

Summary
TIF1β is a transcriptional corepressor that recruits repressive chromatin modifiers to target genes. Its biological function and physiological targets in somatic stem cells remain largely unknown. Here, we show that TIF1β is essential for the maintenance of hematopoietic stem cells (HSCs). Deletion of Tif1b in mice induced active cycling and apoptosis of HSCs and promoted egression of HSCs from the bone marrow, leading to rapid depletion of HSCs. Strikingly, Tif1b-deficient HSCs showed a strong trend of ectopic expression of nonhematopoietic genes. Levels of heterochromatin protein 1 (HP1α, β and γ) proteins, which form a complex with TIF1β, were significantly reduced in the absence of TIF1β and depletion of HP1 recapitulated a part of the phenotypes of Tif1b-deficient HSCs. These results demonstrate that the TIF1β-HP1 system functions as a critical repressive machinery that targets genes not normally activated in the hematopoietic compartment, thereby maintaining the transcriptional signature specific to HSCs.
Highlights
-
•
Deletion of Tif1b in mice causes rapid depletion of HSCs
-
•
Loss of TIF1β leads to reduction in HP1 proteins in HSCs
-
•
The TIF1β-HP1 system represses nonhematopoietic genes in HSCs
-
•
The TIF1β-HP1 system helps maintain the transcriptional integrity of HSCs
Iwama and colleagues show that TIF1β, a transcriptional corepressor that recruits repressive chromatin modifiers to target genes, is essential for the maintenance of hematopoietic stem cells. In collaboration with HP1 proteins, TIF1β functions as a critical repressive machinery that targets genes not normally activated in the hematopoietic compartment, thereby maintaining the transcriptional signature specific to HSCs.
Introduction
During hematopoiesis, hematopoietic stem cells (HSCs) activate specific sets of genes and silence others. Transcription factors fulfill an obvious role in this process. At the same time, chromatin modifiers regulate the accessibility of transcription factors to cis elements by establishing regions of chromatin that are either permissive or repressive to transcription. These epigenetic transcriptional regulations are crucial for the maintenance of multipotency in stem cells (Sauvageau and Sauvageau, 2010; Sashida and Iwama, 2012). Polycomb group (PcG) proteins, which establish a reversible silencing state through repressive histone modifications, maintain multipotency of HSCs by keeping hematopoietic developmental regulator genes poised for activation via bivalent histone domains (Oguro et al., 2010). However, the mechanisms by which nonhematopoietic genes, which should never be activated in the hematopoietic cell lineage, are transcriptionally repressed remain to be elucidated.
TIF1β (also called KAP1 or TRIM28) is a transcriptional corepressor that associates with hundreds of Kruppel-associated box domain-zinc finger proteins (KRAB-ZFPs) that bind DNA in a sequence-specific fashion. TIF1β acts as a scaffold for a multimolecular complex that silences transcription through the formation of heterochromatin by recruiting the histone methyltransferase SETDB1, heterochromatin protein 1 (HP1), or the NuRD-histone deacetylase complex (Nielsen et al., 1999; Schultz et al., 2001, 2002). The KRAB/KAP1 system plays a critical role in the control of endogenous retroviruses during development (Rowe et al., 2010, 2013) but also regulates multiple aspects of mammalian physiology. In hematopoiesis, it functions in erythropoiesis and in the prevention of autoinflammatory T cell development (Chikuma et al., 2012; Barde et al., 2013).
In this study, we demonstrate an essential role for the TIF1β/HP1 system in the maintenance of HSCs and implicate this system in the establishment of transcriptional signature of HSCs by keeping nonhematopoietic genes transcriptionally silent.
Results and Discussion
Deletion of Tif1b Severely Compromises HSC Function in the Fetal Liver
Tif1b-deficient mice show early developmental defects and die by embryonic day 7.5 (E7.5) (Cammas et al., 2000). To delineate function of TIF1β in HSCs, we conditionally deleted Tif1b by crossing Tif1bfl/fl mice with Tie2-Cre mice, which specifically express Cre in hematopoietic and endothelial cells (Tie2-Cre;Tif1bfl/fl). We confirmed efficient deletion of Tif1b in fetal liver lineage-marker (Lin)−c-KIT+ hematopoietic progenitor cells from Tie2-Cre;Tif1b mice by western blot and immunocytochemical analyses in Lin−c-KIT+SCA-1+ (LSK) hematopoietic stem and progenitor cells (HSPCs) (Figures S1A and S1B available online). Tie2-Cre;Tif1bfl/fl embryos were recovered at nearly the expected Mendelian ratio at E13.5, but about 28% (58 out of 207 Tie2-Cre;Tif1bfl/fl embryos) were already dead and no mutant embryos were born alive (data not shown). Tie2-Cre;Tif1bfl/fl embryos were pale and their fetal livers were significantly smaller than those of the littermate controls (Figures 1A and 1B). Flow cytometric analyses demonstrated a significant reduction in TER119+ erythroblasts in Tie2-Cre;Tif1bfl/fl fetal livers (Figure 1B). Detailed analysis revealed a block in differentiation of Tif1bΔ/Δ erythroblasts from CD71+TER119− early erythroblasts to CD71+TER119+ erythroblasts (Figure S1C), resulting in a drastic reduction in the numbers of Tif1bΔ/Δ CD71+TER119+ and CD71−TER119+ mature erythroblasts (Figure 1C). Indeed, Tif1bΔ/Δ erythroblasts were mostly proerythroblasts, while the control erythroblasts were at various differentiation stages from proerythroblasts to mature erythroblasts (Figure 1D). These data are consistent with the recent reports on the impact of deletion of Tif1b on erythropoiesis (Barde et al., 2013), suggesting that severe anemia due to impaired erythroid differentiation could account for embryonic lethality of Tie2-Cre;Tif1bfl/fl embryos.
Figure 1.
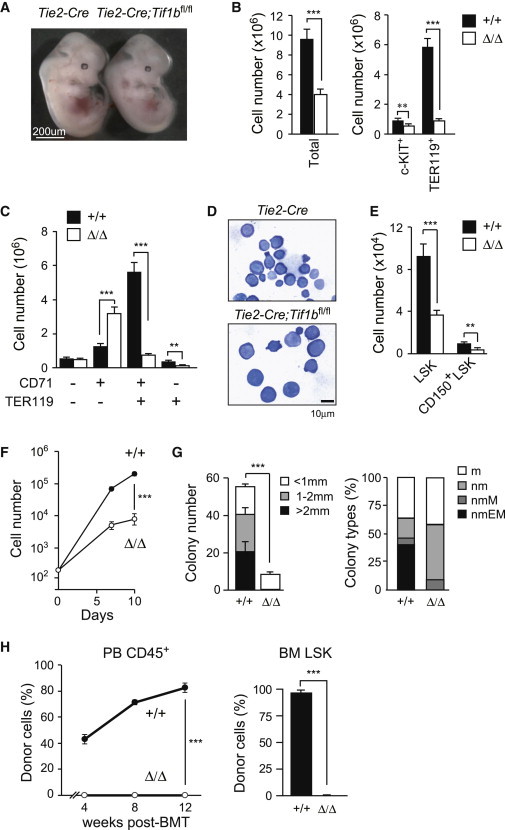
Tie2-Cre;Tif1bfl/fl Embryos Die at Midgestation
(A) Appearance of Tie2-Cre (Tif1b+/+) and Tie2-Cre;Tif1bfl/fl (Tif1bΔ/Δ) embryos at E13.5.
(B) Absolute numbers of whole fetal liver cells, c-KIT+ progenitors, and TER119+ erythroblasts at E13.5. The data are shown as mean ± SEM (Tie2-Cre, n = 16; Tie2-Cre;Tif1bfl/fl, n = 17).
(C) Absolute numbers of erythroid cells at different stages defined by CD71 and TER119 expression in fetal livers at E13.5. The data are shown as mean ± SEM (Tie2-Cre, n = 16; Tie2-Cre;Tif1bfl/fl, n = 17).
(D) Morphology of Tif1b+/+ and Tif1bΔ/Δ fetal liver hematopoietic cells from E13.5 embryos stained with May-Grünwald-Giemsa solution.
(E) Absolute cell numbers of LSK and CD150+LSK cells in the fetal liver at E13.5. The data are shown as mean ± SEM (Tie2-Cre, n = 23; Tie2-Cre;Tif1bfl/fl, n = 21).
(F) Growth of Tif1b+/+ and Tif1bΔ/Δ LSK cells from E13.5 fetal livers in liquid culture in the presence of 50 ng/ml of SCF and TPO. The data are shown as mean ± SEM for triplicate cultures.
(G) Colony-forming assays with Tif1b+/+ and Tif1bΔ/Δ fetal liver cells. Left panel shows the absolute number of colonies with the indicated size per 20,000 fetal liver cells. The data are shown as mean ± SEM for triplicate cultures. Right panel shows the proportion of colony types defined by the composition of colony-forming cells (n, neutrophil; m, macrophage; E, erythroblast; M, megakaryocyte).
(H) Competitive repopulating assays. A total of 2 × 105Tif1b+/+ and Tif1bΔ/Δ fetal liver cells from E13.5 embryos (CD45.2) mixed with the same number of competitor BM cells (CD45.1) were transplanted into CD45.1 recipients. Chimerism of donor-derived CD45.2+ cells in the PB is shown in the left panel. Donor chimerism in the BM LSK cells at 12 weeks posttransplantation is shown in the right panel. The data are shown as mean ± SEM (n = 6). ∗p < 0.05; ∗∗p < 0.005; ∗∗∗p < 0.0005.
Of note, CD150+LSK HSCs and LSK HSPCs were also significantly reduced in Tie2-Cre;Tif1bfl/fl fetal livers at E13.5 compared to the controls (Figures 1E and S1D). Tif1bf/Δ LSK cells proliferated poorly in liquid culture compared to the control cells (Figure 1F). In colony-forming assays, Tif1bΔ/Δ LSK cells gave rise to significantly fewer colonies than control cells and failed to form colonies larger than 2 mm in diameter. Most of the colonies generated from Tif1bΔ/Δ LSK cells consisted of only neutrophils and macrophages with very few megakaryocytes but completely lacked erythroblasts (Figure 1G). We next performed competitive reconstitution assays. Not surprisingly, Tif1bΔ/Δ cells did not contribute to hematopoiesis in the recipient mice in either the peripheral blood (PB) or the bone marrow (BM) (Figure 1H). Further examination revealed that Tif1b+/Δ cells also performed poorly in competitive reconstitution assays, suggesting a haploinsufficient effect with Tif1b heterozygotes (data not shown). Moreover, Tif1bΔ/Δ cells failed to extend the survival of lethally irradiated recipient mice (Figure S1E). These results indicate that loss of TIF1β severely impairs the capacity of HSCs to repopulate hematopoiesis in vivo.
Deletion of Tif1b Causes BM Failure Accompanied by Depletion of HSCs
To evaluate the role of TIF1β in adult BM hematopoiesis, we next analyzed Cre-ERT;Tif1bfl/fl mice. To exclude any influences of deletion of Tif1b on the BM microenvironment, we first transplanted BM cells from Cre-ERT control and Cre-ERT;Tif1bfl/fl mice without competitor cells into lethally irradiated wild-type recipient mice. After confirming engraftment, we deleted Tif1b by inducing nuclear translocation of Cre with intraperitoneal injection of tamoxifen at 8 weeks after transplantation. Deletion of Tif1b was very efficient, with deletion efficiency in BM Lin−c-KIT+ progenitor cells at 92.6% by genomic quantitative PCR (data not shown). As indicated by mild reduction in BM cellularity and mild cytopenia in Cre-ERT control mice, tamoxifen had some toxic effects on hematopoiesis at the early time points postinjection (Figures 2B and S2A). Deletion of Tif1b, however, induced progressive hypoplasia of the BM and the spleen (Figures 2A and 2B) and caused obvious cytopenia in the PB (Figure S2A). Although residual Ly5.1+ host cells gradually overtook Tif1bΔ/Δ cells and eventually restored the blood cell counts in the PB (Figures S2A and S2B), about 70% of the transplanted mice died of BM failure within 40 days of tamoxifen injection (Figure 2C).
Figure 2.
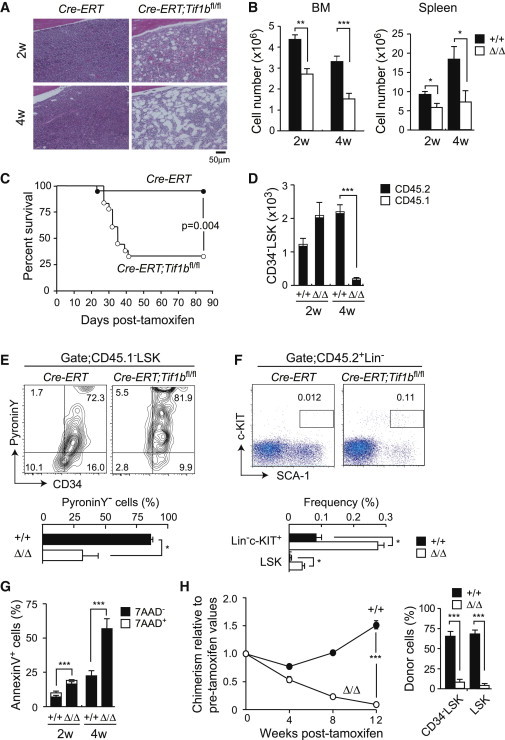
Deletion of Tif1b Leads to Rapid BM Failure
(A) Hematoxylin and eosin staining of BM sections from Cre-ERT (Tif1b+/+) and Cre-ERT;Tif1bfl/fl (Tif1bΔ/Δ) mice at 2 and 4 weeks postdeletion of Tif1b. BM cells from 8-week-old Cre-ERT and Cre-ERT;Tif1bfl/fl mice were transplanted into lethally irradiated recipient mice without competitor cells. At 8 weeks after transplantation, recipient mice were injected with tamoxifen for 5 consecutive days.
(B) Absolute numbers of total BM and spleen cells at the indicated time points postdeletion of Tif1b (2 weeks [2w], n = 8; 4 weeks [4w], n = 8–10).
(C) Survival curve of the recipient mice repopulated with BM cells from Cre-ERT and Cre-ERT;Tif1bfl/fl mice after deletion of Tif1b (n = 12).
(D) Absolute numbers of total Tif1b+/+ and Tif1bΔ/Δ CD34−LSK HSCs in the BM at the indicated time points after deletion of Tif1b (n = 8).
(E) Cell-cycle status of Tif1b+/+ and Tif1bΔ/Δ CD34−LSK HSCs determined by incorporation of Pyronin Y at 2 weeks after the first injection of tamoxifen. Representative flow cytometric profiles are depicted (upper panel) and the proportion of pyronin Y-negative cells in CD34−LSK HSCs is shown as the mean ± SEM (n = 4) (lower panel).
(F) Detection of Tif1b+/+ and Tif1bΔ/Δ HSPCs in the PB at 2 weeks after the first injection of tamoxifen. Representative flow cytometric profiles of LSK cells from CreERT and CreERT;Tif1bfl/fl mice are depicted (upper panel) and the proportions of Lin−c-Kit+ and LSK cells in total mononuclear cells is shown as the mean ± SEM (n = 4) (lower panel).
(G) Apoptosis in BM CD45.2+LSK cells from recipient mice detected with Annexin V and 7-aminoactinomycin D (7-AAD) by flow cytometry at the indicated time points after deletion of Tif1b. The data are shown as mean ± SEM (n = 4).
(H) Competitive reconstitution assays. BM cells from 8-week-old CreERT and CreERT;Tif1bfl/fl mice were transplanted into lethally irradiated recipient mice with the same number of competitor BM cells (1 × 106 cells). At 8 weeks posttransplantation, recipient mice were injected with tamoxifen for 5 consecutive days. The chimerism of CD45.2+ donor-derived cells in the PB of recipient mice is shown as % of chimerism values prior to treatment with tamoxifen (left panel). Donor chimerism in the BM LSK and CD34−LSK cells at 12 weeks after injection of tamoxifen is also shown (right panel). The data are shown as mean ± SEM (n = 6). ∗p < 0.05; ∗∗p < 0.005; ∗∗∗p < 0.0005.
Of interest, the number of Tif1bΔ/Δ HSCs increased transiently at 2 weeks postdeletion and then dramatically decreased by 4 weeks postdeletion (Figure 2D), suggesting enhanced cycling of HSCs. Indeed, 66% of the Tif1bΔ/Δ HSCs had exited the quiescent G0 stage and a significant portion of them were actively cycling compared to the control HSCs (Figures 2E and S2C). A significant number of Tif1bΔ/Δ HSPCs egressed from BM to the periphery as evident from a drastic increase in LSK HSPCs and Lin−c-KIT+ progenitors in the PB (Figure 2F), suggesting a BM niche-interaction defect in Tif1bΔ/Δ HSPCs. In addition, Tif1bΔ/Δ HSCs underwent massive apoptosis (Figure 2G). Among the myeloid progenitors, the numbers of Tif1bΔ/Δ granulocyte-macrophage progenitors (GMPs) and megakaryocyte-erythroid progenitors (MEPs) were also significantly reduced at 4 weeks postdeletion (Figure S2D). Furthermore, Tif1bΔ/Δ LSK cells proliferated poorly in culture (Figure S2E). We next examined the long-term reconstitution capacity of Tif1b-deficient BM cells. To this end, control and Cre-ERT;Tif1bfl/fl BM cells were transplanted along with the same number of BM competitor cells into lethally irradiated wild-type recipients. Upon deletion of Tif1b at 8 weeks after transplantation, donor cells were rapidly outcompeted by the competitor cells and quickly depleted from both the PB and the BM (Figure 2H).
In contrast to its role in erythropoiesis (Barde et al., 2013), the role of TIF1β has never been tested in myeloid cells. We therefore analyzed lysozyme-Cre;Tif1bfl/fl mice, in which Cre is active only in myeloid cells at the maturation stages beyond GMPs. Surprisingly enough, lysozyme-Cre;Tif1bfl/fl mice showed no obvious defects in myeloid differentiation. The hematological data, including the PB cell counts and the lineage composition and the number of BM cells, myeloid progenitor cells and HSCs were almost normal (Figure S3). These findings underline the lineage-specific functions of TIF1β in hematopoiesis.
Loss of HSC Signature in the Absence of TIF1β
To elucidate the changes in gene expression responsible for impaired HSC function in the absence of TIF1β, we performed microarray analysis using LSK cells at 2 weeks postdeletion of Tif1b (Figure 3A). The analysis revealed that 541 genes were upregulated more than 2-fold and 486 genes were downregulated more than 2-fold compared to control cells reproducibly in two independent experiments. We first performed gene set enrichment analysis (GSEA) to see if there was a fundamental loss of stem-cell-like gene expression in Tif1bΔ/Δ HSCs. Using the microarray data taken from various hematopoietic compartments (CD34−LSK HSCs, CD34+LSK MPPs, Lin−c-KIT+ committed progenitors, and Lin+ differentiated cells), we found that a gene signature unique to CD34−LSK HSCs was actually enriched in Tif1bΔ/ΔLSKs (Figure 3B). This suggests that a loss of “stemness” is not responsible for the phenotypes observed in Tif1b-deficient mice.
Figure 3.
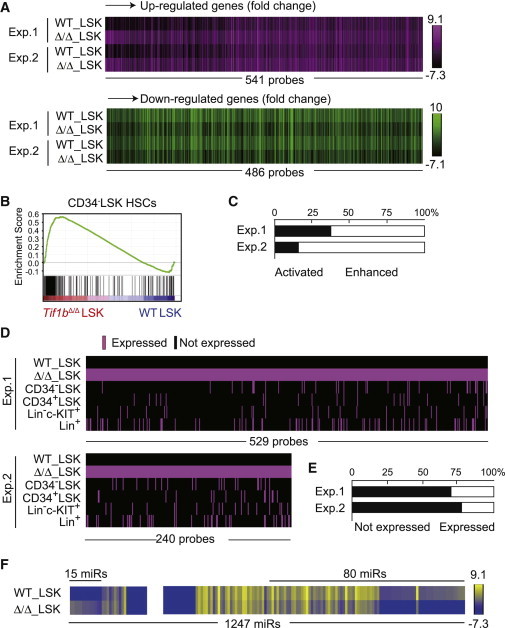
Derepression of Nonhematopoietic Genes in Tif1b-Deficient LSK Cells
(A) Heatmap of the expression of genes that were upregulated (541 probe sets, upper panel) or downregulated (486 probe sets, lower panel) more than 2-fold in Tif1b-deficient BM LSK cells in the microarray analysis.
(B) GSEA of the gene set specific to CD34−LSK HSCs in Tif1bΔ/Δ LSK cells. Using the microarray data from CD34−LSK HSCs, CD34+LSK MPPs, Lin−c-KIT+ committed progenitors, and Lin+ differentiated cells, HSC-specific gene sets were selected and applied to GSEA.
(C) Upregulated genes in (A) were divided into two groups according to their expression levels in Tif1bΔ/Δ LSK cells compared to that in control LSK cells. One group consist of probe sets that were expressed in the control LSK cells and their expression levels were enhanced upon deletion of Tif1b (“enhanced” genes). The other group includes probe sets that were not expressed in the control LSK cells but became activated in Tif1b-deficient LSK cells (“activated” genes). Bar graph shows proportions of these groups.
(D) Schematic representation of expression profiles of “Activated” genes in BM Tif1b-deficient LSK cells in (A) in various hematopoietic cell fractions, including CD34−LSK HSCs, CD34+LSK MPPs, Lin−c-KIT+ committed progenitors, and Lin+ differentiated cells. Expressed genes are depicted as a red bar.
(E) Proportions of probe sets in (D) that were not expressed in any of the hematopoietic cell fractions (Not expressed) and those expressed in at least one of the hematopoietic cell fractions including CD34−LSK HSCs, CD34+LSK MPPs, Lin−c-KIT+ committed progenitors and Lin+ differentiated cells (Expressed).
(F) Heatmap of the expression of miRNAs that were upregulated (15 miRs) or downregulated (80 miRs) more than 2-fold in Tif1b-deficient BM LSK cells in the microarray analysis.
We next focused on the genes derepressed in the absence of TIF1β. Among genes derepressed, a significant portion of genes became transcriptionally detectable from undetectable levels (“activated”) in Tif1bΔ/Δ LSK cells (Figure 3C). We then compared the expression profiles of the “activated” genes in Tif1bΔ/Δ BM LSK cells to various hematopoietic compartments, including CD34−LSK HSCs, CD34+LSK MPPs, Lin−c-KIT+ committed progenitors, and Lin+ differentiated cells (Figure 3D). To our surprise, the majority of “activated” gene transcripts were not detected in any of these hematopoietic compartments (Figure 3E). These results suggest that TIF1β is required for the gene silencing of nonhematopoietic genes in HSPCs.
Among gene sets positively enriched (derepressed) in Tif1bΔ/Δ LSK cells, many genes sets were related to signaling pathways mediated by cell surface receptors or adhesion molecules (e.g., integrin signaling pathway, gap junction, and focal adhesion; see Table S1) and upregulation of such genes was confirmed by quantitative RT-PCR (Figure S4A). These results indicate deregulated expression of nonhematopoietic adhesion molecules in Tif1bΔ/Δ HSCs, which might result in impaired niche interaction in Tif1bΔ/Δ HSCs and enhanced egression of HSCs from the BM to the periphery.
Barde et al. (2013) have recently reported that deletion of Tif1b results in compromised erythropoiesis due to failure in the induction of mitophagy-related genes. They found that the TIF1β together with KRAB-ZNF represses the transcription of microRNAs (miRNAs) targeting mitophagy transcripts to induce mitophagy during the terminal differentiation of erythroblasts. Profiling of miRNAs in Tif1bΔ/Δ LSK cells revealed upregulation of 15 miRNA greater than 2-fold compared to control cells (Figure 3F). However, none of the mitophagy-related miRNAs described by Barde et al. were upregulated in Tif1bΔ/Δ LSK cells. In addition, the expression levels of mitophagy-related genes were not significantly altered in Tif1bΔ/Δ LSK cells (data not shown). Together, these findings highlight the cell-type-specific as well as differentiation stage-specific functions of TIF1β in hematopoiesis. Nonetheless, it should be noted that some of the upregulated miRNAs potentially target genes downregulated in Tif1bΔ/Δ LSK cells, implying involvement of derepressed miRNAs in altered gene expression in Tif1bΔ/Δ LSK cells (data not shown).
TIF1β has been characterized to suppress activation of p53 (Tian et al., 2009). Indeed, p53 target genes, such as proapoptotic genes Bax, Noxa, and p21, were upregulated in fetal Tif1bΔ/Δ LSK cells (Figure S4B). In order to evaluate the contribution of p53, we transplanted Cre-ERT;Tif1bfl/fl CD34−LSK HSCs transduced with the E6 oncoprotein, a potent p53 inhibitor, into lethally irradiated recipients. E6, however, did not have any gross effects on the repopulating capacity of Tif1bΔ/Δ HSCs (Figure S4C), suggesting that the activated p53 response is not the causative event in the hematopoietic failure induced by absence of TIF1β.
Reduction in HP1 Proteins in the Absence of TIF1β
TIF1β physically interacts with all of the HP1 proteins (HP1α, HP1β, and HP1γ; Nielsen et al., 1999; Cammas et al., 2002, 2007) and recruits them in order to establish deep silencing of their target genes (Nielsen et al., 1999; Schultz et al., 2002). Notably, levels of all three members of the HP1 family proteins were reduced approximately 2-fold in fetal liver Tif1bΔ/Δ Lin−c-KIT+ progenitor cells (Figure 4A), though their transcription levels were not significantly changed (data not shown). In accordance with this finding, immunostaining of fetal liver LSK cells revealed a reduction in numbers of chromocenters, a structural component of heterochromatin where HP1α accumulates, in the absence of TIF1β (Figure 4B). These results support the notion that TIF1β functions as a scaffold that stabilizes HP1 proteins. The level of HP1α protein was significantly reduced also in NIH 3T3 cells depleted of TIF1β. However, inhibition of proteasome activity by MG132 in NIH cells failed to stabilize HP1α upon knockdown of Tif1b, leaving the molecular mechanism through which the HP1 proteins are destabilized in the absence of TIF1β still obscure (data not shown). HP1 proteins are recruited to their target loci via physical interaction with TIF1β as well as through direct binding to trimethylated H3K9 to form transcriptionally inactive heterochromatin (Nestorov et al., 2013). The levels of global histone modifications, including H3K9me1/2/3, did not significantly change in Tif1bΔ/Δ Lin−c-KIT+ progenitor cells (Figure S4D). These findings further suggest that TIF1β regulates HP1 function not only by tethering them to their targets but also by securing their posttranslational stability. In order to test whether the reduction in levels of HP1 proteins is involved in the impaired function of Tif1bΔ/Δ HSCs, we knocked down the Hp1 genes in HSCs using small hairpin RNAs (shRNAs) (Figure S4E). Growth of HSCs was significantly impaired upon depletion of Hp1a and Hp1g, but not Hp1b (Figure 4C). The proportion of LSK HSPCs in GFP+ transduced cells was also significantly decreased upon depletion of Hp1a and Hp1g, but not Hp1b, at day 14 of culture (Figure 4D). These results suggest that reduced levels of HP1 proteins play a role in determining the phenotypes of Tif1b-deficient HSCs. Together, our findings highlight the TIF1β-HP1 system as critical transcriptional machinery that keeps nonhematopoietic genes repressed in HSCs, thereby maintaining the transcriptional signature specific to HSCs.
Figure 4.
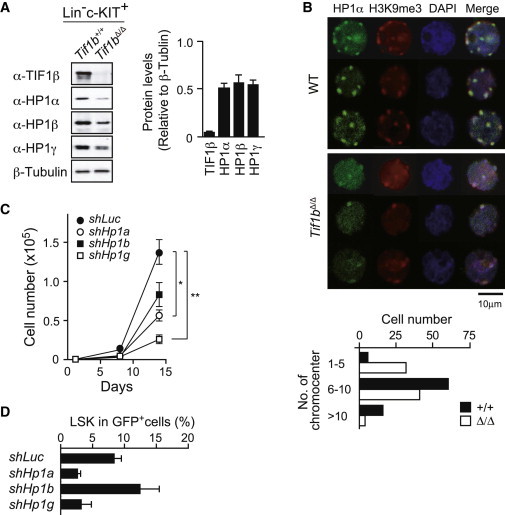
HP1 Depletion Compromises the Proliferation of HSCs
(A) Western blot analysis of Lin−c-Kit+ fetal liver cells from Tie2-Cre and Tie2-Cre;Tif1bfl/fl embryos. Levels of HP1 proteins were normalized to the amount of β-tubulin. The relative levels of HP1 proteins are indicated in the right panel. The data are shown as mean ± SEM (n = 3).
(B) Immunostaining of HP1α and H3K9me3 in LSK cells from Tie2-Cre and Tie2-Cre;Tif1bfl/fl fetal livers at E13.5. Cells were counterstained with DAPI. Representative three individual cell images are depicted. Numbers of chromocenters per cell are shown in the bottom panel. In total, 100 cells were counted for the scoring of the chromocenters.
(C) Growth of CD34−KSL HSCs upon depletion of Hp1 genes in vitro. HSCs were transduced with lentiviruses generating shRNAs against Luciferase (Luc), Hp1a, Hp1b, and Hp1g and then allowed to proliferate in the presence of 100 ng/ml of SCF and TPO for 14 days. The transduction efficiency was around 75% in most experiments as judged from the GFP expression. The data are shown as mean ± SEM for triplicate cultures.
(D) Analysis of CD34−KSL HSC cultures depleted of Hp1 genes in (C) at day 14. The proportion of LSK cells in GFP+ cells is shown. The data are shown as mean ± SEM (n = 3).
TIF1β has been reported to repress the expression of endogenous retrotransposons in embryonic stem cells via the histone methyltransferase ESET/SETDB1 and subsequent H3K9 trimethylation (Matsui et al., 2010; Rowe et al., 2010). However, we did not observe any significant derepression of endogenous retrotransposons in Tif1bΔ/Δ LSK cells (data not shown), indicating that different silencing machineries are operating in embryonic stem cells and somatic stem cells. Of interest, the phenotypes of Tif1bΔ/Δ HSCs are similar to those of HSCs deficient for MI-2β, a component of the NuRD histone deacetylase complex, which directly interacts with TIF1β (Schultz et al., 2001; Yoshida et al., 2008). TIF1β may thus collaborate with the NuRD complex, in addition to HP1 proteins, and stabilize these repressor molecules critical for the transcriptional repression of nonhematopoietic genes in HSCs.
The epigenetic regulation of transcription is crucial for the maintenance of cell-type-specific signatures (Sashida and Iwama, 2012). In the point of view of transcriptional regulation, multipotency of stem cells can be defined as an ability of stem cells to express a set of genes required for differentiation and cell-type-specific function upon cell differentiation. The PcG proteins maintain this “transcriptional competence” by transiently silencing genes that will be activated during differentiation of stem cells (Sauvageau and Sauvageau, 2010). In contrast, our findings in this study indicate that the TIF1β-HP1 system, which collaborates with H3K9me3 and DNA methylation (Rowe et al., 2013), is involved in establishing a more stable silencing state of genes that would never be activated in a given cell lineage. However, the precise mechanisms by which the epigenetic switching between the reversible PcG-mediated system and the TIF1β-HP1 system that we propose here remain to be elucidated. Understanding how these different repressive systems work would prove invaluable to our understanding of the epigenetic regulation of stem cells.
Experimental Procedures
Details regarding experimental procedures are available in Supplemental Experimental Procedures.
Acknowledgments
We thank Drs. P. Chambon and R. Losson for providing Tif1bfl/fl mice and Dr. Nozaki (MAB institute) for modified histone antibodies. This work was supported in part by Grants-in-Aid for Scientific Research (grants 21390289 and 221S0002) and Scientific Research on Innovative Areas “Cell Fate” (grant 22118004) from MEXT (Japan) and by a Grant-in-Aid for Core Research for Evolutional Science and Technology (CREST) from the Japan Science and Technology Corporation (JST).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Accession Numbers
Microarray data have been deposited in the NCBI Gene Expression Omnibus with the accession numbers GSE48929, GSE48986, and GSE52282.
Supplemental Information
References
- Barde I., Rauwel B., Marin-Florez R.M., Corsinotti A., Laurenti E., Verp S., Offner S., Marquis J., Kapopoulou A., Vanicek J., Trono D. A KRAB/KAP1-miRNA cascade regulates erythropoiesis through stage-specific control of mitophagy. Science. 2013;340:350–353. doi: 10.1126/science.1232398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammas F., Mark M., Dollé P., Dierich A., Chambon P., Losson R. Mice lacking the transcriptional corepressor TIF1beta are defective in early postimplantation development. Development. 2000;127:2955–2963. doi: 10.1242/dev.127.13.2955. [DOI] [PubMed] [Google Scholar]
- Cammas F., Oulad-Abdelghani M., Vonesch J.L., Huss-Garcia Y., Chambon P., Losson R. Cell differentiation induces TIF1beta association with centromeric heterochromatin via an HP1 interaction. J. Cell Sci. 2002;115:3439–3448. doi: 10.1242/jcs.115.17.3439. [DOI] [PubMed] [Google Scholar]
- Cammas F., Janoshazi A., Lerouge T., Losson R. Dynamic and selective interactions of the transcriptional corepressor TIF1 beta with the heterochromatin protein HP1 isotypes during cell differentiation. Differentiation. 2007;75:627–637. doi: 10.1111/j.1432-0436.2007.00166.x. [DOI] [PubMed] [Google Scholar]
- Chikuma S., Suita N., Okazaki I.M., Shibayama S., Honjo T. TRIM28 prevents autoinflammatory T cell development in vivo. Nat. Immunol. 2012;13:596–603. doi: 10.1038/ni.2293. [DOI] [PubMed] [Google Scholar]
- Matsui T., Leung D., Miyashita H., Maksakova I.A., Miyachi H., Kimura H., Tachibana M., Lorincz M.C., Shinkai Y. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature. 2010;464:927–931. doi: 10.1038/nature08858. [DOI] [PubMed] [Google Scholar]
- Nestorov P., Tardat M., Peters A.H. H3K9/HP1 and Polycomb: two key epigenetic silencing pathways for gene regulation and embryo development. Curr. Top. Dev. Biol. 2013;104:243–291. doi: 10.1016/B978-0-12-416027-9.00008-5. [DOI] [PubMed] [Google Scholar]
- Nielsen A.L., Ortiz J.A., You J., Oulad-Abdelghani M., Khechumian R., Gansmuller A., Chambon P., Losson R. Interaction with members of the heterochromatin protein 1 (HP1) family and histone deacetylation are differentially involved in transcriptional silencing by members of the TIF1 family. EMBO J. 1999;18:6385–6395. doi: 10.1093/emboj/18.22.6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro H., Yuan J., Ichikawa H., Ikawa T., Yamazaki S., Kawamoto H., Nakauchi H., Iwama A. Poised lineage specification in multipotential hematopoietic stem and progenitor cells by the polycomb protein Bmi1. Cell Stem Cell. 2010;6:279–286. doi: 10.1016/j.stem.2010.01.005. [DOI] [PubMed] [Google Scholar]
- Rowe H.M., Jakobsson J., Mesnard D., Rougemont J., Reynard S., Aktas T., Maillard P.V., Layard-Liesching H., Verp S., Marquis J. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature. 2010;463:237–240. doi: 10.1038/nature08674. [DOI] [PubMed] [Google Scholar]
- Rowe H.M., Friedli M., Offner S., Verp S., Mesnard D., Marquis J., Aktas T., Trono D. De novo DNA methylation of endogenous retroviruses is shaped by KRAB-ZFPs/KAP1 and ESET. Development. 2013;140:519–529. doi: 10.1242/dev.087585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sashida G., Iwama A. Epigenetic regulation of hematopoiesis. Int. J. Hematol. 2012;96:405–412. doi: 10.1007/s12185-012-1183-x. [DOI] [PubMed] [Google Scholar]
- Sauvageau M., Sauvageau G. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 2010;7:299–313. doi: 10.1016/j.stem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz D.C., Friedman J.R., Rauscher F.J., 3rd Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2α subunit of NuRD. Genes Dev. 2001;15:428–443. doi: 10.1101/gad.869501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz D.C., Ayyanathan K., Negorev D., Maul G.G., Rauscher F.J., 3rd SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002;16:919–932. doi: 10.1101/gad.973302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C., Xing G., Xie P., Lu K., Nie J., Wang J., Li L., Gao M., Zhang L., He F. KRAB-type zinc-finger protein Apak specifically regulates p53-dependent apoptosis. Nat. Cell Biol. 2009;11:580–591. doi: 10.1038/ncb1864. [DOI] [PubMed] [Google Scholar]
- Yoshida T., Hazan I., Zhang J., Ng S.Y., Naito T., Snippert H.J., Heller E.J., Qi X., Lawton L.N., Williams C.J., Georgopoulos K. The role of the chromatin remodeler Mi-2β in hematopoietic stem cell self-renewal and multilineage differentiation. Genes Dev. 2008;2:1174–1189. doi: 10.1101/gad.1642808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.