proposed role for MPO-catalyzed oxidation of apoA1/HDL in impairing cholesterol acceptor activity of the lipoprotein in vivo is based upon correlative data comparing the degree of oxidative modifications on apoA1 recovered from artery wall tissues, with both the extent of oxidative modifications formed and the degree of ABCA1-dependent cholesterol efflux impairment observed in model systems ex vivo26,29. Incubation of apoA1 or HDL with the MPO/H2O2/Cl− system can promote oxidative modification of multiple oxidant sensitive residues, including (but not limited to) each of the three Met, each of the seven Tyr, and each of the four Trp. Oxidation of each of these residues has been detected by proteomics analysis of apoA1 or HDL recovered from human atherosclerotic lesions, yet debate exists about the relative abundance of various species and their functional importance26–29,34. We hypothesized that the epitope recognized by mAb r8B5.2 might be an oxidized form of Tyr, Met or Trp resulting from the MPO/H2O2/Cl− system within the appropriate sequence context of apoA1. To test this, we generated recombinant mutant forms of apoA1 in which each of these residues was converted to a relatively non-oxidizable alternative amino acid of similar size and chemical properties – namely: (i) each of the 7 Tyr residues (positions 18,29,100,115,166,192 and 236) were mutated to the relatively non-oxidizable Phe residue (mutant named “7YF”); (ii) each of the 3 Met residues (positions 86,112 and 148) were mutated to the relatively non-oxidizable Val residue (mutant named “3MV”); and (iii) each of the 4 Trp residues (positions 8,50,72 and 108) were mutated to the relatively non-oxidizable Phe residue (mutant named “4WF”). We reasoned that following exposure of the lipoprotein to the MPO/H2O2/Cl− system, the mutant form of apoA1 no longer harboring the oxidant resistant residue, should not be recognized by mAb r8B5.2, whereas recombinant human (wild type sequence) apoA1 (rh-apoA1), would be recognized following oxidation. Exposure of rh-apoA1, 7YF and 3MV to the MPO/H2O2/Cl− system produced an oxidized apolipoprotein recognized by mAb r8B5.2; whereas apoA1 mutant 4WF was not recognized following oxidation (Fig. 2a). Importantly, control studies showed that all apoA1 mutant forms examined retained cholesterol efflux activity in their native form, and all but 4WF lost ABCA1-dependent and total cholesterol efflux activities following exposure to the MPO/H2O2/Cl− system (Fig. 2b). Collectively, these results suggested one or more of the Trp of apoA1 became the epitope recognized following apoA1 oxidation by the MPO/H2O2/Cl− system and plays an important role as an “oxidative switch” for impairment of cholesterol efflux activity.
Figure 2. Epitope mapping of affinity matured mAb r8B5.2.
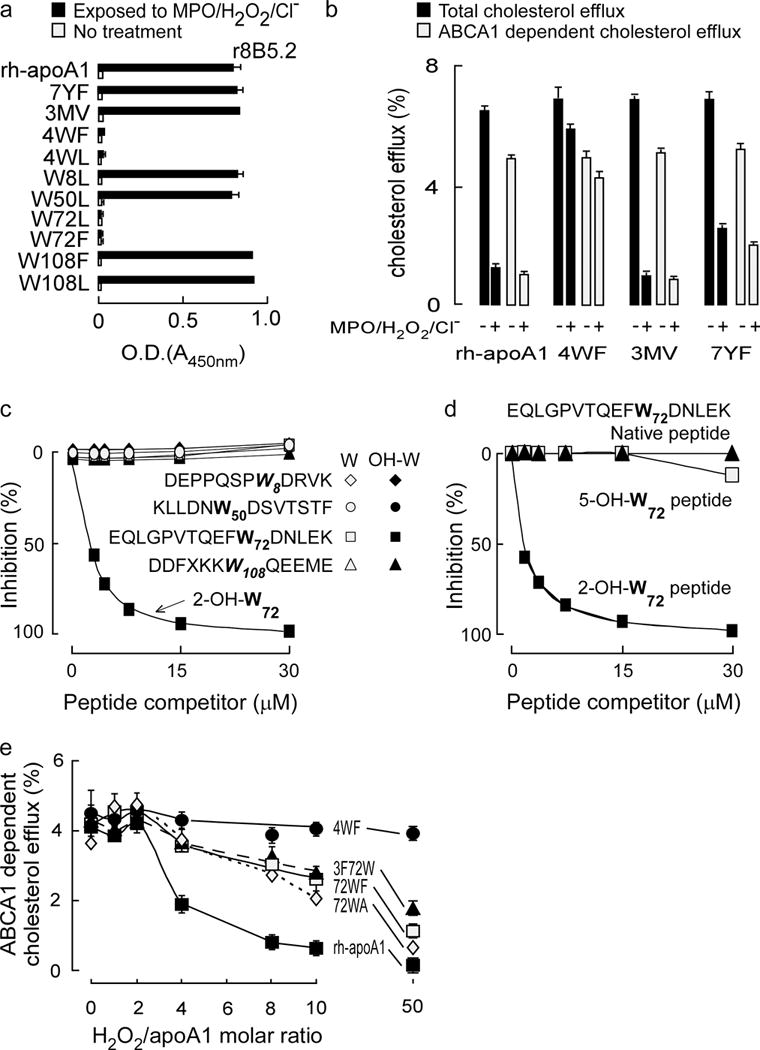
(a) Antibody (r8B5.2) reactivity against the indicated recombinant human apoA1 (rh-apoA1) and mutant forms was quantified before and following exposure to the MPO-H2O2-halide system. The ELISA studies were performed as described in Methods and identify oxidized Trp72 as a recognition epitope. (b) Total and ABCA1-dependent cholesterol efflux activities in RAW macrophages were performed with rh-apoA1 and the indicated mutant apoA1 forms before vs. following exposure to the MPO-H2O2-halide system, as described under Methods. (c,d): Competition ELISAs. Purified human apoA1 oxidized by the MPO/H2O2/Cl− system served as coating antigen. Antibody r8B5.2 binding was competed for with increasing concentrations of the various Trp-containing apoA1 competitor peptides incorporating either native (non-oxidized) Trp or 2-OH-Trp at the indicated positions in c. In d, either the peptide harboring native Trp, 2-OH-Trp or 5-OH-Trp corresponding to position 72 in the indicated apoA1 peptide sequence was used as the competitor. (e) Effect of Trp substitutions on rh-apoA1 ABCA1-dependent cellular cholesterol efflux function. The indicated rh-ApoA1 and mutant apoA1 proteins were exposed to the MPO/H2O2/Cl− oxidation system at the specified molar ratios of H2O2/apoA1 (0 to 10:1, mol:mol). Proteins were assayed for ABCA1-dependent cellular cholesterol acceptor activity as described in Methods. All data shown are means ± S.D. of at least triplicate determinations.
To further define the oxidized Trp residue that serves as the epitope recognized by mAb r8B5.2 and its functional significance, we generated additional site-specific mutant apoA1 forms in which each of the four Trp were converted singly to Leu (W8L, W50L, W72L and W108L) or a subset of sites W72,108 were singly mutated to Phe (W72F and W108F). Examination of each recombinant mutant apoA1 in native and MPO-oxidized forms indicated antibody (mAb r8B5.2) recognition was acquired for every apoA1 mutant following oxidation except for those with site-specific mutation of Trp 72 (Fig. 2a). Parallel competition experiments using molar excess of different oxidized forms of Trp (as free amino acids) suggested an oxygen at the 2-position of the indole ring was important for epitope recognition (Supplementary Fig. 2). Independent confirmation of a 2-oxindolyl alanine (2-OH-Trp moiety at tryptophan 72 of apoA1) (oxTrp72-apoA1) as the recognition site for mAb r8B5.2 was achieved by two additional competition ELISA studies: (i) the first involving synthetic peptides based upon the apoA1 sequences that span each of the 4 different Trp residues of apoA1 in either native form (with Trp), or harboring instead a 2-OH-Trp (Fig. 2c); and; (ii) the second involving synthetic peptides in which the apoA1 sequence spanning Trp72 was examined as native form, or as either 2-OH or 5-OH indole oxidized forms (Fig. 2d).
We next sought to determine the functional significance of Trp72 with respect to cholesterol acceptor activity within apoA1 in native and MPO-oxidized conditions. Exposure of native recombinant human apoA1 to increasing levels of oxidation by the MPO/H2O2/Cl− system initially, at molar ratios of H2O2:apoA1 of ~ 0–2, results in little loss of ABCA1-dependent cholesterol efflux activity (Fig. 2e). Proteomics studies reveal formation of methionine sulfoxide in the three apoA1 Met residues over this interval (data not shown). Upon modest further additions of oxidant exposure, there is abrupt dose-dependent inhibition in ABCA1-dependent cholesterol acceptor activity with virtually no remaining activity at H2O2:apoA1 mol ratio of 10:1 (Fig. 2e), an oxidant exposure within the upper end of that observed within atherosclerotic plaques based upon protein-bound 3-chlorotyrosine content of the apoA1 as monitored by LC/MS/MS analyses (Supplementary Fig. 1d). In contrast, the 4WF apoA1 mutant is remarkably resistant to oxidative inactivation of ABCA1-dependent cholesterol acceptor activity, demonstrating the same activity even in the presence of 50-fold molar excess of oxidant (relative to apoA1; Fig. 2e), consistent with prior reports28. We next examined site-specific mutation of just apoA1 Trp 72 into a relatively oxidant resistant hydrophobic residue (Phe or Ala). An approximate 50% reduction in ABCA1-dependent cholesterol efflux activity was observed at a 10-fold molar excess of oxidant exposure by the MPO/H2O2/Cl− system. Alternatively, mutation of the other three Trp residues of apoA1 to Phe but not Trp 72 (3F72W mutant) showed similar extent (~50%) of functional inactivation under comparable oxidant exposure (10:1, mol:mol of H2O2:apoA1) (Fig. 2e, Supplementary Fig. 1d). Collectively, these studies indicate oxidation of Trp72 of apoA1 yielding a 2-oxindolyl alanine (2-OH-Trp) moiety plays a critical role (approximately 50%) in loss of cholesterol acceptor function of the lipoprotein following exposure to the MPO/H2O2/halide system (Fig. 2e). They also demonstrate mAb r8B5.2 specifically recognizes oxTrp72-apoA1 (henceforth we will call apoA1 detected by mAb r8B5.2 “oxTrp72apoA1” to indicate the epitope recognized).
Human aortic plaque apoA1 harbors a 2-OH-Trp72 group and is “dysfunctional”
Utilizing this novel immunological tool, we first sought to show that oxTrp72-apoA1 was present in human atherosclerotic lesions. Immunohistochemical staining of human atherosclerotic plaque vs. normal human aorta revealed that oxTrp72-apoA1 is only observed within atherosclerotic lesions (Fig. 3a). Parallel staining of tissues with antibody to MPO revealed overall similar staining pattern, though weak MPO staining was also detected along the endothelial surface, within endothelial cells, and the narrow sub-endothelial space of normal human aorta (Supplementary Fig. 3a,b). We next used human aortic plaque as a tissue source for characterization of its oxTrp72-apoA1. An SDS-PAGE analysis of the low speed supernatant of human aortic plaque homogenate (n=5 subjects) that was examined before and after buoyant density ultracentrifugation to separate a global lipoprotein fraction (d ≤ 1.21) from the “lipoprotein-depleted” (LPD) fraction (d > 1.21) is shown in Figure 3b. Parallel Western blot analyses using anti-oxTrp72-apoA1 (mAb r8B5.2) revealed that virtually all detectable oxTrp72-apoA1 resided as a lipid-poor soluble form (i.e. non-lipoprotein associated) (Fig. 3c). More detailed quantification showed recovery of ~80% of the oxTrp72-apoA1 within human aortic plaque homogenate in the LPD fraction, and <1% within the lipoprotein containing fraction (Fig. 3d).
Figure 3. Characterization of oxTrp72-apoA1 recovered from human atherosclerotic plaque.
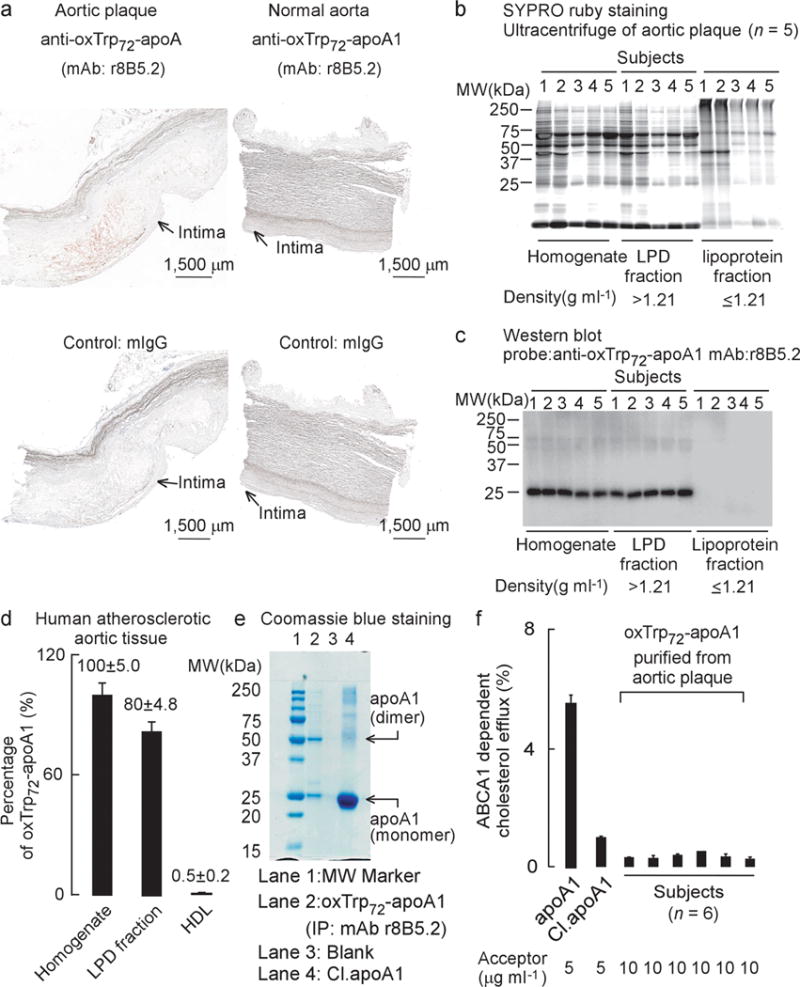
(a) Immunohistochemical staining of fresh frozen normal and atherosclerotic sections of human aorta. OxTrp72-apoA1 was detected with mAb r8B5.2. Mouse IgG was used as control. Positive staining: Red; Blue, haematoxylin counter stain. (b) SDS-PAGE, protein (SYPRO ruby) staining and (c) Western blot of human aortic plaque homogenate and the indicated fractions separated by buoyant density ultracentrifugation. (d) Level of oxTrp72-apoA1 (as a percentage relative to total apoA1) recovered in human aortic tissue homogenate vs. the lipoprotein (d ≤ 1.21 g/ml) and lipoprotein depletion (LPD) fractions (d > 1.21 g/ml). N.A. = non-detectable. Means ± S.D. of triplicate determinations are shown. (e) SDS-PAGE and Coomassie blue (protein) staining of mAb r8B5.2 immunopurified oxTrp72-apoA1 from human aortic tissue (lane 2). As positive control, apoA1 exposed to the MPO/H2O2/Cl− system (at 10:1 molar ratio of H2O2 to apoA1), Cl.apoA1, was loaded in lane 4. ApoA1 monomer and dimer (MS sequence confirmed) are indicated. (f) ABCA1-dependent cholesterol efflux activity of oxTrp72-apoA1 immunoaffinity purified from individual human aortic plaque. Native apoA1 and Cl.apoA1 served as controls.
We next sought to characterize the function of the oxTrp72-apoA1 recovered from human atherosclerotic plaque. Affinity columns with covalently linked mAb r8B5.2 were used to immuno-affinity isolate ox-apoA1 harboring the 2-OH-Trp72 moiety from human artery lesion homogenate. Examination of the affinity isolated oxTrp72-apoA1 by SDS-PAGE showed it was pure, with major visible bands at the molecular weight anticipated for the apoA1 monomer and dimer. Proteomics analyses directly detected the tryptic peptides containing oxTrp72(OH) (Supplementary Fig. 3c), and virtually no recovered tryptic peptides harboring native (unoxidized) Trp72 were observed. Further, proteomics studies of excised bands corresponding to the presumed monomer, dimer, trimer, and higher molecular weight cross-linked forms of apoA1 revealed all contained predominantly apoA1 (Supplementary Table 1a and 1b). The cholesterol acceptor function of oxTrp72-apoA1 recovered from aortic lesions from multiple subjects (n=6) was next examined. Despite using over twice the concentration (10 μg ml−1) of oxTrp72-apoA1 recovered from human atherosclerotic lesions as cholesterol acceptor compared to native human apoA1, only negligible ABCA1-dependent cholesterol efflux activity was observed (Fig. 3f). Collectively, these results directly demonstrate that a MPO-modified apoA1 form present within human atheroma, oxTrp72-apoA1, is predominantly non-HDL (lipoprotein) associated, and dysfunctional (with respect to cholesterol efflux activity).
OxTrp72-apoA1 is pro-inflammatory and formed during inflammation
In parallel studies we examined the distribution of oxTrp72-apoA1 in the circulation. Plasma from multiple healthy subjects was fractionated by buoyant density ultracentrifugation, and the level of oxTrp72-apoA1 determined within each fraction. Notably, similar to human atherosclerotic plaque, virtually all detectable oxTrp72-apoA1 in plasma was observed to reside in the LPD fraction (d > 1.21), with only nominal levels recovered in HDL (d = 1.063–1.21) (Fig. 4a–c). Because of the possibility that the high shear forces of ultracentrifugation might “knock off” oxTrp72-apoA1 from HDL, we performed independent studies to verify these findings. Plasma was recovered from 10 healthy (> 50 yr old) subjects with no known CAD and used for immuno-isolation of HDL with polyclonal antibodies (anti-HDL IgY) raised in chickens against human HDL and covalently attached to resin. Separate characterization studies of the IgY antibody binding characteristics revealed preferential recognition of lipidated (HDL-associated) apoA1, but also clear recognition of lipid-free apoA1 too (data not shown). Coomassie blue staining of native gels performed using the immune-affinity recovered material from plasma confirmed recovery of HDL from each subject (Supplementary Fig 4a). Western blot analyses with a mAb that equally recognizes both native and oxidized forms of apoA1 and HDL similarly confirmed the presence of apoA1 within the immuno-affinity isolated HDL/apoA1 from each subject (Supplementary Fig 4b). In contrast, Western blot analyses of native gels with mAb r8B5.2 did not detect any recovered material within the HDL fractions even with extensive over-exposure, despite detecting the positive control (Supplementary Fig 4c). If the larger amounts of the samples were instead examined by SDS-PAGE and Western blot using mAb r8B5.2 and extensive over-exposure, bands were observed (see below). Together, these results indicate that oxTrp72-apoA1, while present within the circulation, does not reside to any major extent on an HDL particle, but rather, is present as a low abundance lipid-poor apoA1 form in the LPD fraction.
Figure 4. Characterization of oxTrp72-apoA1 recovered from human plasma.
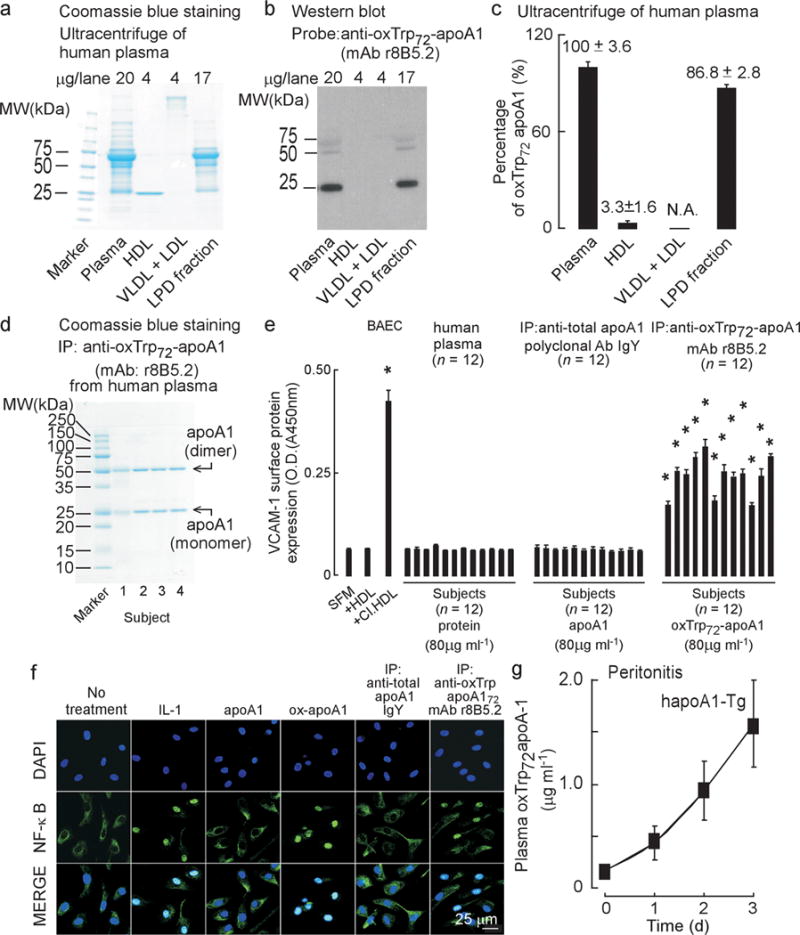
(a) SDS-PAGE, Coomassie blue staining and (b) oxTrp72-apoA1 (mAb 8B5.2) western blot of human plasma proteins before and after buoyant density ultracentrifugation separation into the indicated fractions. (c) Distribution of oxTrp72-apoA1 in plasma. Results shown are mean ± S.D. of triplicate determinations of four pooled healthy donor plasma pools. (d) SDS-PAGE with Coomassie blue staining of immuno-purified (using mAb r8B5.2) oxTrp72-apoA1 from plasma from four donors. (e) Bovine aortic endothelial cell (BAEC) were incubated with aliquots of individual subjects’ plasma, total apoA1 (recovered using immobilized anti-total apoA1 IgY) or oxTrp72-apoA1 (using immobilized mAb r8B5.2) and surface VCAM-1 protein expression was quantified by cell-based ELISA as described under Methods. Serum free media (SFM), HDL, or HDL previously exposed to the MPO/H2O2/Cl− system (Cl.HDL, at H2O2:apoA1, 10:1, mol:mol) were used as vehicle, negative and positive controls, respectively. *, P < 0.05. (f) Nuclear translocation of NF-κB: Confocal immunofluorescent analysis of BAEC cells, untreated or treated with the following: IL-1 (positive control) native (apoA1) or MPO-oxidized human apoA1 (ox-apoA1), plasma-derived total apoA1 immunopurified using IgY to apoA1; plasma oxTrp72-apoA1 immuno-purified using r8B5.2. NF-κB p65 (E498) staining is green and nuclear counterstaining (DAPI) is blue. (g) Human apoA1-Tg mice were injected i.p. with zymosan (T=0). Plasma oxTrp72-apoA1 levels were quantified at the indicated times by sandwich ELISA using mAb r8B5.2 as capture antibody, and mAb 10G1.5 (anti-total apoA1) as detecting antibody, as described in Methods.
To characterize its biological activity, we next used mAb 8B5.2 to immuno-affinity isolate oxTrp72-apoA1 from the circulation. As shown in Fig. 4d, Coomassie Blue staining of the recovered protein revealed it was quite pure, with the major visible bands corresponding to the apoA1 monomer and oxidative cross-linked dimer. Proteomics analyses confirmed the recovered bands as apoA1, with multiple peptides containing oxidized residues noted (predominantly Trp and Met; Supplementary Table 1c). Parallel Western blot analyses of the immunopurified apoA1 readily showed bands at the anticipated molecular weight of the apoA1 monomer and dimer using an mAb that recognizes native and oxidized apoA1 forms alike (mAb 10G1.5, “anti-total” apoA1)25, as well as with anti-oxTrp72-apoA1 (i.e. mAb r8B5.2; Supplementary Fig. 4d). Parallel control Western blot analyses showed no significant light or heavy chain immunoglobulin contamination under the conditions employed (Supplementary Fig. 4d). We and others have shown that HDL isolated from healthy volunteers promotes anti-inflammatory effects when incubated with bovine aortic endothelial cells, whereas apoA1 or HDL modified by MPO ex vivo converts the lipoprotein into a pro-inflammatory particle29. We therefore examined the pro-inflammatory properties of oxTrp72-apoA1 recovered from the circulation of apparently healthy middle aged (>50 yr old) donors (n=12). For comparison, total apoA1 was isolated from the same donors using polyclonal antibodies raised in chickens (IgY) against native (total) human apoA1 (Fig. 4e). Notably, when oxTrp72-apoA1 recovered from each subject’s plasma was incubated with bovine aortic endothelial cells, a marked pro-inflammatory effect was observed as monitored by substantial increases in surface VCAM1 protein. In contrast, when apoA1 recovered from plasma with IgY immunoprecipitation was used or a comparable amount of plasma protein was used, no pro-inflammatory response in the endothelial cells was noted (Fig. 4e). Parallel studies confirmed the ability of immuno-isolated lipid-free (organic solvent extracted) oxTrp72-apoA1 recovered from plasma to promote endothelial cell NF-κB activation and nuclear localization, similar to in vitro oxidized lipid-free apoA1 (by MPO-H2O2-Cl− system) and IL-1 exposure as positive controls. The pro-inflammatory behavior observed with lipid-free oxTrp72-apoA1 contrasts with the lack of NF-κB activation observed using lipid-free apoA1 isolated from plasma HDL recovered during either buoyant density ultracentrifugation or an affinity column generated with anti-human apoA1 IgY and employed under conditions that quantitatively removed apoA1 from plasma (Fig. 4f).
We next investigated whether formation of oxTrp72-apoA1 is specific to atherosclerotic plaque only or if it is formed in other sub-acute inflammatory conditions, such as peritonitis (induced by i.p. injection of yeast cell wall protein carbohydrate components, zymosan). We have previously shown MPO serves as a major enzymatic catalyst for both protein and lipid oxidation within the model35. Formation of oxTrp72-apoA1 was readily detected over time in both peritoneal lavage fluid (not shown) and plasma in human apoA1-Tg mice, demonstrating endogenous human apoA1 may be targeted for post translational modification at Trp72 during sub-acute inflammation (Fig. 4g). Thus, apoA1 may be oxidized at a local site of inflammation, whether it be the artery wall or an inflamed peritoneal cavity, and then diffuse into the circulation.
OxTrp72-apoA1 has impaired ABCA1-dependent HDL biogenesis activity in vivo
ABCA1 is critical for HDL biogenesis and hepatocyte ABCA1 expression accounts for nearly 80% of plasma HDL production36. Given that MPO potently inhibits ABCA1-dependent cholesterol efflux activity of apoA1/HDL in vitro, and our site-directed mutagenesis studies support a critical role for apoA1 Trp72 in ABCA1-dependent efflux activity (Fig. 2e), we wanted to demonstrate that oxTrp72-apoA1 recovered from human plasma has impaired capacity to stimulate HDL biogenesis in vivo. We first focused our attention on development of an animal model in which to monitor nascent HDL particle formation in vivo via an ABCA1-dependent pathway. Injection of lipid-free human apoA1 sub cutaneously (s.c). into apoA1-KO mice was shown to result in appearance of apoA1 into the vascular compartment (i.e. plasma), incremental increases in circulating HDL particle cholesterol content, and incorporation of human apoA1 into the HDL particle pool (Figs. 5a–d). To determine if these were ABCA1-dependent phenomenon, we performed parallel studies injecting lipid-free human apoA1 s.c. into hepatocyte-specific ABCA1-KO mice vs. the appropriate WT controls36. The time-dependent increases in HDLc observed in plasma following apoA1 injection demonstrated an absolute requirement for ABCA1, and the majority of human apoA1 appearance within the HDL fraction was attenuated in the hepatocyte-specific ABCA1-KO mouse (Supplementary Figs. 5a–c).
Figure 5. OxTrp72-apoA1 has impaired function in vivo and is associated with CVD.
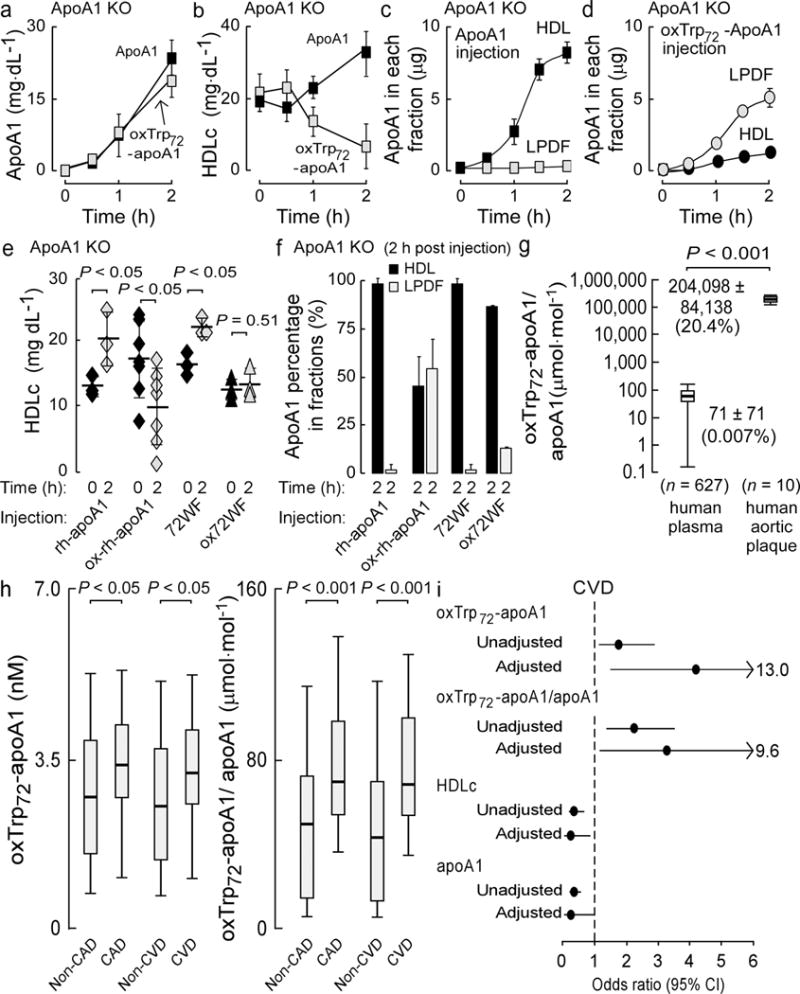
ApoA1 KO mice (6–8 weeks old) were injected s.c. with comparable amounts of apoA1 or oxTrp72apoA1 as described in Methods. Total apoA1 levels (a), HDLc levels (b), and apoA1 distribution within HDL or LPD fractions of plasma (c,d) were quantified at the indicated times. In (e), plasma HDLc levels are shown from apoA1-KO mice injected s.c. with equivalent amounts of either rh-apoA1, 72WF, or their corresponding MPO-oxidized forms (formed at H2O2:apoA1, 10:1, mol:mol) beflore and 2h following apoa1 injections. ApoA1 distribution within plasma HDL or LPD fractions (f) were also quantified pre-injection (0h) and 2h post injection. (g) Levels of oxTrp72-apoA1 (relative to total apoA1) in plasma and human aortic plaque homogenate from the indicated number of subjects were quantified by sandwich ELISA using mAb r8B5.2 as capture antibody and mAb 10G1.5 (anti-total apoA1) as detecting antibody. (h) Box whisker plots of oxTrp72-apoA1 concentration, and oxTrp72-apoA1/total apoA1 ratio (as percent) within plasma. (i) Odds ratio and 95% confidence intervals for CVD risk (unadjusted and following multilogistic regression analyses) are shown. The fully loaded model included oxTrp72-apoA1, apoA1, age, sex, hypertension, diabetes, smoking, HDLc, LDLc, TG, hsCRP, HgbA1C, MPO and lipid lowering medicine.
Using this in vivo model of “ABCA1-dependent HDL biogenesis”, we next examined whether oxTrp72-apoA1 recovered from human plasma was indeed dysfunctional in vivo. Injection of apoA1-KO mice s.c. with equivalent amounts of either lipid-free native human apoA1 or immunoaffinity isolated oxTrp72-apoA1 resulted in comparable time-dependent appearance of total apoA1 in the circulation (Fig. 5a). However, whereas native human apoA1 s.c. injection induced incremental increases in HDLc levels over a short period of time (2h), injection of the oxTrp72-apoA1 did not increase HDLc levels (despite being in the vascular compartment), and even reproducibly slightly reduced HDLc levels (Fig. 5b). Native human apoA1 appearing in the circulation resided entirely within HDL particles (Fig. 5c), whereas oxTrp72-apoA1 remained predominantly within the LPDF (Fig. 5d). Of note, while lipid-free apoA1 modified by the MPO/H2O2/Cl− system in vitro is capable of forming a reconstituted nascent HDL particle using the cholate dialysis method (data not shown), it similarly (to oxTrp72-apoA1 recovered from human plasma) fails to interact productively with ABCA1 facilitating cholesterol efflux and nascent HDL formation (Fig. 2b,2e).
We next tested whether an apoA1 form in which Trp72 is mutated to Phe (72WFapoA1), which is more resistant to MPO-catalyzed oxidative inactivation in vitro (Fig 2e), demonstrated improved biological function relative to native human apoA1 following oxidation. Native “wild type” recombinant human apoA1 (rh-apoA1) and 72WFapoA1 were each examined for in vivo HDL biogenesis activity in native and MPO-oxidized form. Within 2h of s.c. injection of native rh-apoA1 into apoA1-KO mice, incremental increases in HDLc were observed within plasma, consistent with de novo HDL biogenesis (Fig. 5e). In contrast, s.c. injection of rh-apoA1 exposed to the MPO-H2O2-Cl− system (apoA1:H2O2, 10:1) was accompanied by time (within 2h) dependent decrease in HDLc level (Fig. 5e). Parallel studies using the 72WFapoA1 mutant revealed comparable levels of total apoA1 within the vascular compartment following s.c. injection of either native or oxidized forms of rh-apoA1 and 72WFapoA1 (Supplementary Fig. 6a). Further, similar magnitude increase in HDLc was observed following s.c. injection of the unmodified mutant apoA1, and no reduction in HDLc levels following s.c. injection of ox72WFapoA1 (Fig. 5e). Examination of the HDL particle distribution of the injected apoA1 forms revealed more significant differences (Fig. 5f, Supplementary Fig. 6b). Whereas MPO-catalyzed oxidation of rh-apoA1 produced an oxapoA1 form that in majority failed to become incorporated into an HDL particle, mutation of apoA1 Trp72 to Phe completely protected the mutant apoA1 form, permitting virtually all circulating ox72WFapoA1 to become incorporated into HDL particles (Fig. 5f). Thus, site-specific mutation of apoA1 at Trp 72 into the more oxidant resistant residue Phe produced a mutant apoA1 form with some protection from oxidative loss in HDL biogenesis activity, consistent with in vitro data demonstrating improved interaction with ABCA1 following exposure to MPO-catalyzed oxidation (Fig. 2e).
OxTrp72-apoA1 is abundant in human atheroma and plasma levels predict CVD risk
To quantify the content of oxTrp72-apoA1 in human plasma and aortic atherosclerotic plaque homogenate we developed a sandwich immunoassay using mAb r8B5.2 as the capture antibody, and as detecting antibody we used a mAb previously developed that recognizes native vs. oxidized and lipid-free vs. HDL-associated apoA1 forms to a comparable extent (anti-total apoA1, mAb 10G11.5;25). Sequential consenting subjects (n = 627) undergoing elective risk factor evaluation and cardiac examination in an outpatient preventive cardiology clinic were examined. Supplementary Table 2a shows the clinical and demographic characteristics of the cohort, of which 35% had clinical evidence of CVD. Quantification of oxTrp72-apoA1 and total apoA1 levels in plasma of the entire cohort revealed that on average only 0.007% of the apoA1 in plasma harbors the 2-OH-Trp72 moiety, and that there was wide variation in levels among the subjects (Fig. 5g). In contrast, oxTrp72-apoA1 represented 20% of total apoA1 within atherosclerotic plaque-laden aorta (n = 10), over 1000-fold more abundant (relative to total apoA1) than observed in plasma (Fig. 5g; Supplementary Fig. 7a,b). For further illustrative purposes, plasma from a random sampling of non-CVD (n=8) and CVD subjects (n=8) are shown following SDS PAGE and either Coomasie Blue staining for protein, or Western blot analyses using each mAb employed in the sandwich immunoassay (Supplementary Fig. 7c–e). Examinations of the distribution of HDLc and apoA1 levels within the entire cohort reveal the expected inverse correlations with CAD and CVD in the cohort (Supplementary Fig. 7f–i). In contrast, the distribution of both the absolute concentration of oxTrp72-apoA1 and the ratio of oxTrp72-apoA1/total apoA1 within the entire cohort showed significantly higher levels in subjects with either CAD or CVD compared to their corresponding controls (i.e. non-CAD or non-CVD; Fig. 5h). Further analyses of circulating oxTrp72-apoA1 levels (both absolute concentration and as ratio relative to total apoA1) within the entire cohort revealed that higher levels, particularly within the 4th quartile relative to 1st, were independently associated with increased CAD and CVD risks (Fig. 5i, Supplementary Table 2b). The positive association with increased CAD and CVD risks remained significant even following adjustments for traditional CVD risk factors, apoA1, MPO and high sensitivity C-reactive protein levels, and lipid lowering medication use (Fig. 5i, Supplementary Table 2b). The improvement in model performance introduced by the inclusion of oxTrp72-apoA1 was evaluated using net reclassification index (NRI) and integrated discrimination improvement (IDI). AUC was calculated using the area under ROC curve. Though there is no significant gain in AUC, inclusion of oxTrp72-apoA1 to the fully adjusted model resulted in a significant improvement in risk estimation, based on NRI (CAD: 4.1%, P < 0.001 CVD: 5.9%, P < 0.001) and IDI (20% for both CAD and CVD, P < 0.001). Of note, when the prognostic value of HDLc and apoA1 were similarly examined within the cohort, each demonstrated the expected inverse association with CVD and CAD risks (Fig. 5i, Supplementary Table 2b). Comparisons of the odds ratio (95% confidence interval) for oxTrp72-apoA1 versus traditional risk factors for CVD are shown in Supplementary Table 2c, and illustrate oxTrp72-apoA1 levels are comparable in magnitude to numerous established risk factors within the cohort.
DISCUSSION
The atheroprotective role of HDL has been questioned due to recent failures of multiple HDLc raising drugs and mendelian randomization studies showing genetic variants that control HDLc levels are not associated with risk of CAD16–18,37. Yet there is overwhelming preclinical data showing apoA1 promotes anti-atherosclerotic functions1,3,5–6. This apparent contradiction has spurred interest in better understanding the role of the HDL particle in human coronary artery disease. Ideally, a means of monitoring HDL function in the circulation should reflect what is occurring within the diseased artery wall. Studies thus far of apoA1 recovered from human atheroma reveal it is highly oxidized, cross-linked, dysfunctional (with respect to cholesterol efflux and LCAT activities), and lipid-poor since it is not associated with an HDL particle25. Despite the recognition that apoA1 in lesions is heavily oxidized, direct functional interrogation of a structurally specific oxidative modification on apoA1 recovered from the artery wall has not yet been reported. The present studies identify for the first time the structural basis of an abundant dysfunctional form of apoA1/HDL within the artery wall – MPO-catalyzed site-specific oxidation of Trp72 of apoA1 forming an oxindolyl alanine (2-OH-Trp) moiety. Quantification in human atheroma reveals it is highly prevalent, accounting for one in five apoA1 within the lesion. Remarkably, the oxidized apoA1 form, oxTrp72-apoA1, was only nominally HDL particle associated, with virtually all found in both the artery wall and within plasma as a lipid-poor form. Thus, studies of HDL-like particles recovered from human arterial lesion homogenates using buoyant density ultracentrifugation, an approach typically used by some studying apoA1 from lesions38–39, would essentially miss the vast majority of this abundant modified apoA1 form in vivo. Of note, lipid-poor apoA1 modified by the MPO/H2O2/Cl- system acquires pro-inflammatory activities, and lipid-free oxTrp72-apoA1 recovered from human plasma was capable of activating endothelial cell NF-κB. Whether differences in lipid binding capability of oxidized vs. native apoA1 forms contribute to pathophysiological functions is unclear.
An overall scheme of our findings is illustrated in Fig. 6a. Circulating HDL readily diffuses into the artery wall. Within atherosclerotic lesions, MPO is enriched and promotes site-specific oxidative modification of apoA1 at residue Trp72. Site-directed mutagenesis studies show that this residue is essential for ABCA1-dependent cholesterol efflux activity of apoA1 in vitro, and in vivo animal model studies with the 72WFapoA1 mutant demonstrate the importance of this residue in loss of apoA1 association with HDL particles in vivo following MPO-catalyzed oxidation. Recovery of apoA1 from lesions using mAb specific for recognition of oxTrp72-apoA1 revealed virtually no detectable ABCA1-dependent cholesterol acceptor activity of the recovered ox-apoA1. Further biological characterization of the modified lipoprotein revealed it was lipid-poor, not associated with the HDL particle, harbored potent pro-inflammatory activity including promotion of endothelial cell NF-κB activation, nuclear localization and surface expression of VCAM-1 protein, an adhesion protein with known pro-atherogenic function40. Importantly, direct injection of apoA1 harboring the 2-OH-Trp moiety into apoA1-KO mice confirmed this post-translationally modified apoA1 form possesses impaired HDL biogenesis activity since it both failed to elicit any incremental increases in circulating HDLc levels or appear on HDL particles, despite being at comparable level as native apoA1 within the plasma compartment in the HDL biogenesis model.
Figure 6. Formation of a dysfunctional apoA1 form, oxTrp72-apoA1, within human atherosclerotic lesions.
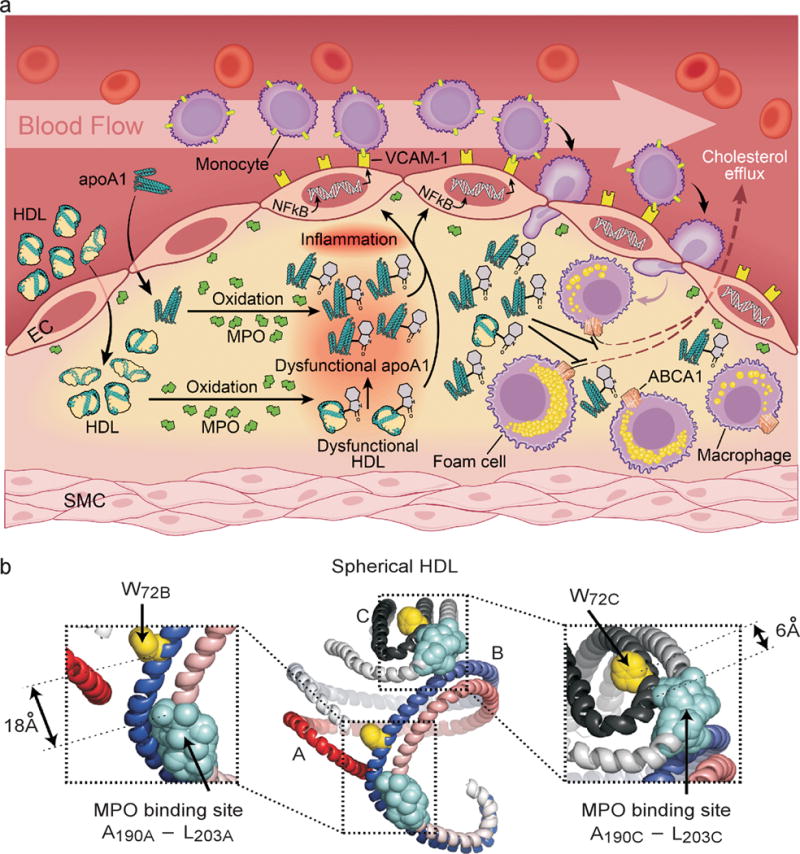
(a) Schematic summary illustrating formation of an abundant dysfunctional apoA1 form in human atheroma generated by MPO. (b) Model of spherical HDL demonstrating close spatial proximity between MPO binding site on one apoA1 chain, and Trp72 on the contralateral anti-parallel apoA1 chain. Shown are the longest (18Å) and shortest (6Å) predicted distances among two of the three apoA1 chains43. The apoA1 polypeptide chains are depicted in red and slate gray. W72, yellow; MPO interaction site with apoA1, light blue.
Another intriguing finding is the specificity observed for the MPO/H2O2/Cl− oxidative system in generating a 2-oxindolyl alanine (2-OH-Trp) moiety on apoA1 at Trp72. Detailed structural analyses reveals a remarkable specificity for oxidation on the indole ring at the 2 position, as well as at residue Trp72 of apoA1. Examination of the mechanism accounting for the selective targeting of the Trp72 site of apoA1 by the MPO system and not alternative oxidation systems appears to have, in part, a structural basis – the close proximity between the MPO binding site on the anti-parallel apoA1 chain and Trp72 of apoA1 (Fig. 6b). It is widely believed that apoA1 is configured as anti-parallel amphipathic helices within HDL, either between two anti-parallel chains, or in some regions, as a hairpin27,41–45. We recently used small angle neutron scattering with contrast variation to directly visualize the protein shape of apoA1 within spherical HDL, the most abundant form of HDL in the circulation44. Coupled with cross-linking mass spectrometry studies and molecular modeling, we developed a model of apoA1 within spherical HDL44. When the location of apoA1 residues involved in MPO binding as determined by hydrogen/deuterium exchange (residues A190–L203)26 are highlighted on this model along with apoA1 Trp72, the distance between Trp72 and the MPO binding region on each of the apoA1 chains is remarkably short, ranging between a maximum predicted length of 18 Å to a minimal distance of only 6 Å (Fig. 6b). The predicted distance between Trp72 of one apoA1 chain and the MPO binding site in nascent HDL similarly shows a short diffusion distance (14Å) (Supplementary Fig. 8). The oxidant formed by the MPO/H2O2/Cl- system, HOCl, is highly reactive, and is rapidly scavenged by the first susceptible target it encounters (which is virtually any amino acid, though Met, Cys, His, Trp, Lys and Tyr are most reactive46). The short diffusion distance between the MPO binding site and Trp72 of all HDL models examined no doubt contributes to the remarkable selective targeting of Trp72 for oxidative modification by MPO (Fig. 6b).
While quantification of oxTrp72-apoA1 reveals it to be highly enriched within the atherosclerotic plaque, no detectable immunostaining with mAb 8B5.2 was observed within normal aortic tissue. Thus, within the artery wall, MPO-dependent oxidative modification of apoA1 appears to be highly correlated with the atherosclerotic disease process. However, our studies also observed increases in circulating oxTrp72-apoA1 levels in human apoA1-Tg mice during a sub-acute inflammation model. Thus, apoA1 may be oxidized at a local site of inflammation, whether it be the artery wall or an inflamed peritoneal cavity, and then diffuse into the circulation. Based upon its mechanism of formation, one would not anticipate that detection of oxTrp72-apoA1 in the circulation would serve as a specific indicator of atherosclerosis. None-the-less, multilogistic regression analyses within the clinical cohort examined demonstrates that elevated levels of the dysfunctional apoA1 form are sufficiently selectively associated with CAD and CVD risks so as to remain statistically significant following adjustments for multiple traditional risk factors including apoA1, MPO and other markers of inflammation. It is tempting to speculate that the pro-inflammatory biological activity observed with oxTrp72-apoA1 may render it pro-atherogenic, thus possibly serving as a contributor to the well-recognized phenomenon of increased CVD risk during chronic inflammatory conditions. However, further studies are needed to test this intriguing hypothesis.
Given the 1000-fold enrichment of oxTrp72-apoA1 content (relative to total apoA1) in atheroma and our histological studies, our studies suggest that formation of the modified apoA1 form occurs within the protected environment of the subendothelial space (Fig. 6a), and the oxTrp72-apoA1 observed within the circulation reaches the intravascular compartment by diffusion out of the artery wall. MPO has been shown to accumulate within the subendothelial compartment47, and it has been mechanistically linked to formation of vulnerable plaque, such as through catalytic consumption of nitric oxide48–49, activation of matrix metalloproteinase pathways50, or promotion of endothelial cell apoptosis and superficial erosions within culprit lesions51. Indeed, it is of interest that MPO is being examined as a potential target for functional imaging of vulnerable plaque52–53. The present studies suggest that quantification of oxTrp72-apoA1 within the circulation may serve as a gauge of molecular processes that impair apoA1/HDL function in the artery wall, and thus serve as a new diagnostic and therapeutic target in CAD.
Online methods
Materials
Human MPO was isolated and characterized as described54. [3H]Cholesterol was obtained from Amersham Biosciences (Pisc-ataway, NJ). L-[13C6]tyrosine, L-[13C9,15N1]tyrosine, [13C6,15N1] Leucine, L-[13C9,15N1]Phenylalanine, and L-[13C5,15N1]Valine were purchased from Cambridge Isotope Laboratories Inc. (Andover, MA). Mouse immunoglobulin isotyping kit was obtained from GIBCO BRL (Life Technologies GmbH). Trypsin was from Promega (Madison, WI) and all cell lines were from the American Type Culture Collection (Rockville, MD). 5-hydroxy-L-tryptophan was purchased from MP Biomedicals (Solon, OH). 6-Hydroxyindole and 7-hydroxyindole were purchased from Chem-Impex International Inc. (Wood Dale, IL). All peptides were synthesized by the CS Bio Company, Inc. (Menlo Park, CA). All other reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise specified.
General procedures
All study protocols were approved by the Institutional Review Board of the Cleveland Clinic, and all subjects gave written informed consent. All mouse studies were performed under protocols approved by the Institutional Animal Care and Use Committee at the Cleveland Clinic.
Circulating HDL and plasma-derived purified apoA1 were obtained from healthy volunteer donors as previously described55. Protein content was determined by the Markwell-modified Lowry protein assay56 with BSA as standard. Recombinant human apoA1 was generated in an Escherichia coli expression system and isolated by sequential column chromatographies as described26,57. Briefly, rh-apoA1 and mutants were expressed in Escherichia Coli strain BL21 (DE-3) pLysS. All the mutations were made by using the QuickChange Mutagenesis Kit (Stratagene, La Jolla, CA) and confirmed by DNA sequencing. 4WF and 4WL represent recombinant human apoA1 in which all of the endogenous tryptophans (residues 8, 50, 72, and 108) were converted to phenylalanine or leucine, respectively. W8L, W50L, W72L, W72F, W108F and W108L are recombinant human apoA1 mutants with the indicated single tryptophan mutation. 3MV is an apoA1 mutant in which all three methiones (at residues 86, 112, and 148) were mutated to valine. 7YF is an apoA1 mutant in which all seven tyrosines (at residues 18, 29, 100, 115, 166, 192, and 236) were converted to phenylalanine. Reconstituted nascent HDL was prepared using the modified sodium cholate dialysis method58 at an initial molar ratio of 100:10:1 of POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine):cholesterol:apoA1. To totally remove all endotoxin from purified recombinant apoA1, all mutants were lipidated (using POPC:apoA1, 100:1 mol:mol) forming nascent HDL, and then delipidated by organic solvent extraction for 6 consecutive rounds. Endotoxin levels in all recombinant apoA1 were confirmed to be nominal (< 0.5 EU/mg/ml protein) by Limulus amoebocyte lysate assay (Charles River Laboratories, Wilmington, MA). Concentrations of apoA1 mutants were determined by both their predicted extinction coefficient at 280nm and amino acid composition analyses following HCl hydrolysis (in pure form). Accuracy of protein concentrations of isolated pure apoA1 mutants was determined by stable isotope dilution LC/MS/MS analyses on an AB SCIEX 5000 triple quadrupole mass spectrometer using synthetic L-[13C9,15N1]tyrosine, [13C6,15N1]leucine, L-[13C9,15N1]phenylalanine, and L-[13C5,15N1]valine as internal standards, and predicted sequence of the mutant. The concentration of H2O2 was determined spectrophotometrically (ε240 = 39.4 M−1cm−1). HOCl was added as NaOCl into buffered reaction mixtures. The concentration of NaOCl was determined spectrophotometrically (ε292=350 M−1cm−1). Peroxynitrite(ONOO−) was quantified spectrophotometrically (ε302= 1670 M−1cm−1). Protein-bound ClTyr was quantified by stable isotope dilution LC/MS/MS analyses using synthetic 3-Cl[13C6] tyrosine and L-[13C9,15N1] tyrosine as internal standards on an AB SCIEX 5000 triple quadrupole mass spectrometer as described26,35. Monoclonal antibodies specific for apoA1 or MPO modified apoA1 (exposed to MPO/H2O2/Cl− or MPO/H2O2/NO2− system; H2O2:apoA1 molar ratio of 5:1) were generated via hybridoma technology59. Cholesterol efflux (ABCA1-dependent and total) activity of apoA1 was determined as described26. Specifically, RAW264.7 cells (ATCC,Rockville, MD) were treated with 2 μg ml−1 of apoA1 (Fig. 2b,e) or indicated amount of proteins in (Fig. 3f) for 6 hours. Efflux to cholesterol acceptor was calculated as described26. 2,3,-Dioxindolyl alanine and 2-oxindoly lalanine were synthesized as described in a prior publication60. Western blot quantification was done using either ImageJ software (version 1.46, http://rsbweb.nih.gov/ij/index.html) or Image Studio software (version 2, LI-COR Biosciences, Lincoln, NB). HDL containing lipoprotein fraction (d ≤ 1.21 g ml−1) and LPD fractions (d > 1.21 g ml−1) were recovered from plasma or tissue homogenate by sequential buoyant density ultracentrifugation using sucrose and D2O to avoid high ionic strength associated alterations to protein compositions of lipoprotein particles observed with KBr use61. Mice plasma (10–40μl) was diluted to 500μl with 1XPBS buffer (pH 7.0) and 887 μl of sucrose D2O buffer(d =1.325 g ml−1; 0.1742 g K2HPO4, 0.1361 g KH2PO4, 0.8182 g NaCl, 111.1 g sucrose and 100 ml of D2O). The fraction with density less than 1.21 g ml was obtained after a 48 h ultracentrifugation at 40,000 rpm at 20°C (Beckman rotor 50.4Ti, 8×49mm ultra-clear centrifuge tubes). The lipoprotein fraction (d ≤ 1.21 g ml−1) was recovered by slicing the upper 0.25 ml of the tube. HDL cholesterol precipitating reagent (StanBIO Laboratory, Cat. No. 0599) was added into mice plasma to precipitate LDL, and HDL cholesterol was quantified by stable isotope dilution gas chromatography mass spectrometry analyses62.
Phage display and affinity maturation of parental antibody
A single chain (scFv) phage display library was constructed by using the cDNA derived from the 8B5.2 hybridoma cell line using methods as described by Barbas et al63. The murine Kappa variable light chain families VK1 to VK17 and the variable heavy chain families VH1 to VH19 were amplified using 26 pairs of family-specific primers and then the variable light chain and variable heavy chain sequences “stitched” together via a flexible linker (SSGGGGSGGGGGGSSRSS) using overlap PCR and cloned into phagemid pCOMB3 as described63. Recombinant phages were prepared from the library with the helper phage M13K07 at a multiplicity of infection of ~10. The library was selected using immunotubes (Costar #3690) coated with 0.5 ml 25 μg ml−1 Cl.apoA1 (apoA1 exposed to the MPO/H2O2/Cl− system at 10:1 molar ratio of H2O2 to apoA1) in 50 mM carbonate buffer, pH 9.6 overnight at 4°C64 The rescue-selection-plating cycle was repeated three times. For affinity maturation, another phage display scFv library was constructed by error-prone PCR and followed by passaging the library in XL1-red mutator E. coli (Stratagene, LaJolla, CA) for four times. Screening of scFv by dissociation rate constant (Koff) was performed using real-time biospecific interaction analysis based on surface plasmon resonance (SPR) in a Biacore 2000 (GE Healthcare Life Sciences, Piscataway, NJ). A third library was generated by introducing all combinations of mutant residues found in the nine individual clones into variable regions of the scFv gene. The scFv gene was subcloned to a dicistronic baculoviral shuttle vector (a generous gift from Mifang Liang, China CDC) then recloned into a mammalian expression vector and the dicistronic alleles expressed in DG44 CHO cells as a chimeric human IgG antibody.
Surface plasmon resonance
In a Biacore flow cell, approximately 2,000 resonance units (RU) of Cl.apoA1 (5 μg ml−1) in 10 mM sodium acetate, pH 4.5 was coupled to a CM5 sensor chip. Association and dissociation rates of original 8B5.2 and recombinant affinity matured antibody were measured under continuous flow of 30 ml min−1 using a concentration range from 0.7 to 1000 nM. The Ka1 and Kd1 values were calculated using BIAeveluation software and sensorgrams were fitted by a bivalent model. The apparent KD was calculated from the ratio of Kd1/Ka1.
Immunopurification of oxTrp72apoA1
Antibodies were covalently coupled to UltraLink Hydrazide Resin (Pierce Chemical Co., Rockford, IL) for immunoaffinity isolations of native or oxidized apoA1 forms from plasma. Before immune-affinity isolations of apoA1 forms, human albumin was first depleted from plasma as follows. Human plasma was dialyzed in a binding buffer (20 mM MOPS, pH7.0, 100 mM NaCl) and diluted 4-fold in the binding buffer before it was loaded onto 500 ml column bed of Q Sepharose XL resin (GE Healthcare Life Sciences, Piscataway, NJ). Albumin depleted plasma was collected as flow through (and Western analyses with various anti-apoA1 mAb confirmed complete recovery of all apoA1 forms as well). This flow through fraction was delipidated, and to more completely remove contaminating immunoglobulins during the immune-affinity isolation step, reduced and denatured with guanidine hydrochloride (3 M final, pH7.8) and dithiothreitol (1mM final). After the denatured fraction was incubated for 1 h at room temperature, the protein was alkylated by the addition of iodoacetamide (2.5 mM final) and incubated for 1 h at room temperature in a foil wrapped container. The denatured and alkylated fraction was dialyzed against 20 mM Tris.Cl, 150 mM NaCl and oxTrp72-apoA1 was purified from this solution using a covalently coupled r8B5.2 affinity column. Total apoA1 was similarly purified using a covalently coupled chicken polyclonal IgY that recognizes total apoA1 (Genway Biotech, San Diego, CA).
Cell Surface VCAM-1 protein expression
Cell surface VCAM-1 protein levels were determined in BAEC. The BAEC cells were starved in DMEM for 4 h and then incubated with 80 μg of protein per ml of HDL, or 80 μg of protein per ml of HDL exposed to MPO/H2O2/Cl− system (Cl.HDL) or, where indicated, individual protein and plasma for 16 h. After one wash with phosphate-buffered saline, the cells were fixed in 4% paraformaldehyde for 20 min and blocked in 0.5% casein in PBS with 0.02% NaN3 for 4 h. Surface VCAM-1 protein was determined using goat polyclonal anti-VCAM-1 primary antibody (1:4000, catalog # sc-1504, Santa Cruz Biotechnology, Santa Cruz, CA,) with anti-goat IgG antibody conjugated with horseradish peroxidase (1:5000, #sc-2020, Santa Cruz Biotechnology, Santa Cruz, CA,) as a secondary antibody, detection by SureBlue tetramethylbenzidine peroxidase substrate (KPL Inc., Gaithersburg, MD) and measuring absorbance at 450 nm on a 96-well plate reader (Spectramax 384 Plus; Molecular Devices, Sunnyvale, CA) after the addition of 1 M HCl to stop the reaction.
Confocal immunofluorescent analysis
BAEC cells (2 × 104 cells per well) were inoculated into Lab-Tek Chamber 16 well glass slides (Electron Microscopy Sciences, Hatfield, PA) for 16 h. The cells were treated with drugs (Fig. 4 panel f) 80 μg ml−1 each at 37°C for 20 mins. The cells were then washed with PBS and fixed with 4% paraformaldehyde in PBS (15 min, room temperature) and permeabilized by immersion in 0.5% Triton-X100 (10 min, room temperature). Cells were quenched with 100mM glycine (10 min, room temperature) and blocked in 1%BSA / 10% normal goat serum in PBS, pH 7.0 (1 h at room temperature). Blocked cells were incubated sequentially with primary antibody NF-κB p65 (E498) (cat # 3987, Cell Signaling Technology, Danvers, MA) diluted in blocking buffer (overnight, 4 °C) and secondary antibody goat anti rabbit IgG-FITC (cat # ab6717-1, Abcam, Cambridge, MA) diluted in blocking buffer (100:1, 1 h, room temperature). Coverslips were mounted on to glass slides using Vectashield DAPI mounting media (Vector Laboratories Inc., Burlingame, CA). Images were viewed on a Leica DMRXE confocal microscope (Leica Microsystems Inc. Buffalo Grove, IL) using a 63 X oil-immersion objective with a 1.40 numeric aperture. Confocal images were acquired using the Leica Confocal Software Version 2.61.
Immunochemistry staining of atherosclerotic plaques
Fresh human aortic tissue was obtained as discarded surgical material, both at time of organ harvest from transplant donors, and during valve/aortic arch (“elephant trunk”) replacement surgery. Immunohistochemistry of atherosclerotic plaque and normal aorta was carried out on fresh frozen sections. The anti-oxTrp72apoA1 antibody (r8B5.2 Fab) version and polyclonal rabbit anti human MPO (#A0398, Dako North America, Inc., Carpinteria, CA) were used at a dilution of 1:400. Mouse normal IgG1 (X0931, Dako North America, Inc., Carpinteria, CA) was used as control for antibody staining. The staining procedure followed the protocol of the DakoCytomation Envision+ system HRP(AEC) (Dako North America, Inc., Carpinteria, CA).
Aortic tissue homogenization
At time of harvest, all tissues were immediately rinsed in ice-cold PBS supplemented with 100 μM diethylenetriamine pentaacetic acid (DTPA) and frozen at −80°C in argon-sparged buffer A (65 mM sodium phosphate, pH 7.4, 100 μM DTPA, 100 μM butylated hydroxytoluene) in gas tight containers overlaid with argon, which were kept within larger argon flushed sealed containers. At time of use, samples sealed in gas-tight containers were placed in ice/water bath. Immediately before complete thaw, the tissues were rinsed five times with ice-cold argon sparged Ca2+ and Mg2+-free PBS supplemented with 100 μM DTPA to remove any residual visible hemaglobin. The aorta segment was rapidly cleaned of fatty tissues and placed in a Petri dish containing ice-cold PBS buffer. The adventitial layer and outer portion of the media were carefully peeled from the vessel to give a clean “smooth” aorta. The aorta was rinsed with ice-cold PBS three times and the wet weight of the aorta was determined. The tissue was then cut into small pieces (about 3 to 5 mm in diameter) in a Petri dish cover on ice. Tissue pieces were suspended in ice-cold Ca2+ and Mg2+-free PBS containing a protease inhibitor cocktail. The protease cocktail PMSF (1 mM) and Sigma-Aldrich protease inhibitor cocktail (catalog no. P8340) were included in all subsequent solutions used for lipoprotein isolation. Aortic tissue was homogenized on ice with a Polytron Tissue Homogenizer (Model PT 10/35, Brinkman Instruments Inc., Westbury, NY) for 1 min followed by 5 × 30 sec intervals in ice/water bath. The homogenate was then centrifuged at 12,000 × g for 30 min. The pellet was discarded and the supernatant stored on ice until use. HDL and LPD fractions were separated by ultracentrifugation using a sucrose and D2O protocol61.
Indirect ELISA
Nunc MaxSorp ELISA plates (#446612, Thermo Fisher Scientific, Waltham, MA) were coated with the indicated purified native or oxidized protein (0.2 μg ml−1) in Carbonate and Bicarbonate buffer, pH 9.6. Purified human apoA1, rh-apoA1 and mutant apoA1’s were modified by either the MPO/H2O2/Cl− system, the MPO/H2O2/NO2− system or by multiple other oxidation systems as indicated at 10:1 molar ratio of oxidants/apoA1, unless otherwise specified, details of oxidation conditions are described in reference 25. Primary antibody r8B5.2 (50 ng ml−1) (Fig. 1d and Fig. 2a,e) was added into the 5% BSA blocked ELISA wells and incubated for 1 hr at room temperature. The plates were then washed with PBS, 0.05% Tween 20 (PBST) 4 times. The signal was detected by Peroxidase AffiniPure Goat Anti-Mouse IgG, F(ab’)2 Fragment Specific (1:10,000 dilution, Code # 115-036-006, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA)
Competitive ELISA
Nunc MaxSorp ELISA plates (#446612, Thermo Fisher Scientific, Waltham, MA) were coated with 0.5 μg ml−1 Cl.apoA1 in Carbonate and Biocarbonate buffer, pH 9.6. (human apoA1 modified by MPO/H2O2/Cl− complete oxidative system at 10:1 molar ratio of H2O2/apoA1). r8B5.2 (25 ng ml−1) and peptides were added into the 5% BSA blocked ELISA wells and incubated for 1 hr at room temperature. The signal was detected by Peroxidase AffiniPure Goat Anti-Mouse IgG, F(ab’)2 Fragment Specific (1:10,000 dilution, Code # 115-036-006, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA).
Sandwich ELISA
Costar ELISA plates (#3690) were coated with capture antibody r8B5.2 (50 μl per well, 7 μg ml−1 in 0.1M Tris.Cl pH8.5) at 4°C for overnight. Except for the capture antibody coating process, all other reactions were performed at room temperature (20 °C). Coated plates were washed with PBS buffer twice, and then washed plates were blocked with 170 μl of blocking buffer [1% fish peptone (Fluka Biochemika, CAT# 93490), 3% BSA (Sigma cat# A7030), 2% Poly Vinyl Alcohol, 0.05% Tween 20 in PBS, pH 7.4] for 2 h. A calibration curve was constructed with Cl.apoA1 (8 pM – 8nM) in sample dilution buffer (1% salmon serum, 0.05% Tween 20 in PBS, pH 7.4). Pooled plasma samples which had been aliquoted into single use samples were thawed once and used as controls on every plate. Plasma samples were diluted in sample dilution buffer (1% salmon serum, 0.05% Tween 20 in PBS, pH 7.4). Diluted samples were added into the blocked EIA wells at 50μl per well and incubated for 45 min. The plates were then washed with PBST 4 times. Detecting mAb 10G1.5 (anti-total apoA125) was diluted (1:40,000) in antibody dilution buffer (0.03%BSA (Sigma cat# A7030) in PBST) and incubated in EIA well at 50 μl per well for 30 min. After four washes with PBST, HRP conjugated neutravidin (Cat #31001, Pierce Chemical Co. Rockford, IL; 1:10,000 diluted in antibody dilution buffer,) was added into the ELISA plates (50 μl per well) and incubated for 30 min. The ELISA was developed with SureBlue TMB substrate (Cat. #52-00-0, KPL, Inc., Gaithersburg, MD) and read at O.D. 450nm.
Research subjects
Levels of oxTrp72-apoA1 were determined in sequential consenting subjects presenting to the Preventive Cardiology outpatient clinic of the Cleveland Clinic as part of an observational study called BioBank. Fasting plasma samples and associated clinical data were collected at time of patient visit. CAD definition included adjudicated diagnoses of stable or unstable angina, myocardial infarction, angiographic evidence of > 50% stenosis of one or more major epicardial vessels or history of coronary revascularization. PAD was defined as any evidence of extra-coronary atherosclerosis or ABI <0.9. Atherosclerotic CVD was defined as the presence of either CAD or PAD. Patients who had experienced a myocardial infarction or stroke within 1 month preceding enrollment were ineligible. All subjects gave written informed consent and the Institutional Review Board of the Cleveland Clinic approved all study protocols.
Mice models
Breeders of all conventional mice (C57BL/6J, apoA1 Tg+/+ and apoA1−/− mice on a C57BL/6J background) were obtained from Jackson Laboratories. Wildtype fl/fl (WT fl/fl) and hepatocyte-specific ABCA1 knockout apoA1 (ABCA1 KO) mice on a C57BL/6J background have been described36,65. All animal studies were performed under approval of the Animal Research Committee of the Cleveland Clinic. To induce a subacute peritonitis model, apoA1 Tg+/+ animals were injected i.p. with zymosan. Plasma was obtained at baseline before injection, and then serially (1, 2 and 3 days) following injection. OxTrp72-apoA1 levels in plasma were measured by sandwich ELISA using mAb r8B5.2 as capture antibody, and mAb 10G1.5 (anti-total apoA125) as detecting antibody. To examine the role of hepatocyte ABCA1 in de novo HDL biogenesis in vivo following apoA1 injection, WT fl/fl and hepatocyte-specific ABCA1-KO mice (6–7 weeks old) were injected with the indicated apoA1 forms s.c. at 400 mg kg−1. Blood was collected by saphenous vein puncture one week prior to apoA1 injection (baseline) as well as 0.5h, 1h, 1.5h and 2h post apoA1 injection time. ApoA1-KO mice were similarly s.c. injected with the indicated apoA1 forms s.c. at 400 mg kg−1. and the appearance and distribution of total human apoA1 and oxTrp72-apoA1 levels in blood monitored both at baseline and 2h post apoA1 injection time.
Statistical analysis
Data are presented as median (first quartile–third quartile) for continuous measures and as number (percentage) for categorical measures. The Student’s t-test or Wilcoxon-Rank sum test for continuous variables and chi-square test for categorical variables were used to examine the difference between participants who had cardiovascular disease events and those who did not. Logistic regression models with adjustment for traditional Framingham cardiac risk factors (age, sex, hypertension, diabetes mellitus, current or former smoker, LDLc, HDLc), triglycerides (TG), apoA1, high sensitivity C-reactive protein (hsCRP), MPO, hemoglobin A1C (HgbA1C) and history of lipid lowering medicine were used to estimate odds ratio and 95% confidence interval for CVD or CAD. All analyses were performed using R 2.15.0 (Vienna, Austria) and P-values <0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
This study was supported by National Institutes of Health grants P01HL098055 and HL119962. BioBank, the clinical study from which samples were analyzed, was supported in part from P01HL098055, P01HL076491, R01HL103866, P20HL113452 and R01HL103931. This work was also supported in part by a grant from the LeDucq Fondation. SLH is also partially supported by a gift from the Leonard Krieger Fund. Mass spectrometry instrumentation used was housed within the Cleveland Clinic Mass Spectrometry Facility with partial support through a Center of Innovation by AB SCIEX.
Footnotes
AUTHOR CONTRIBUTIONS
YH participated in all laboratory, animal and human studies, assisted in statistical analyses, helped design the experiments, and drafted the manuscript. BL, GG, VG, CK, ZW and XF assisted with various laboratory and mass spectrometry studies; DS, JB, MC, SB, and CC helped perform various animal experiments; JD, DS, TK, XG, MC, JH, AD, and DP helped make various bacterial expression clones and produce and purify recombinant proteins used; JD and SL helped with mAb generation and screening; TK and TN helped with ELISA assays; LL and YW provided statistical analyses of clinical data; JD, LC, EP, PF, VG, WT, JP, EF, JS and SH provided experimental analysis and expertise; All authors took part in critical review of the manuscript; the project was scientifically conceived and directed by SH.
COMPETING FINANCIAL INTERESTS
Dr. Tang has previously received research grant support from Abbott Laboratories. Drs. Hazen, Wang, Levison and Smith report being listed as co-inventor on pending and issued patents held by the Cleveland Clinic relating to cardiovascular diagnostics or therapeutics. Dr. Hazen reports having been paid as a consultant for the following companies: AstraZeneca Pharmaceuticals LP, Cleveland Heart Lab, Esperion, Lilly, Liposcience Inc., Merck & Co., Inc., Pfizer Inc., Procter & Gamble, and Takeda. Dr. Hazen reports receiving research funds from Cleveland Heart Lab, Liposcience Inc., Procter & Gamble, and Takeda. Dr. Smith reports having the right to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics or therapeutics from Cleveland Heart Lab and Esperion, and being paid as a consultant for Esperion. Dr. Hazen reports having the right to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics or therapeutics and the companies shown below: Cleveland Heart Lab., Esperion, Frantz Biomarkers, LLC, Liposcience Inc. Drs. Levison and Wang report having the right to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics from Liposcience Inc.
References
- 1.Barter PJ, et al. Antiinflammatory properties of HDL. Circulation research. 2004;95:764–772. doi: 10.1161/01.RES.0000146094.59640.13. [DOI] [PubMed] [Google Scholar]
- 2.Duffy D, Rader DJ. Update on strategies to increase HDL quantity and function. Nat Rev Cardiol. 2009;6:455–463. doi: 10.1038/nrcardio.2009.94. [DOI] [PubMed] [Google Scholar]
- 3.Navab M, Reddy ST, Van Lenten BJ, Fogelman AM. HDL and cardiovascular disease: atherogenic and atheroprotective mechanisms. Nat Rev Cardiol. 2011;8:222–232. doi: 10.1038/nrcardio.2010.222. [DOI] [PubMed] [Google Scholar]
- 4.Khera AV, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. The New England journal of medicine. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nature cell biology. 2011;13:423–433. doi: 10.1038/ncb2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fisher EA, Feig JE, Hewing B, Hazen SL, Smith JD. High-density lipoprotein function, dysfunction, and reverse cholesterol transport. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:2813–2820. doi: 10.1161/ATVBAHA.112.300133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. The American journal of medicine. 1977;62:707–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 8.Badimon JJ, Badimon L, Galvez A, Dische R, Fuster V. High density lipoprotein plasma fractions inhibit aortic fatty streaks in cholesterol-fed rabbits. Laboratory investigation; a journal of technical methods and pathology. 1989;60:455–461. [PubMed] [Google Scholar]
- 9.Badimon JJ, Badimon L, Fuster V. Regression of atherosclerotic lesions by high density lipoprotein plasma fraction in the cholesterol-fed rabbit. The Journal of clinical investigation. 1990;85:1234–1241. doi: 10.1172/JCI114558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rubin EM, Krauss RM, Spangler EA, Verstuyft JG, Clift SM. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature. 1991;353:265–267. doi: 10.1038/353265a0. [DOI] [PubMed] [Google Scholar]
- 11.Plump AS, Scott CJ, Breslow JL. Human apolipoprotein A-I gene expression increases high density lipoprotein and suppresses atherosclerosis in the apolipoprotein E-deficient mouse. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:9607–9611. doi: 10.1073/pnas.91.20.9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hughes SD, Verstuyft J, Rubin EM. HDL deficiency in genetically engineered mice requires elevated LDL to accelerate atherogenesis. Arteriosclerosis, thrombosis, and vascular biology. 1997;17:1725–1729. doi: 10.1161/01.atv.17.9.1725. [DOI] [PubMed] [Google Scholar]
- 13.Nissen SE, et al. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA : the journal of the American Medical Association. 2003;290:2292–2300. doi: 10.1001/jama.290.17.2292. [DOI] [PubMed] [Google Scholar]
- 14.Sacks FM, et al. Selective delipidation of plasma HDL enhances reverse cholesterol transport in vivo. Journal of lipid research. 2009;50:894–907. doi: 10.1194/jlr.M800622-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tardif JC, et al. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA : the journal of the American Medical Association. 2007;297:1675–1682. doi: 10.1001/jama.297.15.jpc70004. [DOI] [PubMed] [Google Scholar]
- 16.Barter PJ, et al. Effects of torcetrapib in patients at high risk for coronary events. The New England journal of medicine. 2007;357:2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 17.Nissen SE, et al. Effect of torcetrapib on the progression of coronary atherosclerosis. The New England journal of medicine. 2007;356:1304–1316. doi: 10.1056/NEJMoa070635. [DOI] [PubMed] [Google Scholar]
- 18.Boden WE, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. The New England journal of medicine. 2011;365:2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 19.Voight BF, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–580. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhattacharyya T, et al. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA: the journal of the American Medical Association. 2008;299:1265–1276. doi: 10.1001/jama.299.11.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Besler C, et al. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. The Journal of clinical investigation. 2011;121:2693–2708. doi: 10.1172/JCI42946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sorci-Thomas MG, Thomas MJ. High density lipoprotein biogenesis, cholesterol efflux, and immune cell function. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:2561–2565. doi: 10.1161/ATVBAHA.112.300135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shih DM, et al. Combined serum paraoxonase knockout/apolipoprotein E knockout mice exhibit increased lipoprotein oxidation and atherosclerosis. The Journal of biological chemistry. 2000;275:17527–17535. doi: 10.1074/jbc.M910376199. [DOI] [PubMed] [Google Scholar]
- 24.Tang WH, et al. Clinical and genetic association of serum paraoxonase and arylesterase activities with cardiovascular risk. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:2803–2812. doi: 10.1161/ATVBAHA.112.253930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Didonato JA, et al. Function and distribution of apolipoprotein A1 in the artery wall are markedly distinct from those in plasma. Circulation. 2013;128:1644–1655. doi: 10.1161/CIRCULATIONAHA.113.002624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng L, et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. The Journal of clinical investigation. 2004;114:529–541. doi: 10.1172/JCI21109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Z, et al. The refined structure of nascent HDL reveals a key functional domain for particle maturation and dysfunction. Nature structural & molecular biology. 2007;14:861–868. doi: 10.1038/nsmb1284. [DOI] [PubMed] [Google Scholar]
- 28.Peng DQ, et al. Apolipoprotein A-I tryptophan substitution leads to resistance to myeloperoxidase-mediated loss of function. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:2063–2070. doi: 10.1161/ATVBAHA.108.173815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Undurti A, et al. Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. The Journal of biological chemistry. 2009;284:30825–30835. doi: 10.1074/jbc.M109.047605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hadfield KA, et al. Myeloperoxidase-derived oxidants modify apolipoprotein A-I and generate dysfunctional high-density lipoproteins: comparison of hypothiocyanous acid (HOSCN) with hypochlorous acid (HOCl) The Biochemical journal. 2013;449:531–542. doi: 10.1042/BJ20121210. [DOI] [PubMed] [Google Scholar]
- 31.Van Lenten BJ, et al. Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. The Journal of clinical investigation. 1995;96:2758–2767. doi: 10.1172/JCI118345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ansell BJ, et al. Inflammatory/antiinflammatory properties of high-density lipoprotein distinguish patients from control subjects better than high-density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation. 2003;108:2751–2756. doi: 10.1161/01.CIR.0000103624.14436.4B. [DOI] [PubMed] [Google Scholar]
- 33.Charles-Schoeman C, et al. Effects of high-dose atorvastatin on antiinflammatory properties of high density lipoprotein in patients with rheumatoid arthritis: a pilot study. The Journal of rheumatology. 2007;34:1459–1464. [PubMed] [Google Scholar]
- 34.Shao B, Pennathur S, Heinecke JW. Myeloperoxidase targets apolipoprotein A-I, the major high density lipoprotein protein, for site-specific oxidation in human atherosclerotic lesions. The Journal of biological chemistry. 2012;287:6375–6386. doi: 10.1074/jbc.M111.337345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brennan ML, et al. A tale of two controversies: defining both the role of peroxidases in nitrotyrosine formation in vivo using eosinophil peroxidase and myeloperoxidase-deficient mice, and the nature of peroxidase-generated reactive nitrogen species. The Journal of biological chemistry. 2002;277:17415–17427. doi: 10.1074/jbc.M112400200. [DOI] [PubMed] [Google Scholar]
- 36.Timmins JM, et al. Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. The Journal of clinical investigation. 2005;115:1333–1342. doi: 10.1172/JCI23915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barter PJ, Kastelein JJ. Targeting cholesteryl ester transfer protein for the prevention and management of cardiovascular disease. Journal of the American College of Cardiology. 2006;47:492–499. doi: 10.1016/j.jacc.2005.09.042. [DOI] [PubMed] [Google Scholar]
- 38.Bergt C, et al. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proc Natl Acad Sci U S A. 2004;101:13032–13037. doi: 10.1073/pnas.0405292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shao B, et al. Tyrosine 192 in apolipoprotein A-I is the major site of nitration and chlorination by myeloperoxidase, but only chlorination markedly impairs ABCA1-dependent cholesterol transport. The Journal of biological chemistry. 2005;280:5983–5993. doi: 10.1074/jbc.M411484200. [DOI] [PubMed] [Google Scholar]
- 40.Cybulsky MI, et al. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. The Journal of clinical investigation. 2001;107:1255–1262. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Segrest JP, et al. A detailed molecular belt model for apolipoprotein A-I in discoidal high density lipoprotein. The Journal of biological chemistry. 1999;274:31755–31758. doi: 10.1074/jbc.274.45.31755. [DOI] [PubMed] [Google Scholar]
- 42.Wu Z, et al. Double superhelix model of high density lipoprotein. The Journal of biological chemistry. 2009;284:36605–36619. doi: 10.1074/jbc.M109.039537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gogonea V, et al. Congruency between biophysical data from multiple platforms and molecular dynamics simulation of the double-super helix model of nascent high-density lipoprotein. Biochemistry. 2010;49:7323–7343. doi: 10.1021/bi100588a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu Z, et al. The low resolution structure of ApoA1 in spherical high density lipoprotein revealed by small angle neutron scattering. The Journal of biological chemistry. 2011;286:12495–12508. doi: 10.1074/jbc.M110.209130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gogonea V, et al. The low-resolution structure of nHDL reconstituted with DMPC with and without cholesterol reveals a mechanism for particle expansion. Journal of lipid research. 2013;54:966–983. doi: 10.1194/jlr.M032763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pattison DI, Davies MJ. Absolute rate constants for the reaction of hypochlorous acid with protein side chains and peptide bonds. Chemical research in toxicology. 2001;14:1453–1464. doi: 10.1021/tx0155451. [DOI] [PubMed] [Google Scholar]
- 47.Baldus S, et al. Endothelial transcytosis of myeloperoxidase confers specificity to vascular ECM proteins as targets of tyrosine nitration. The Journal of clinical investigation. 2001;108:1759–1770. doi: 10.1172/JCI12617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abu-Soud HM, Hazen SL. Nitric oxide is a physiological substrate for mammalian peroxidases. The Journal of biological chemistry. 2000;275:37524–37532. doi: 10.1074/jbc.275.48.37524. [DOI] [PubMed] [Google Scholar]
- 49.Eiserich JP, et al. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science. 2002;296:2391–2394. doi: 10.1126/science.1106830. [DOI] [PubMed] [Google Scholar]
- 50.Wang Y, et al. Myeloperoxidase inactivates TIMP-1 by oxidizing its N-terminal cysteine residue: an oxidative mechanism for regulating proteolysis during inflammation. The Journal of biological chemistry. 2007;282:31826–31834. doi: 10.1074/jbc.M704894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sugiyama S, et al. Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:1309–1314. doi: 10.1161/01.ATV.0000131784.50633.4f. [DOI] [PubMed] [Google Scholar]
- 52.Nahrendorf M, et al. Activatable magnetic resonance imaging agent reports myeloperoxidase activity in healing infarcts and noninvasively detects the antiinflammatory effects of atorvastatin on ischemia-reperfusion injury. Circulation. 2008;117:1153–1160. doi: 10.1161/CIRCULATIONAHA.107.756510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ronald JA, et al. Enzyme-sensitive magnetic resonance imaging targeting myeloperoxidase identifies active inflammation in experimental rabbit atherosclerotic plaques. Circulation. 2009;120:592–599. doi: 10.1161/CIRCULATIONAHA.108.813998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hazen SL, Heinecke JW. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. The Journal of clinical investigation. 1997;99:2075–2081. doi: 10.1172/JCI119379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zamanian-Daryoush M, et al. The cardioprotective protein apolipoprotein A1 promotes potent anti-tumorigenic effects. J Biol Chem. 2013;288:21237–21252. doi: 10.1074/jbc.M113.468967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Markwell MA, Haas SM, Bieber LL, Tolbert NE. A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Analytical biochemistry. 1978;87:206–210. doi: 10.1016/0003-2697(78)90586-9. [DOI] [PubMed] [Google Scholar]
- 57.Ryan RO, Forte TM, Oda MN. Optimized bacterial expression of human apolipoprotein A-I. Protein expression and purification. 2003;27:98–103. doi: 10.1016/s1046-5928(02)00568-5. [DOI] [PubMed] [Google Scholar]
- 58.Matz CE, Jonas A. Micellar complexes of human apolipoprotein A-I with phosphatidylcholines and cholesterol prepared from cholate-lipid dispersions. The Journal of biological chemistry. 1982;257:4535–4540. [PubMed] [Google Scholar]
- 59.Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 60.Todorovski T, Fedorova M, Hennig L, Hoffmann R. Synthesis of peptides containing 5-hydroxytryptophan, oxindolylalanine, N-formylkynurenine and kynurenine. Journal of peptide science : an official publication of the European Peptide Society. 2011;17:256–262. doi: 10.1002/psc.1322. [DOI] [PubMed] [Google Scholar]
- 61.Stahlman M, et al. Proteomics and lipids of lipoproteins isolated at low salt concentrations in D2O/sucrose or in KBr. Journal of lipid research. 2008;49:481–490. doi: 10.1194/jlr.D700025-JLR200. [DOI] [PubMed] [Google Scholar]
- 62.Robinet P, Wang Z, Hazen SL, Smith JD. A simple and sensitive enzymatic method for cholesterol quantification in macrophages and foam cells. Journal of lipid research. 2010;51:3364–3369. doi: 10.1194/jlr.D007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barbas CF, III, Burton DR, Scott JK, Silverman GJ. Phage display: a laboratory manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. [Google Scholar]
- 64.Marks JD, et al. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. Journal of molecular biology. 1991;222:581–597. doi: 10.1016/0022-2836(91)90498-u. [DOI] [PubMed] [Google Scholar]
- 65.Chung S, et al. Targeted deletion of hepatocyte ABCA1 leads to very low density lipoprotein triglyceride overproduction and low density lipoprotein hypercatabolism. The Journal of biological chemistry. 2010;285:12197–12209. doi: 10.1074/jbc.M109.096933. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.