

Abstract
Nuclear processing and quality control of eukaryotic RNA is mediated by the RNA exosome, which is regulated by accessory factors. However, the mechanism of exosome recruitment to its ribonucleoprotein (RNP) targets remains poorly understood. Here we disclose a physical link between the human exosome and the cap-binding complex (CBC). The CBC associates with the ARS2 protein to form CBC-ARS2 (CBCA), and then further connects together with the ZC3H18 protein to the nuclear exosome targeting (NEXT) complex, forming CBC-NEXT (CBCN). RNA immunoprecipitation using CBCN factors as well as the analysis of combinatorial depletion of CBCN and exosome components underscore the functional relevance of CBC-exosome bridging at the level of target RNA. Specifically, CBCA suppresses read-through products of several RNA families by promoting their transcriptional termination. We suggest that the RNP 5′cap links transcription termination to exosomal RNA degradation via CBCN.
Introduction
Processing by ribonucleolytic enzymes is essential for the nuclear maturation of eukaryotic RNA. Moreover, RNA turnover-based quality control systems prevent the unwanted accumulation of spurious transcripts. Central here is the 3′-5′ exo- and endo-nucleolytic RNA exosome complex, conserved in all studied eukaryotes1,2. To exert its multitude of processing and degradation reactions, the catalytically inactive exosome core complex associates with active ribonucleases; such as, in human nuclei, hRRP6 and hDIS3 (refs. 3,4). In addition, the exosome utilizes cofactors that directly stimulate its enzymatic activity and serve as adapters to its many substrates5. Several of these cofactors are not well conserved between yeast and man, indicating key differences in RNA metabolism6. Specifically, while the function of the yeast nuclear exosome depends largely on the activities of the trimeric Trf4p-Air1p-Mtr4p polyadenylation (TRAMP) complex7–9, such dependence is only seen in the nucleoli of human cells6. Instead, the non-nucleolar pool of the human homolog of yeast Mtr4p, hMTR4 (also known as SKIV2L2), associates with the metazoan-specific RBM7 and ZCCHC8 proteins to form the trimeric NEXT complex, recently shown to aid the exosomal degradation of so-called PROMoter uPstream Transcripts (PROMPTs)6,10.
The mechanism underlying NEXT complex targeting of RNPs destined for exosomal decay remains elusive. In yeast, PROMPT-like cryptic unstable transcripts (CUTs) and other short RNA polymerase II (RNAPII) products harbor binding sites for the Nrd1p-Nab3p-Sen1p (NNS) complex. Although not fully characterized, it is believed that NNS terminates RNAPII transcription and mediates a ‘handover’ of RNA to the TRAMP-exosome complex for subsequent trimming and degradation11–14. Human cells harbor a homolog of Sen1p, Senataxin (also known as SETX), but no obvious homologs of Nrd1p and Nab3p. Interestingly, the co-immunoprecipitation (co-IP) experiments that identified the NEXT complex6 also yielded detectable amounts of all three components of what we call the CBC-ARS2 (CBCA) complex: cap-binding proteins 20 (CBP20) and 80 (CBP80) as well as the arsenic resistance protein 2 (ARS2). These factors have previously been shown to associate with the 5′methyl-guanosine cap of RNAPII-derived RNA15,16. While this suggests that the ubiquitously present RNA 5′ cap may be a means to recruit the exosome, any physical links involved in such potential bridging and their functional consequences remain unexplored.
The 5′ capping of the ~20nt long nascent RNA chain17 is a hallmark of RNAPII transcription. The cap coordinates an array of regulatory events, including RNA splicing18, 3′ end formation19, turnover20 and subcellular localization21–23. These functions are presumably mediated by the CBC16,24. However, how a simple heterodimer is capable of controlling such a diversity of RNA metabolic events is confounding, as the impact of CBC interaction has only been explained for a few complexes or factors15,22,23. Best characterized are interactions mediating the functions of the CBC in RNA localization. Here, the phosphorylated adaptor for RNA export (PHAX) protein has been shown to couple the CBC with the transport receptor CRM1 to mediate the nuclear export and the intra-nuclear transport of small nuclear RNA (snRNA)23 and small nucleolar RNA (snoRNA)25, respectively. Moreover, the ALY/REF RNP factor bridges CBC to the hTREX mRNA export complex22. Less characterized are the connections facilitating CBC-directed RNA stabilization26 and its stimulation of mRNA 3′end processing19.
Here we set out to characterize and quantify the composition of human NEXT and CBC sub-complexes and elucidate their functional relevance in RNA metabolism. To this end we applied an improved affinity capture (AC) mass spectrometry (ACMS) approach27 to demonstrate a strong physical link between the CBCA and NEXT complexes also including the uncharacterized zinc finger CCCH domain-containing protein 18 (ZC3H18, also known as NHN1). We name this protein complex assembly CBC-NEXT (CBCN) and show, by combinatorial depletion of CBCN and RNA exosome components, that CBCN forms a functionally relevant connection from the RNA 5′cap to the ribonucleolytic activity of the exosome. Furthermore, we show that CBCA promotes transcription termination at U snRNA loci and in promoter upstream regions expressing PROMPTs. This rationalizes the involvement of CBCA in exosome-targeted RNA metabolism and, despite the lack of any sequence homology, provides a functional resemblance to the yeast NNS complex.
Results
A strong physical link between the NEXT and CBCA complexes
In addition to RNA exosome association, our previous ACMS characterization of NEXT complex components hMTR4 and ZCCHC8 also revealed their binding to CBCA components6. To get a comprehensive understanding of such a possible connection between protein complexes engaged at RNA 5′- and 3′-ends, we employed an optimized procedure for protein AC and identification27. This practice allows for the division of interacting proteins into sub-complexes based on their abundance in the AC eluate (see below). We performed ACMS of affinity-tagged proteins using ‘localization and affinity purification’ (LAP) or FLAG tags from the CBCA (CBP20-3xFLAG, CBP80-3xFLAG, LAP-ARS2) and NEXT (hMTR4-LAP, RBM7-LAP) complexes. LAP-tagged proteins were expressed as C- or N-terminal fusions from stably integrated genes harboring their naturally occurring promoter and terminator sequences28, and C-terminally 3xFLAG tagged proteins were expressed from single-copy genes driven by a tetracycline-inducible promoter. All tagged proteins were expressed at near-endogenous levels (Supplementary Fig. 1). Moreover, each ACMS experiment was performed in triplicate, enabling an assessment of the statistical significance of the individual protein interaction data29. To focus on the link between the CBCA and NEXT complexes, we intersected the statistically significant interaction partners of all three CBCA components. This resulted in a list of fourteen proteins, containing CBCA, the entire NEXT complex, the previously identified NEXT complex-associated protein ZC3H18 (ref. 6), four RNA or protein transport factors and three proteins unrelated to RNA metabolism (Supplementary Table 1). Of these, only CBCA and NEXT components, together with ZC3H18, could be identified in both hMTR4-LAP and RBM7-LAP ACMS experiments (Fig. 1a,c and Supplementary Table 1). This confirms the specific association between the NEXT complex and ZC3H18, and indicates that the link between the NEXT and CBCA complexes does not involve nucleocytoplasmic transport factors. Hence, to fully complement our interaction studies we also performed ACMS analyses using a ZC3H18-3xFLAG cell line. In the following, each set of the conducted ACMS experiments is presented with special emphasis on sub-complex compositions and relative abundance relating to the RNA exosome, NEXT, ZC3H18 and CBCA.
Figure 1.

AC of NEXT complex components hMTR4 and RBM7 reveal stoichiometrically interacting ZC3H18 and CBCA complex (a) Volcano plot displaying the result of triplicate hMTR4-LAP ACMS experiments. The plot shows log2 ratios of averaged peptide MS intensities (normalized to the protein molecular weight (MW)) between bait and reference (‘bait-less’ cell line) eluate samples (x-axis) plotted against the negative log10 p-values (y-axis) calculated across the triplicate data (t-test). A dashed red hyperbolic curve separates specific hMTR4-interacting proteins (upper right-hand side of plot) marked in gray or color-coded from background (light blue dots) and from enriched proteins from the reference cell line (upper left-hand side of plot). Major hMTR4-interacting protein groups are color-coded as indicated in the legend and protein names relevant for this study are denoted. The full data set of specific co-precipitating proteins is given in Supplementary Table 2. (b) Column chart displaying abundance of selected proteins from hMTR4-LAP AC eluates. Y axis shows peptide intensities divided by protein molecular weight taken from (a) and normalized to results for the bait protein. Note disruption of the axis to reveal intensities of all plotted factors. Error bars represent standard deviation triplicates. (c) Volcano plot displaying the result of triplicate RBM7-LAP ACMS experiments plotted and labeled as in a. The full data set of specific co-precipitants is given in Supplementary Table 3. (d) Column chart displaying abundance of selected proteins from RBM7-LAP AC eluates plotted and labeled as in b.
Among the highly specific and abundant interaction partners, the hMTR4-LAP ACMS experiments identified the entire RNA exosome core complex, including the ribonuclease hRRP6 (also known as EXOSC10) and excluding the more loosely attached hDIS3 (also known as hRRP44) enzyme4,6. Moreover, the CBCA complex, the two other NEXT components (ZCCHC8 and RBM7), as well as the ZC3H18 protein were found in notable yields (Fig. 1a, Supplementary Table 2). Other proteins present in this purification are factors engaged in RNP transport, snoRNP formation, pre-mRNA splicing and other RNA binding proteins (RBPs) such as RNA helicases (Supplementary Table 2). The identified proteins are very similar to those obtained by an alternative AC approach6. To estimate the relative abundance of sample constituents we calculated averaged peptide intensities normalized to the molecular weight (MW) of the relevant protein6 (see Fig. 1b legend). hMTR4 co-precipitated the exosome complex most abundantly, followed by the two other NEXT components, CBCA and ZC3H18, which were all present sub-stoichiometrically to the exosome (Fig. 1b). This result is consistent with previous evidence that nucleolar hMTR4 interacts directly with the RNA exosome in the absence of other NEXT components6. Instead, non-nucleolar hMTR4 can exist in the nucleoplasmic NEXT complex, which became obvious when conducting the ACMS of non-nucleolar RBM7-LAP: The abundances of purified RNA exosome and NEXT complexes changed to higher relative yields of the NEXT complex (Fig. 1c,d). In addition, the RBM7-LAP AC experiments also revealed apparently similar abundances of CBCA and ZC3H18 (Fig. 1d). Other protein groups present in this purification were mostly splicing factors and RBPs or helicases (Supplementary Table 3).
The high yields of ZCCHC8 and hMTR4 in the RBM7-LAP IP confirmed our earlier notion that cellular RBM7 is primarily engaged within NEXT6. However, more surprisingly, we detected CBCA and ZC3H18 factors in higher amounts than RNA exosome components (Fig. 1d). Thus, despite its first identification as an RNA exosome cofactor complex, these data indicate that a major part of the purified NEXT is associated with CBCA-ZC3H18.
CBCA and ZC3H18 IPs identify a CBC-NEXT sub-complex
Having revealed substantial and similar levels of CBCA components and ZC3H18 in NEXT purifications, we next analyzed the ACMS data from the CBP80-3xFLAG, CBP20-3xFLAG and LAP-ARS2 IPs. The CBP80-3xFLAG purifications revealed the CBCA complex and further identified similar levels of ZC3H18 and NEXT complex components (Fig. 2a,b). We also identified components of the human exosome, RNP transport factors (PHAX being one of the most prominent), the entire negative elongation factor (NELF) complex, pre-mRNA splicing factors, RBPs or helicases and most known components of the human nuclear pore complex (NPC) (Supplementary Table 4). Albeit the IP was less efficient, the CBP20-3xFLAG purifications largely confirmed such a CBC interaction network (Fig. 2c,d and Supplementary Table 5). Importantly, these experiments again demonstrated similar amounts of co-precipitating ZC3H18 and NEXT complex components as well as a robust association with ARS2 (Fig. 2d). Thus, based on results presented so far, we suggest the existence of a NEXT-containing CBC sub-complex, comprising CBP20, CBP80, ARS2, ZC3H18 and the NEXT complex, which we coin CBC-NEXT (CBCN). LAP-ARS2 ACMS experiments confirmed CBCN integrity (Fig. 2e, Supplementary Table 6); i.e. protein abundance analysis suggested the purification of a near stoichiometric CBCN complex (Fig. 2f). Among other proteins identified in this IP were RNP transport factors, pre-mRNA splicing factors and RBPs or helicases. We note that PHAX was identified as a specific interaction partner in IPs of all CBCA components, suggesting a strong interaction between CBCA and PHAX. We were not able to identify other proteins previously reported to interact with ARS2 like DROSHA15 or FLASH30.
Figure 2.


AC of CBCA complex components CBP80, CBP20 and ARS2 reveals stoichiometric CBCN complex (a,c,e) Volcano plots displaying the results of triplicate CBP80-3xFLAG, CBP20-3xFLAG and LAP-ARS2 ACMS experiments plotted and labeled as in Fig. 1a. The full data sets of specific co-precipitants are given in Tables S4, S5 and S6. (b,d,f) Column charts displaying abundance of selected proteins from CBP80-3xFLAG, CBP20-3xFLAG and LAP-ARS2 AC eluates plotted and labeled as in Fig. 1b.
Finally, we analyzed ACMS data from ZC3H18-3xFLAG purifications. The ACMS profile (Fig. 3a, Supplementary Table 7) was very similar to that obtained for the RBM7-LAP construct (Fig. 1c, Supplementary Table 3), suggesting that a notable portion of cellular ZC3H18 is engaged in the CBCN complex. This was substantiated by protein abundance analysis placing CBC and NEXT components at similar levels (Fig. 3b). Consistently, indirect immunofluorescence microscopy of ZC3H18-3xFLAG revealed the protein within the nucleoplasm, outside nucleoli (Fig. 3c). A similar cellular distribution was previously observed for RBM7 and ZCCHC8 (ref. 6). Thus, both ZC3H18 ACMS and immunoflourescence data confirm the existence of CBCN.
Figure 3.

AC of ZC3H18 identifies stoichiometric CBCN complex (a) Volcano plot displaying the result of triplicate ZC3H18-3xFLAG ACMS experiments plotted and labeled as in Fig. 1a. The full data set of specific co-precipitants is given in Supplementary Table 7. (b) Column chart displaying abundance of selected proteins from ZC3H18-3xFLAG AC eluates plotted and labeled as in Fig. 1b. (c) α-FLAG immunofluorescence microscopy analysis of HEK293 Flp-In T-Rex cells stably expressing ZC3H18-3xFLAG. DAPI stain is included as nuclear marker. Scale bar represents 50 μm.
CBCN components contact PROMPTs and 3′extended transcripts
What then are the functional implications of the physical linkage between the RNA exosome and the CBC, established via CBCN? To investigate this question, we first performed RNA-IP (RIP) analysis employing cell lines expressing tagged CBCN components, CBP20, ARS2 and RBM7, as baits, to uncover RNAs associated with this complex. Bait-captured RNAs were identified by the use of tiling microarrays covering human chromosomes 1 and 6. Consistent with earlier observations that PROMPTs are NEXT-exosome substrates6,10, all three RIP experiments captured RNA derived from promoter-upstream regions of a large number of the 3131 interrogated genes; i.e. substrates captured commonly by any two of the investigated CBCN components demonstrated a highly significant overlap (p < 1e–36, Fisher’s exact test) (Fig. 4a). In addition, focused analyses of the 430 genes yielding a significant promoter upstream ARS2-RIP signal, revealed a considerable positional overlap of RNA from this region in all three RIP experiments (Fig. 4b–d). Compared to ARS2, CBP20 and RBM7 more robustly purified transcripts originating downstream the TSSs of these genes, the majority of which is likely to be pre-mRNA or mRNA. The lowered ARS2 RIP-signal downstream of TSSs may reflect a predominant ARS2-binding to nascent RNA at these loci (see Discussion).
Figure 4.

CBCN components share common RNA targets (a) Venn diagram showing overlap of CBP20, RBM7 and ARS2 RIP signals within promoter upstream regions enriched in each set of duplicate experiments. (b–d) Tiling array signals from RBM7 (b), CBP20 (c) and ARS2 (d) RIP experiments. Regions 1 kb down- and upstream of the TSSs of 430 genes with significant promoter upstream ARS2-RIP signal are displayed. Dotted black lines indicate TSS position. (e) Tiling array signals from RBM7, CBP20 and ARS2 RIP experiments. Mean RIP signal is displayed over regions around the 3′ends of 75 replication-dependent histone genes on chromosome 1 and 6 (solid lines) as well as 100 highly expressed protein coding genes as controls (dotted lines). Areas marked by dotted grey lines roughly indicate RDH gene body position. Note that the plotted RIP signal has been subtracted the signal from the control (no tag) IP, likely explaining the relatively low RIP signal in the RDH gene body region. (f–h) Tiling array signals from RBM7 (e), CBP20 (f) and ARS2 (g) RIP experiments. Regions 2 kb down- and upstream of the four U1 genes on chromosome 1 are displayed. Areas marked by dotted grey lines indicate U1 gene position.
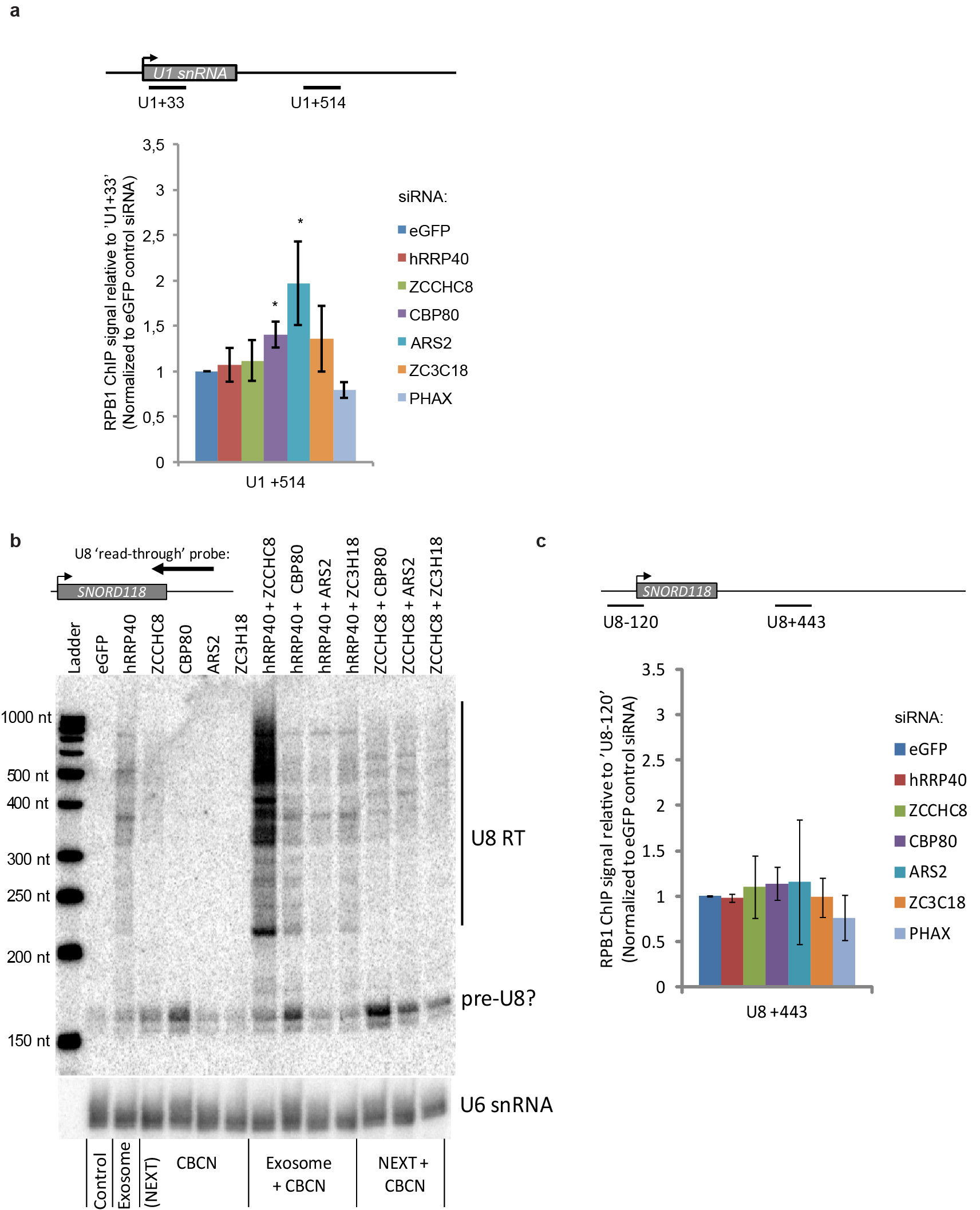
As additional common substrate classes for all three CBCN components, the tiling array data disclosed 3′end extended products of two groups of short genes, namely replication-dependent histone (RDH) genes (Fig. 4E) and U1 snRNA genes (Fig. 4f–h), both of which depend on polyadenylation (pA)-site-independent mechanism for their 3′end processing. A control sample of mRNA-genes did not display factor binding in their 3′end extended regions (Fig. 4e, dotted lines). For RDH RNAs, the region downstream the mature 3′end was bound by all tested CBCN components, whereas only CBP20 and ARS2 RIPs gave appreciable signal over the RDH RNA body (Fig. 4e). This suggests early recruitment of CBCA followed by subsequent assembly of CBCN. While the presence of several U1 snRNA genes (and pseudogenes) on chromosomes 1 and 6 caused the filtering away of the mature U1 snRNA sequence from the utilized microarrays, both regions up- and down-stream were present and showed evidence of CBCN-binding (Fig. 4f–h). We surmise that the signal upstream of U1 transcription start sites (TSSs) is reminiscent of the previously recognized U6 PROMPTs31, whereas the approximately 1 kb area of signal detection downstream U1 roughly equals the region predicted to elicit RNAPII termination (see below).
Functional implications of the exosome-CBCN connection
Having established transcript classes bound by the CBCN complex, we next studied the functional relevance of the CBCN-exosome connection by assaying the effects of depletion of factor(s) on the steady-state levels of selected PROMPTs. To this end, we treated cells with siRNAs targeting i) the core exosome (hRRP40 (EXOSC3)), ii) NEXT (ZCCHC8), iii) CBCA (CBP80 or ARS2) and iv) ZC3H18 either alone, or in combination with siRNAs against hRRP40 or ZCCHC8. We verified knockdown efficiencies by western blotting analyses (Fig. 5a). As previously reported6, RT-qPCR analyses revealed that levels of PROMPTs originating upstream of the EXT1, IFNAR1, MGST3 and POGZ genes all increased upon depletion of hRRP40 or ZCCHC8 (Fig. 5b). We note that the utilized random hexamer priming of cDNA synthesis results in somewhat lower PROMPT levels as compared to the dT-priming previously used6. Depletion of CBP80 and ARS2 yielded slightly smaller effects, whereas ZC3H18 depletion had no effect and was comparable to the eGFP siRNA control. Interestingly, however, co-depletion of hRRP40 and ZCCHC8 caused a synergistic accumulation of PROMPTs to 2–6 fold the levels of the hRRP40 single knockdown. We also observed such hyper-accumulation when co-depleting hRRP40 or ZCCHC8 with CBP80 or ARS2, whereas we detected no effect in the hRRP40 + ZC3H18 and ZCCHC8 + ZC3H18 co-depletions (Fig. 5b). To assay an additional CBCN-bound substrate class, we employed U snRNAs. Due to the heterogeneity of human U1 genes, we chose to assay U2 snRNA (RNU2-2) transcript accumulation. Indeed, using a northern probe designed to target U2 3′extended RNA, we observed a pattern of hyper-accumulation in co-depletions, reminiscent to that of PROMPTs (Fig. 5c). We note that co-depletion of ZC3H18 with hRRP40 resulted in a more prominent RNA accumulation than detected for PROMPTs, and speculate that efficient recruitment of the RNA exosome through the CBCN may be more of a bottleneck for highly transcribed loci like U2. In any case, the data collectively argue that CBP80, ARS2 and ZCCHC8 are unlikely to merely serve to target PROMPTs and 3′extended snRNAs in a sequential fashion for exosomal degradation, as such a scenario would not be expected to yield additional target RNA in combinatorial depletion with hRRP40 as compared to that of the single hRRP40 depletion. Instead, hyper-accumulation of the investigated substrates in the co-depletions suggests that part of CBCN, CBP80, ARS2 and ZCCHC8, is involved in RNA metabolism through a different mechanism(s), the delineation of which might help uncover the functional implication of the CBCN-exosome connection (see below).
Figure 5.

Hyper-accumulation of PROMPTs upon co-depletion of CBCA with NEXT or RNA exosome complex components (a) Western blotting analysis showing protein depletion upon the indicated siRNA administrations. Anti-XRN2 antibody was used as loading control. (b) RT-qPCR analysis of total RNA from HeLa cells subjected to the indicated siRNA transfections and using amplicons for the indicated PROMPT regions (Supplementary Table 10). Data are displayed as mean values normalized to the control (eGFP siRNA) and normalized to GAPDH RNA as an internal control. Error bars represent standard deviations (n=3 biological replicates). (c) Stabilization of 3′-extended U2 RNA upon the indicated single- and co-depletions of CBCA, NEXT and exosome components as assessed by northern blotting analysis. Position of the utilized probe is shown at the top. U6 snRNA was probed as loading control. Detected RNA species and sizes of molecular ladder are displayed to the right and left of the blot, respectively. (d,e) Increased PROMPT ‘read-trough’ RNA levels upon CBP80 and ARS2 depletion. Ratios of proRBM39 (d) and proPOGZ (e) RNA levels relative to their 5′ends (RT-qPCR using amplicons positioned as displayed below the column charts). PROMPT levels relative to those derived from the 5′region are displayed normalized to the eGFP siRNA control. Error bars represent standard deviations (n=3 biological repeats). * P < 0.05 (two tailed one-sample t-test).
CBP80 and ARS2 mediate transcription termination activity
We first considered the possibility that CBCN components mediate a 5′-3′ degradation pathway of PROMPTs perhaps functioning in the absence of, or in parallel with, exosomal decay. If this is the case, depletion of both major 5′-3′ exonucleases, hXRN1 and XRN2, should yield an expected PROMPT hyper-accumulation when combined with hRRP40 depletion. This, however, was not the case, as hXRN1+XRN2 depletions alone, or in combination with hRRP40 (Supplementary Fig. 2a), had no effect on the investigated PROMPTs (Supplementary Fig. 2b).
In an alternative scenario, components of CBCN could be involved in the early termination of PROMPT transcription. Depletion of the responsible activity would then cause an increased production of ‘read-through PROMPTs’ long enough for detection by the utilized RT-qPCR amplicons. As 3′extended PROMPTs are predicted exosome substrates, their accumulation upon exosome-depletion would explain the observed hyper-accumulation upon co-depletion of factors (Fig. 5b). To test this idea, we first analyzed RNA from individual depletions of exosome and CBCN components for the abundance of 5′- vs. 3-′ends of ‘proRBM39’ PROMPT RNA derived from upstream the RBM39 gene promoter. Taking advantage of our mapping of PROMPT transcription initiation sites32, RT-qPCR amplicons could be designed to selectively measure RNA levels from the proRBM39 ‘downstream’ region and compare these to levels derived from a region closer to the proRBM39 TSS. Serving as an indirect measure of transcriptional read-through effects, these analyses showed significant increases of RNA from the proRBM39 3′-region upon depletions of both CBP80 and ARS2 relative to the other knockdowns (Fig. 5d). We observed similar trends when analyzing an artificial PROMPT locus constructed by inserting a ~700 bp region downstream of the proPOGZ PROMPT TSS in between a CMV promoter and a BGH polyadenylation signal (32; Fig. 5e). Moreover, the stabilization of U2 read-through RNA (Fig. 5c) is consistent with the idea that CBCN depletion causes appearance of exosome-targeted 3′extended species.
To directly analyze whether transcription termination is impacted by CBCN components, we turned to chromatin IP (ChIP) assays. The high transcription activity of the U2 locus readily allowed for the analysis of its RNAPII occupancy upon depletion of various CBCN components (Fig. 6a). As a measure of read-through transcription, we compared RNAPII ChIP signal in the U2 read-through region ~1 kb downstream of the U2 RNA 3′end processing site (Fig. 6b, amplicon ‘U2+957’) to that of the beginning of the U2 gene body (Fig. 6b, amplicon ‘U2-44’). Depletion of both ARS2 and CBP80 elicited a robust read-through transcription phenotype, whereas hRRP40, ZCCHC8 and ZC3H18 depletions yielded more modest effects (Fig. 6b). As neither ARS2 nor CBP80 depletion changed levels of candidate U2 transcription termination factors XRN2, NELF-E, CPSF73 or CPSF73L (Fig. 6a), the measured effects are likely direct. This finding therefore explains the observed hyper-accumulation of 3′extended U2 RNA upon hRRP40+CBP80 and hRRP40+ARS2 co-depletions, because CBP80 and ARS2 inactivation leads to the increased synthesis of read-through transcripts, which further accumulate in the absence of exosome activity (Fig. 5c).
Figure 6.

CBCA depletion causes inefficient transcription termination and exosome-dependent removal of read-through transcripts (a) Western blotting analysis of whole cell extracts used for ChIP analysis. Display and labeling as in Fig. 5a. (b) Transcriptional read-through of the U2 gene as assessed by monitoring the occupancy by ChIP of the large RNAPII subunit (RPB1) in the U2 read-through (amplicon ‘+957’, top schematics) and the U2 gene promoter (amplicon ‘−44’, top schematics) regions. Amplicon names denote the genomic distance from the respective gene TSS to the nearest end of the amplicon. ChIP analyses were performed with cells previously transfected with control or experimental siRNAs (indicated by color-code; depletion levels shown in a) and results were displayed as the ratio of ‘+957’ to ‘−44’ signals normalized to the eGFP siRNA control.. Error bars represent standard deviations (n=4 biological repeats) * P < 0.05 (two-tailed one-sample t-test). (c,d) CBCA-dependent termination of promoter upstream transcription. RNAPII (RPB1) ChIP analysis on cells containing the proRBM39 (c) and proPOGZ (d) loci from Fig. 5d,e and using the indicated PCR amplicons. Cells were factor-depleted as in a. Displayed results are the signal ratio from a given amplicon relative to the 5′-most amplicon. All signals were normalized to the eGFP siRNA control. Error bars represent standard deviations (n=3 biological repeats). * P < 0.05 and # P < 0.1 (two-tailed one-sample t-test).
To substantiate these data, we also analyzed transcription over a U1 snRNA (RNU1-1) locus in similar conditions and found significant transcription read-through upon both ARS2 and CBP80 depletion (Supplementary Fig. 3a). In addition, and consistent with a transcription termination phenotype, RNAPII ChIP levels showed increases in the proRBM39 downstream region upon depletion of ARS2 relative to the eGFP siRNA control (Fig. 6c). Likewise the assay revealed a dependency on CBP80 and ARS2 for normal transcription termination within the proPOGZ PROMPT region (Fig. 6d). In contrast, northern blotting analysis of the U8 snoRNA (SNORD118) locus did not show hyper-accumulation of 3′extended U8 snoRNAs upon hRRP40 + CBP80, hRRP40 + ARS2, ZCCHC8 + CBP80 and ZCCHC8 + ARS2 co-depletions (Supplementary Fig. 3b), and consistent with this we also could not detect any transcription termination phenotypes of the U8 locus upon neither CBP80 nor ARS2 depletion (Supplementary Fig. 3c). Thus, CBP80 and ARS2 (i.e. CBCA) specifically partake in the transcription termination of U1 and U2 genes. Finally, the hyper-accumulation of 3′extended U2 transcripts upon co-depleting ZCCHC8 and hRRP40 probably arises from a different mechanism as ZCCHC8 depletion only has a marginal effect on transcription termination of U2 (Fig. 6b) and as 3′extended U8 RNAs are dramatically elevated upon hRRP40 + ZCCHC8 depletion (Supplementary Fig. 3b) in the absence of any discernible transcription defect (Supplementary Fig. 3c).
CBCA globally impacts PROMPT transcription termination
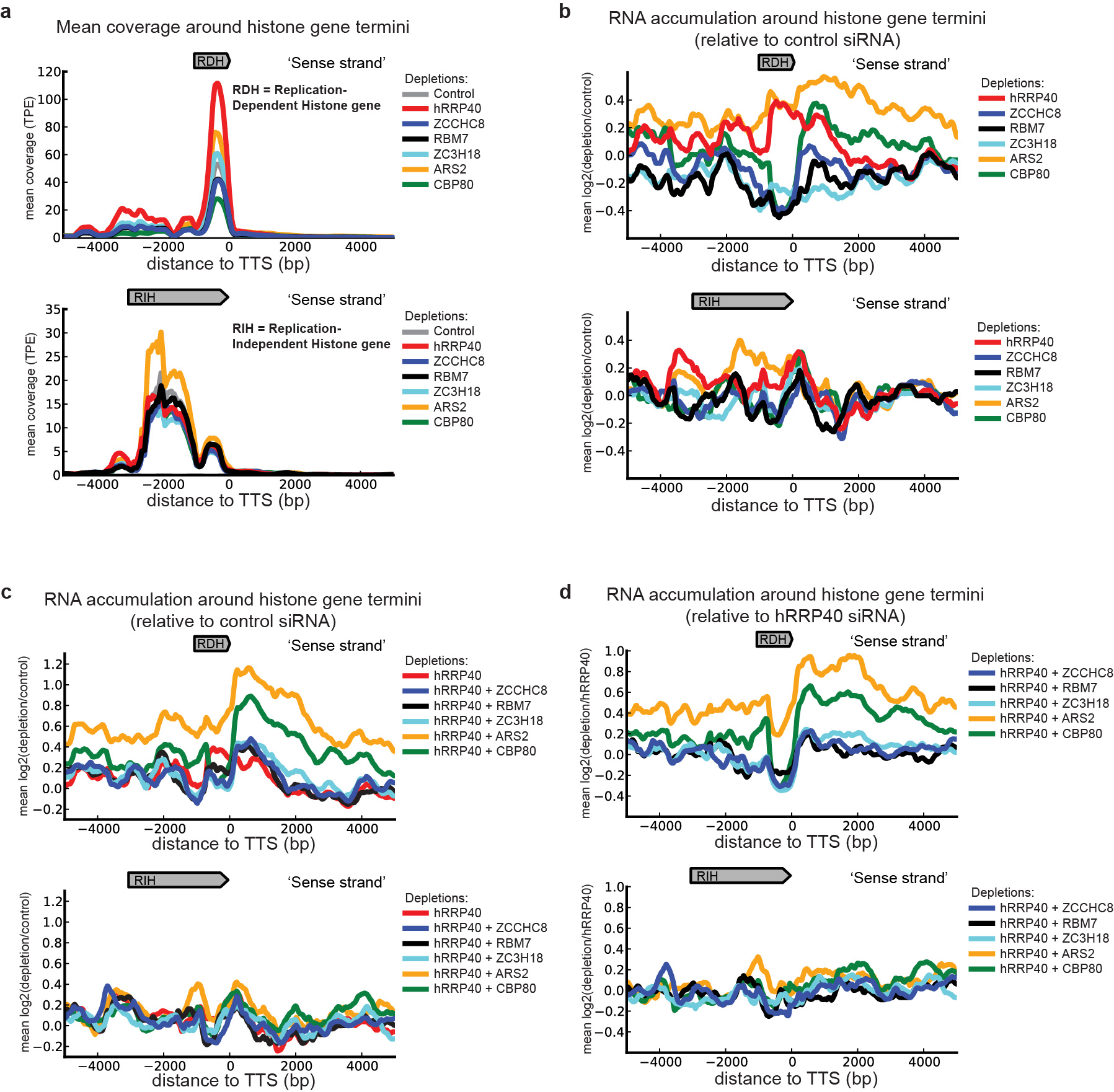
To assess the generality of the functions of CBCA and CBCN described above, we performed RNA sequencing (RNA-seq) of samples derived from cells depleted of CBCN and exosome components both alone and in combination. We analyzed PROMPT accumulation profiles for an unbiased and broad selection of 1733 TSSs from active genes with no confounding upstream annotations and plotted the global effects of single depletions on PROMPTs as the mean log2(ratio) between factor-and control- (eGFP siRNA) depleted samples over a 10 kb window around the selected TSSs. Consistent with our RT-qPCR data, a moderate general accumulation of PROMPTs was evident upon single CBCN component depletions, whereas we observed a more prominent stabilization upon hRRP40 depletion (Fig. 7a). Interestingly, while most of the single depletions resulted in PROMPT peak accumulations of around −500 bp relative to the TSS, depletion of ARS2 and CBP80 led to somewhat more elongated stabilization profiles with peaks at −645 and −1052 bp, respectively. This is consistent with global functions of CBCA in PROMPT transcription termination. Analyzing PROMPT stabilization profiles from the combinatorial depletions of hRRP40 and CBCN components confirmed the hyper-accumulation patterns previously observed by single loci RT-qPCR analyses (compare Supplementary Fig. 4a and Fig. 5b) and again indicated that co-depletions of CBP80 and ARS2 with hRRP40 led to the appearance of 3′extended PROMPTs. This phenotype was even more striking when plotting results from co-depleted samples relative to the hRRP40 single-depletion. Here, marked differences in the PROMPT position profiles were obvious between the different CBCN components (Fig. 7b). Specifically, while co-depletion of hRRP40 with ZC3H18 or the two NEXT components, ZCCHC8 and RBM7, resulted in higher PROMPT coverage around the same position as depletion of the RNA exosome component hRRP40, depletion of CBP80, and ARS2 of the CBCA complex led to additional coverage further upstream of the TSS with peaks at −1362 and −1381 bps, respectively. Plotting sequencing data from the combinatorial depletions of ZCCHC8 with CBP80 or ARS2 reached similar conclusions (Supplementary Fig. 4b,c). These results therefore support a global function of the CBCA complex in promoting transcription termination of PROMPTs.
Figure 7.

CBCA depletion displays a genome-wide termination defect of PROMPT transcription (a) Coverage profiles of mean log2(ratio) between indicated factor depletion (color coded) and control (eGFP siRNA) sample plotted on the antisense strand ± 5 kb relative to the annotated gene TSSs. Peak positions of each curve smoothened over 501-bp sliding windows are noted in the left side of the diagram. The direction of PROMPT transcription is indicated by grey arrow-box on top. Faded areas represent 95% confidence intervals for each curve (bootstrap analysis with 1000 resampling replicates). (b) Coverage profiles plotted and presented as in a, but with indicated co-depletions relative to the hRRP40-depleted sample. Dotted lines indicate peak positions of each curve. (c) Model for assembly and utility of CBCN in human nuclear RNA metabolism. i) The CBCA complex associates with the nascent RNA cap and impacts TSS-proximal transcript termination. ii) CBCN assembles on the RNA-CBC platform of these transcripts and recruits the RNA exosome. See text for details.
Around the annotated 3′ends of mature RDH RNAs, we observed a strong CBP80-and ARS2-depletion-dependent accumulation of 3′extended species. This we found when plotting the coverage of sequence tags derived from both single (Supplementary Fig. 5a and Fig. 4b) and combinatorial depletions relative to control siRNA (Supplementary Fig. 5c) or the hRRP40 single-depletion (Supplementary Fig. 5d) In addition, the mature forms of these RNAs were upregulated upon hRRP40 depletion, while ZCCHC8, RBM7 and in particular CBP80 depletion diminished their levels (Supplementary Fig. 5a and Fig. 4b). We found products from the pA-site-dependent and relatively longer replication-independent histone (RIH) genes not to be appreciably affected by the tested depletions (Supplementary Fig. 5). Together, the data provide additional support that CBCA, also at replication-dependent histone loci, exerts a general function in promoting transcription termination and furthermore suggest that CBCN and the RNA exosome control replication-dependent histone expression at multiple levels.
Discussion
While the RNA 5′cap is well established as a hub for mediating key metabolic events in the life of RNAPII transcripts, the biochemical consequences of only a few of its physical interactions have been characterized. Here, we outline CBCN as an unprecedented link from the cap to the major human nuclear ribonucleolytic machinery, the RNA exosome. Furthermore, part of the CBCN complex, CBCA, is shown to be involved in the biogenesis of these RNAs by helping to facilitate their transcription termination. This positions CBCA at the interface between transcription and RNA metabolism.
Our ACMS approach also identified many additional interaction partners of the CBC, constituting other biologically important interactions in RNA metabolism. Central among these are RNA transport factors, including components of the NPC, the entire human transcription-export (hTREX) complex and PHAX, all previously shown to associate with the CBC22,23. Indeed, Hallais et al.33 report the identification of a complex consisting of CBC, ARS2 and PHAX (termed CBC-ARS2-PHAX (CBCAP)). Consistently, we find PHAX in all our ACMS experiments of CBCA components (Supplementary Table 1). We note that though many of the identified proteins are RNA binding, the described interactions are most likely RNA-independent as all samples were RNAse-treated. Collectively, the data suggest that separate CBC sub-complexes exist that target substrates toward a degradation and/or processing route (via CBCN) or toward productive NPC translocation (via CBC-associated RNA transport factors).
The CBCN complex provides a platform for exosome targeting
While both S. cerevisiae TRAMP and human NEXT cofactor complexes have been shown to be important for exosome function via their ‘built-in’ polyadenylation and/or helicase activities6–9, it has been unclear how they directly link to their substrates. We demonstrate here that NEXT connects the human nuclear exosome to RNAPII-derived RNA via its direct coupling to the 5′cap-bound CBC of these RNPs (Fig. 7c). Such CBC connection may contribute to the conserved association of the RNA exosome with active transcription34–36. Interestingly, functional interactions between the S. cerevisiae CBC and the RNA exosome have previously been suggested37,38 and the Nrd1p factor of the NNS complex was found to co-purify both the CBC and components of the nuclear exosome13. Moreover, the NNS complex links transcription termination of CUTs and sn(o)RNAs to exosome recruitment11,12. It therefore appears that S. cerevisiae CBC-NNS-exosome and human CBCN-exosome connections display biochemical and functional similarities. This is noteworthy because no sequence homologs of the Nrd1p and Nab3p components of the NNS complex have been identified in human cells.
CBCA complex activity in transcription termination
The transcription termination phenotypes at both PROMPT and U snRNA loci upon CBP80 and ARS2 depletion strongly argue that CBCA acts co-transcriptionally (Fig. 7ci). Consistent with this idea, the CBC contacts the cap shortly after transcription initiation39–41 and ARS2 associates with chromatin of the SOX2 gene in an RNA-dependent fashion42. Although we cannot tell whether the entire CBCN complex assembles on capped nascent RNA, data suggesting that the exosome can be recruited co-transcriptionally34,36 imply that, at least in some cases, the CBCN-mediated cap-connection may be established early during transcription. Alternatively, NEXT could be recruited via the RNA 3′end processing apparatus and then link to cap-bound CBCA (Fig. 7cii). The latter idea receives some support from previous characterization of accessory proteins associated with core mRNA 3′end processing factors, which included considerable amounts of the entire NEXT complex43.
CBCA transcription termination activity measured in the present work is likely mediated through ARS2. First, ARS2 depletion resulted in the strongest and most consistent read-through phenotype at the investigated loci. Second, ARS2 RNA binding is enriched in PROMPT- and terminator-regions of snRNA and RDH genes. Nrd1p and Nab3p are proposed to elicit transcription termination through their binding to consensus sites within target RNAs11,12. Whether this similarity extends to ARS2 remains to be investigated. Previous data have linked the CBC and ARS2 to RNA 3′end formation. The CBC in cooperation with NELF was suggested to aid the 3′end formation of human replication-dependent histone RNA44, which was later shown also to involve ARS2 (ref. 45). A reported RNA-independent interaction between the CBC and the histone 3′end stem-loop binding protein (SLBP)44 indicates that direct protein interactions may functionally connect CBC to the histone 3′end processing machinery. Consistently, ARS2 was found to interact with FLASH30, an essential histone RNA 3′end processing factor46. Despite these connections, a coupling between cap-bound proteins and transcription termination has not previously been shown.
The exact mechanism underlying CBCA-mediated transcription termination still needs investigation and may include possible influences on transcription elongation processes as well as effects on RNA 3′end cleavage42,45,47. We see at least two plausible scenarios: (i) direct interaction between CBCA and factors involved in transcription termination (e.g. 3′end processing factors), facilitating their recruitment or (ii) competitive binding to the CBC of ARS2 vs. transcription elongation factors. The first possibility is supported by experimental evidence (Hallais et al.33) as well as by previously published interactions between the CBC and 3′end processing factors19. Though our CBCA purifications do not contain RNAPII subunits, we note that several proteins involved in transcriptional regulation do not exhibit strong, or direct, enough interactions with RNAPII to be captured by ACMS48. This can be due to either short-lived interactions, involving only a minor pool of the relevant factor(s), or because regulation is mediated indirectly through another activity.
It also remains to be explored the types of transcriptional terminators that respond to CBCA, which may have more widespread activities, including the possibility to exert fail-safe termination of read-through transcripts as also suggested for the S. cerevisiae NNS complex11,12,49,50. Interestingly, PROMPT regions and sequences downstream of snRNA- and RDH-genes most often harbor mRNA-like pA sites32,51, providing a possible node through which CBCA could exert its effect. Notably, promoter-proximal pA sites efficiently couple to decay by the RNA exosome, whereas RNAs expressed from longer transcription units appear less affected32. The loci found here to depend on CBCA for their 3′end formation (PROMPTs, read-through snRNAs and read-through RDH RNAs) are all relatively short (<1kb). Consistently, an artificial extension of the distance between a TSS and a pA signal makes transcription termination less dependent of CBCA (Hallais et al.33). How promoter-distal and -proximal pA sites are distinguished at the molecular level has yet to be resolved. The NNS complex functions via promoter-proximal binding sites in the nascent RNA and such distance dependence is linked to the phosphorylation-state of the RNAPII C-terminal domain52,53, which may also be relevant for CBCA function. Alternatively, CBCA activity could diminish with the growing distance between the 5′cap and the travelling RNAPII or with an increased maturation state of the transcription elongation complex. These issues set aside, we conclude that of the various sub-complexes based on the same CBC platform and exercising specific functions in nuclear RNA metabolism; one such, the CBCN complex, plays a key role in suppressing ncRNA expression in human cells.
ONLINE METHODS
ACMS and AC-western analyses
For ACMS and AC-western analysis, epitope-tagged proteins were expressed in two cell systems: (i) HEK293 Flp-In T-Rex cells stably expressing C-terminally 3xFLAG tagged proteins under control of a tetracycline-inducible promoter or (ii) HeLa Kyoto cells stably expressing LAP-tagged proteins from BACs (Bacterial Artificial Chromosomes). Cell lines were established as previously described 27,28. Plasmids and BAC clones used to establish cell lines are listed in Tables S8 and S9, respectively. Oligonucleotides used for amplification of protein coding sequences are listed in Supplementary Table 10.
Cryogenic disruption of cells as well as utilized GFP- and 3xFLAG-affinity capture methodologies have previously been described (Lubas et al. 2011; Domanski et al., 2012). IPs were done in extraction buffer consisting of 150mM NaCl, 0.5% Triton X-100, 20mM HEPES pH7.4 and supplemented with protease inhibitor. All ACMS experiments were performed label-free and in triplicates. Prior to AC, cell extracts were treated with 100μg/ml RNase A. Elution of bait-captured proteins was performed by mixing in 50μl of 0.5M Acetic Acid for 10 min at RT. Collected eluates were neutralized with 5μl of 5.5M Ammonium Hydroxide. Samples were concentrated to 30μl and processed using the FASP protocol 54. Trypsinized samples were acidified with 0.1% TFA, desalted using C18 stage tips and analyzed by MS using an LTQ Orbitrap Velos instrument (Thermo Scientific). Data acquisition, processing and plotting were performed as described 29. Note, that in this analysis, reference values were not subtracted from bait values as the reference procedure yielded more background material binding to unshielded antibody epitopes sometimes obscuring analysis (unpublished data, M.D.)
For western blotting analyses of tagged protein expression levels (Supplementary Fig. 1), 3xFLAG constructs were induced using 10 ng/mL tetracycline and both HEK293 and HeLa cells were harvested at 90% confluency. Ten micrograms WCE (whole cell extract) were run on a 6% (HeLa hMTR4-LAP, HeLa LAP-ARS2, HEK293 ZC3H18-3xFLAG), 8% (HEK293 CBP80-3xFLAG) and 15% (HeLa RBM7-LAP, HEK293 CBP20-3xFLAG) Tris-Glycine gel and transferred to a PVDF membrane. The membranes were probed with antibodies against endogenous proteins.
ZC3H18-3xFLAG localization studies
Protein localization analysis was performed essentially as described4. In brief, ZC3H18-3xFLAG containing cells were seeded on 6-well Lab-Tek Chamber Slides (Nunc). Protein expression was induced by media exchange and tagged protein was visualized in formaldehyde-fixed cells by anti-FLAG antibodies (SIGMA, F7425) and subsequent incubation with goat anti-rabbit Alexa Fluor 488 (Life Technologies). Nuclei were stained with DAPI.
RIP-tiling array analyses
For RIP-tiling array analyses, HeLa Flp-in cells stably expressing CBP20-3xFLAG and ARS2-3xFLAG as well as HeLa Kyoto cells expressing RBM7-GFP from a BAC were used. HeLa control cells expressed no tagged protein. Cells were extracted in HNTG (20 mM HEPES (pH 7.9150 mM NaCl, 1% Triton, 10% glycerol, 1 mM MgCl2, 1 mM EGTA, and protease inhibitors (Roche))) and tagged proteins were precipitated with M2 and GFP binder beads (Sigma and Chromotek, respectively). RNA was prepared from the pellets using TRIzol reagent (Sigma) and RNeasy kit (Qiagen), amplified with random primers and labeled with fluorophores before hybridization to Affymetrix GeneChip Human tiling 2.0R arrays covering human chromosome 1 and 6 as recommended by the manufacturer. The log2 ratio of signal intensities of the control (no tag) IP were subtracted from any specific IP and analyzed as follows: For snRNAs, the four U1 genes present on Chr1 were aligned at their TSSs (coordinates: 16713367, 16939596, 17095061 and 16866030), and probe signals at same relative positions were averaged. For replication-dependent histone (RDH) genes the 75 genes present on chromosome 1 and 6 were aligned with respect to their annotated 3′ end processing site and RIP tiling array signals were averaged. As a control group for RDH genes we analyzed the most 100 highly expressed coding genes located on chromosome 1 and 6, selected by a microarray analysis of total RNA extracted from these cells. For regions upstream of promoters, protein coding genes were aligned at their TSSs, and probe signals were averaged and pooled in bins of 100 bp relative to the TSS, to cover 1kb upstream and downstream, respectively. Overlapping genes were eliminated from the analysis, leaving 3131 loci. These were sorted by the enrichment of signal in their promoter upstream regions (here defined as the region from the TSS and 1kb upstream), and signals were considered significant only if enrichment was above the mean of signal from the 1kb upstream region of the 3131 genes + 1.5 STD. Gene lists were then intersected to create the Venn diagram of Fig. 4a. Genes with upstream regions (−1kb to the TSS) specifically enriched (> mean of signal from the 1kb upstream region of the 3131 genes +1.5STD) in the ARS2 IP (430 genes) were averaged together, for each IP, to create the profiles shown in Fig. 4b–D. Areas of the genome, randomly chosen, were used as negative control regions. P values for difference between test and control regions were calculated using a Wilcoxon test for all plots. These analyses showed statistical significance of the enrichment of transcripts from promoter upstream regions over control regions (p value range from 10−8 to 10−40) as well as enrichment of transcripts from RDH gene downstream regions over control gene downstream regions (‘downstream regions’ tested are from the 3′ processing sites and 500 bp downstream; p value range from 0.06 to 10−9). All experiments were performed in duplicates.
Cell culture and RNAi
HeLa cells were grown in Dulbecco’s modified eagle medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% Penicilin/Streptavidin, at 37°C, 5% CO2. For RNAi assays, cells were seeded at low confluence (approximately 3300 cells/cm2), and grown for 24 hr, before the first transfection. Transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions using siRNA at a final concentration of 15 nM of each siRNA (Supplementary Table 11) and Lipofectamine 2000 (final dilution1:1000) in RPMI 1640 media. Two days after the first transfection, the media was changed, and the transfection was repeated. At least five hours after each transfection, the media was replaced with fresh media. Cells were harvested 24–48 hr post the second transfection, preventing plates from being more than 90% confluent.
Factor depletion efficiencies were monitored by western blotting analysis. Cell pellets were resuspended in RSB100 (100 mM Tris pH 7.5, 100 mM NaCl, 2.5 mM MgCl2)/0.5% TritonX100 with proteinase inhibitor and sonicated. Cell debris was removed by centrifugation at 4000×g, 4°C for 15 min. Samples were separated by 10% denaturing PAGE and transferred to nitrocellulose membranes (Whatman). Membranes were blocked in 5% skimmed milk powder (SMP) in PBS for 30 min and incubated with primary antibodies (Supplementary Table 12) in 5% SMP in PBS. The membranes were subsequently washed three times for 10 min in PBS with 0.05% Tween20 and incubated in horse-radish-peroxidase (HRP) conjugated goat-anti-rabbit or -mouse secondary antibody (Dako) in 5% SMP in PBS, washed again and exposed using Supersignal West Femto Substrate (Thermo Scientific).
RT-qPCR
RNA was purified using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. For RT-qPCR analyses, RNA was DNase I-treated and cDNA was synthesized using random hexamers according to standard procedures. qPCR was performed using primers in Supplementary Table 13 on a Stratagene MX3005P QPCR System. Data were processed using the ΔΔCt method, normalizing to both GAPDH mRNA levels and eGFP siRNA control samples. Error bars represent standard errors calculated from at least three independent experiments.
Northern blotting analyses
For northern blotting analyses, precipitated aliquots of 20 μg of total RNA were redissolved in UREA load buffer and separated by standard denaturing PAGE (6% gels). RNA was then transferred to nylon membranes by wet-blotting (15V, overnight at 8°C in a BioRad TRANS-BLOT CELL). RNA was UV-cross-linked to the membranes, which were blocked by incubation with ULTRAhyb-Oligo hybridization buffer (Ambion) for 1 hr at 42°C. Northern probes were 5′end-labeled with γ-P32-ATP using T4 Polynucleotide Kinase (Fermentas), purified on columns and hybridized to the blots in 10 mL ULTRAhyb-Oligo hybridization buffer overnight at 42°C. Blots were washed three times in 2X SSC with 0.1% SDS, exposed to phosphoimager screens and scanned using a Typhoon TRIO+ scanner (GE Healthcare). Probes:
U2_3′ext: 5′-AATCTTCCATTAAACAACGGTTGTTCTCTCCCCGAAGGGAGAGTGCACCG
U8_3′ext: 5′-GTGGATATCTGCTAATCAGCATAACACAAATGTAAGTGATCGTCAGAAAG
U6: 5′-GCTAATATTCTCTGTATCG.
ChIP analyses
Following double siRNA transfection of HeLa cells, DNA and protein were cross-linked in 1% formaldehyde and rocking at room-temperature for 10 min. Reactions were quenched by the addition of glycine to 0.125M. Cells were then washed in ice-cold PBS and lysed 10 min in ChIP Lysis Buffer (20 mM Tris-HCl pH 8.0, 85 mM KCl, 0.5% NP40) on ice. Aliquots of cell extracts were taken for western blotting analysis and nuclei were pelleted and lysed >1 hr on ice in 1 mL Nuclei Lysis Buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl pH 8.0). Nuclear extracts (NE) were sonicated using a Covaris sonicator for 15 min at intensity 8, 20% burst and 200 cycles per burst. Debris was then pelleted by >20 min centrifugation at 15.700 × g at 4°C and aliquots were taken for estimation of DNA fragmentation efficiency by agarose gel electrophoresis. DNA concentration in sonicated NEs was measured and adjusted in Nuclei Lysis Buffer and equal amounts of DNA was diluted to 0.2 μg/μL in ChIP Dilution Buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl pH 8.0, 167 mM NaCl). 1% aliquots were taken for ‘input’ samples and 1 mL NEs aliquots were incubated with either RNAPII antibody (PolII (H-224), 2.5 μL from the 200 μg/0.1 ml stock, Cat#: sc-9001 X, Santa Cruz Biotechnology, Inc., validation of specificity is available on the manufacturer’s website) or no antibody overnight at 8°C with rotation. 20 μL blocked Sepharose Protein A beads (Invitrogen) were added to each NE sample and incubated 1 hr at 8°C with rotation. Beads were then washed once in Low Salt Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.0, 150 mM NaCl), once in High Salt Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.0, 500 mM NaCl), once in LiCl Immune Complex Wash Buffer (0.25 M LiCl, 1% NP40, 1% Sodiumdeoxycholate, 1 mM EDTA, 10 mM Tris-HCl pH 8.0) and twice in TE (10 mM Tris-HCl pH 8.0, 1 mM EDTA). DNA was eluted twice in 100 μL Elution Buffer (1% SDS, 0,1M NaHCO3) by shaking for 10 min at 65°C. Eluted DNA and ‘input’ samples were reverse cross-linked by treatment with RNase A (overnight at 65°C) and Proteinase K (3 hr at 45°C) and DNA was purified on PCR purification columns (Fermentas) and diluted to 300 μL in nfH2O. qPCR reaction were set up in a total of 15 μL with 5 μL DNA sample, 2.5 μM primer pair (see Supplementary Table 13) and 7.5 μL 2X Plaitnum SYBR Green qPCR SuperMix-UDG and run as a standard short PCR programme with an annealing temperature of 60°C.
RNA-seq library construction
5 μg total RNA from each sample was depleted of rRNA using the RiboMinus kit following manufacturer’s instructions. Multiplexed RNA-seq libraries were then prepared using the ScriptSeq v2 kit (Epicentre). The libraries were sequenced on an Illumina HiSeq 2000 platform.
RNA-seq data analyses
Reads were de-multiplexed and then mapped to the hg19 human reference genome using the STAR software 55. Mapped reads were used to construct coverage files, which in turn were used to plot the mean coverage around selected anchor points from annotated genomic features. Coverage values were smoothened over 501-bp sliding windows to reduce noise and normalized to the number of mapped reads for each sample. To enable comparisons of knock-downs against control or hRRP40 siRNA, a log2 coverage ratio between samples and the given control was calculated for each anchor point, and then the mean log2 ratio for the selected group of anchor points was plotted over a defined genomic window. For the plotting of PROMPT coverage profiles, gene lists were prepared as follows: RPKM values in the 02_Ctrl sample were calculated for all genes annotated by ENSEMBL. Genes with RPKMs>4 were selected and those with overlapping annotation in a region of 5 kb upstream of their TSSs were filtered away. Bootstrap analysis (1000 re-samplings) was used to produce 95% confidence intervals for the plotted mean values at each nucleotide position. These intervals are indicated as faded areas around the plotted curves. For the analysis of histone genes complete lists of annotated replication-dependent- and replication-independent genes were obtained from (http://www.genenames.org/genefamilies/histones). To avoid confounding transcription units in the downstream region where read-through transcripts would be detected, we removed all genes with annotations in the region 5 kb downstream of the mature histone RNA 3′ends. Due to the prevalence of tandem positioning of histone genes this left 31 RDH and 9 RIH genes for the coverage analysis.
Original images of northern blots used in this study can be found in Supplementary Fig. 6.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank M. Schmid, S. Lykke-Andersen, M. Lubas, A. Dziembowski and D. Libri for critical comments to the manuscript. Special thanks to Estelle Marchal for excellent technical assistance. E. Izaurralde (Max Planck Institute for Developmental Biology) and J. Lykke-Andersen (Division of Biological Sciences at UCSD) are acknowledged for sharing reagents. This work was supported by the Danish National Research Foundation (grant DNRF58), the Danish Cancer Society and the Lundbeck- and Novo Nordisk Foundations (to T.H.J.); the US National Institutes of Health (grant U54 GM103511) (to M.P.R.), the l’ARC and La Ligue Contre Le Cancer (to E.B.), Foundation for Strategic Research grant FFL09-0130 (R.S.) and the European Community’s Seventh Framework Programme (FP7/2007-2013) under grant No. 241548 (MitoSys) (to A.H.). The authors declare that they have no competing financial interests.
Footnotes
Accession Codes
RNA-seq data have been deposited in the Sequence Read Archive (SRA) database under accession code SRP031620. RIP–tiling array data can be accessed through GEO accession code GSE52132.
Author Contributions
M.D., J.B. performed ACMS experiments and analyzed the data together with J.S.A., J.L. and M.P.R., while I.P. and A.H. contributed tagged cell lines. C.V., M.H. and E.B performed and analyzed RIP experiments. P.R.A, M.S.K and A.S. performed experimental RNA analyses. P.R.A. and E.N. performed and analyzed ChIP experiments. P.R.A., H.S. and R.S. analyzed RNA-seq data. P.R.A., M.D., M.S.K. and T.H.J. wrote the manuscript.
References
- 1.Chlebowski A, Lubas M, Jensen TH, Dziembowski A. RNA decay machines: The exosome. Biochim Biophys Acta. 2013;1829:552–60. doi: 10.1016/j.bbagrm.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 2.Houseley J, Tollervey D. The many pathways of RNA degradation. Cell. 2009;136:763–76. doi: 10.1016/j.cell.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 3.Staals RH, et al. Dis3-like 1: a novel exoribonuclease associated with the human exosome. Embo J. 2010;29:2358–67. doi: 10.1038/emboj.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tomecki R, et al. The human core exosome interacts with differentially localized processive RNases: hDIS3 and hDIS3L. Embo J. 2010;29:2342–57. doi: 10.1038/emboj.2010.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lykke-Andersen S, Brodersen DE, Jensen TH. Origins and activities of the eukaryotic exosome. J Cell Sci. 2009;122:1487–94. doi: 10.1242/jcs.047399. [DOI] [PubMed] [Google Scholar]
- 6.Lubas M, et al. Interaction profiling identifies the human nuclear exosome targeting complex. Mol Cell. 2011;43:624–37. doi: 10.1016/j.molcel.2011.06.028. [DOI] [PubMed] [Google Scholar]
- 7.LaCava J, et al. RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell. 2005;121:713–24. doi: 10.1016/j.cell.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 8.Wyers F, et al. Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell. 2005;121:725–37. doi: 10.1016/j.cell.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 9.Vanacova S, et al. A new yeast poly(A) polymerase complex involved in RNA quality control. PLoS Biol. 2005;3:e189. doi: 10.1371/journal.pbio.0030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Preker P, et al. RNA exosome depletion reveals transcription upstream of active human promoters. Science. 2008;322:1851–4. doi: 10.1126/science.1164096. [DOI] [PubMed] [Google Scholar]
- 11.Arigo JT, Eyler DE, Carroll KL, Corden JL. Termination of cryptic unstable transcripts is directed by yeast RNA-binding proteins Nrd1 and Nab3. Mol Cell. 2006;23:841–51. doi: 10.1016/j.molcel.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 12.Thiebaut M, Kisseleva-Romanova E, Rougemaille M, Boulay J, Libri D. Transcription termination and nuclear degradation of cryptic unstable transcripts: a role for the nrd1-nab3 pathway in genome surveillance. Mol Cell. 2006;23:853–64. doi: 10.1016/j.molcel.2006.07.029. [DOI] [PubMed] [Google Scholar]
- 13.Vasiljeva L, Buratowski S. Nrd1 interacts with the nuclear exosome for 3′ processing of RNA polymerase II transcripts. Mol Cell. 2006;21:239–48. doi: 10.1016/j.molcel.2005.11.028. [DOI] [PubMed] [Google Scholar]
- 14.Steinmetz EJ, Conrad NK, Brow DA, Corden JL. RNA-binding protein Nrd1 directs poly(A)-independent 3′-end formation of RNA polymerase II transcripts. Nature. 2001;413:327–31. doi: 10.1038/35095090. [DOI] [PubMed] [Google Scholar]
- 15.Gruber JJ, et al. Ars2 links the nuclear cap-binding complex to RNA interference and cell proliferation. Cell. 2009;138:328–39. doi: 10.1016/j.cell.2009.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Izaurralde E, et al. A nuclear cap binding protein complex involved in pre-mRNA splicing. Cell. 1994;78:657–68. doi: 10.1016/0092-8674(94)90530-4. [DOI] [PubMed] [Google Scholar]
- 17.Rasmussen EB, Lis JT. In vivo transcriptional pausing and cap formation on three Drosophila heat shock genes. Proc Natl Acad Sci U S A. 1993;90:7923–7. doi: 10.1073/pnas.90.17.7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gornemann J, Kotovic KM, Hujer K, Neugebauer KM. Cotranscriptional spliceosome assembly occurs in a stepwise fashion and requires the cap binding complex. Mol Cell. 2005;19:53–63. doi: 10.1016/j.molcel.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 19.Flaherty SM, Fortes P, Izaurralde E, Mattaj IW, Gilmartin GM. Participation of the nuclear cap binding complex in pre-mRNA 3′ processing. Proc Natl Acad Sci U S A. 1997;94:11893–8. doi: 10.1073/pnas.94.22.11893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hosoda N, Kim YK, Lejeune F, Maquat LE. CBP80 promotes interaction of Upf1 with Upf2 during nonsense-mediated mRNA decay in mammalian cells. Nat Struct Mol Biol. 2005;12:893–901. doi: 10.1038/nsmb995. [DOI] [PubMed] [Google Scholar]
- 21.Izaurralde E, et al. A cap-binding protein complex mediating U snRNA export. Nature. 1995;376:709–12. doi: 10.1038/376709a0. [DOI] [PubMed] [Google Scholar]
- 22.Cheng H, et al. Human mRNA export machinery recruited to the 5′ end of mRNA. Cell. 2006;127:1389–400. doi: 10.1016/j.cell.2006.10.044. [DOI] [PubMed] [Google Scholar]
- 23.Ohno M, Segref A, Bachi A, Wilm M, Mattaj IW. PHAX, a mediator of U snRNA nuclear export whose activity is regulated by phosphorylation. Cell. 2000;101:187–98. doi: 10.1016/S0092-8674(00)80829-6. [DOI] [PubMed] [Google Scholar]
- 24.Kataoka N, Ohno M, Moda I, Shimura Y. Identification of the factors that interact with NCBP, an 80 kDa nuclear cap binding protein. Nucleic Acids Res. 1995;23:3638–41. doi: 10.1093/nar/23.18.3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boulon S, et al. PHAX and CRM1 are required sequentially to transport U3 snoRNA to nucleoli. Mol Cell. 2004;16:777–87. doi: 10.1016/j.molcel.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 26.Balatsos NA, Nilsson P, Mazza C, Cusack S, Virtanen A. Inhibition of mRNA deadenylation by the nuclear cap binding complex (CBC) J Biol Chem. 2006;281:4517–22. doi: 10.1074/jbc.M508590200. [DOI] [PubMed] [Google Scholar]
- 27.Domanski M, et al. Improved methodology for the affinity isolation of human protein complexes expressed at near endogenous levels. BioTechniques. 2012;0:1–6. doi: 10.2144/000113864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poser I, et al. BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nature methods. 2008;5:409–15. doi: 10.1038/nmeth.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hubner NC, Mann M. Extracting gene function from protein-protein interactions using Quantitative BAC InteraCtomics (QUBIC) Methods. 2011;53:453–9. doi: 10.1016/j.ymeth.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 30.Kiriyama M, Kobayashi Y, Saito M, Ishikawa F, Yonehara S. Interaction of FLASH with arsenite resistance protein 2 is involved in cell cycle progression at S phase. Molecular and cellular biology. 2009;29:4729–41. doi: 10.1128/MCB.00289-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Preker P, et al. PROMoter uPstream Transcripts share characteristics with mRNAs and are produced upstream of all three major types of mammalian promoters. Nucleic Acids Res. 2011;39:7179–93. doi: 10.1093/nar/gkr370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ntini E, et al. Polyadenylation site-induced decay of upstream transcripts enforces promoter directionality. Nat Struct Mol Biol. 2013;20:923–8. doi: 10.1038/nsmb.2640. [DOI] [PubMed] [Google Scholar]
- 33.Hallais M, et al. CBC-ARS2 stimulate 3′-end maturation of multiple RNA families and favor cap-proximal processing. Nat Struct Mol Biol. 2013 doi: 10.1038/nsmb.2720. [DOI] [PubMed] [Google Scholar]
- 34.Andrulis ED, et al. The RNA processing exosome is linked to elongating RNA polymerase II in Drosophila. Nature. 2002;420:837–41. doi: 10.1038/nature01181. [DOI] [PubMed] [Google Scholar]
- 35.Hessle V, et al. The exosome associates cotranscriptionally with the nascent pre-mRNP through interactions with heterogeneous nuclear ribonucleoproteins. Molecular biology of the cell. 2009;20:3459–70. doi: 10.1091/mbc.E09-01-0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hieronymus H, Yu MC, Silver PA. Genome-wide mRNA surveillance is coupled to mRNA export. Genes & development. 2004;18:2652–62. doi: 10.1101/gad.1241204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Das B, Butler JS, Sherman F. Degradation of normal mRNA in the nucleus of Saccharomyces cerevisiae. Mol Cell Biol. 2003;23:5502–15. doi: 10.1128/MCB.23.16.5502-5515.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuai L, Das B, Sherman F. A nuclear degradation pathway controls the abundance of normal mRNAs in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2005;102:13962–7. doi: 10.1073/pnas.0506518102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Visa N, Izaurralde E, Ferreira J, Daneholt B, Mattaj IW. A nuclear cap-binding complex binds Balbiani ring pre-mRNA cotranscriptionally and accompanies the ribonucleoprotein particle during nuclear export. The Journal of cell biology. 1996;133:5–14. doi: 10.1083/jcb.133.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glover-Cutter K, Kim S, Espinosa J, Bentley DL. RNA polymerase II pauses and associates with pre-mRNA processing factors at both ends of genes. Nat Struct Mol Biol. 2008;15:71–8. doi: 10.1038/nsmb1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Listerman I, Sapra AK, Neugebauer KM. Cotranscriptional coupling of splicing factor recruitment and precursor messenger RNA splicing in mammalian cells. Nat Struct Mol Biol. 2006;13:815–22. doi: 10.1038/nsmb1135. [DOI] [PubMed] [Google Scholar]
- 42.Andreu-Agullo C, Maurin T, Thompson CB, Lai EC. Ars2 maintains neural stem-cell identity through direct transcriptional activation of Sox2. Nature. 2012;481:195–8. doi: 10.1038/nature10712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi Y, et al. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol Cell. 2009;33:365–76. doi: 10.1016/j.molcel.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Narita T, et al. NELF interacts with CBC and participates in 3′ end processing of replication-dependent histone mRNAs. Mol Cell. 2007;26:349–65. doi: 10.1016/j.molcel.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 45.Gruber JJ, et al. Ars2 promotes proper replication-dependent histone mRNA 3′ end formation. Mol Cell. 2012;45:87–98. doi: 10.1016/j.molcel.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang XC, Burch BD, Yan Y, Marzluff WF, Dominski Z. FLASH, a proapoptotic protein involved in activation of caspase-8, is essential for 3′ end processing of histone pre-mRNAs. Mol Cell. 2009;36:267–78. doi: 10.1016/j.molcel.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lenasi T, Peterlin BM, Barboric M. Cap-binding protein complex links pre-mRNA capping to transcription elongation and alternative splicing through positive transcription elongation factor b (P-TEFb) J Biol Chem. 2011;286:22758–68. doi: 10.1074/jbc.M111.235077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Das R, et al. SR proteins function in coupling RNAP II transcription to pre-mRNA splicng. Mol Cell. 2007;26:867–81. doi: 10.1016/j.molcel.2007.05.036. [DOI] [PubMed] [Google Scholar]
- 49.Grzechnik P, Kufel J. Polyadenylation linked to transcription termination directs the processing of snoRNA precursors in yeast. Mol Cell. 2008;32:247–58. doi: 10.1016/j.molcel.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rondon AG, Mischo HE, Kawauchi J, Proudfoot NJ. Fail-safe transcriptional termination for protein-coding genes in S. cerevisiae. Mol Cell. 2009;36:88–98. doi: 10.1016/j.molcel.2009.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Almada AE, Wu X, Kriz AJ, Burge CB, Sharp PA. Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature. 2013 doi: 10.1038/nature12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gudipati RK, Villa T, Boulay J, Libri D. Phosphorylation of the RNA polymerase II C-terminal domain dictates transcription termination choice. Nat Struct Mol Biol. 2008;15:786–94. doi: 10.1038/nsmb.1460. [DOI] [PubMed] [Google Scholar]
- 53.Vasiljeva L, Kim M, Mutschler H, Buratowski S, Meinhart A. The Nrd1-Nab3-Sen1 termination complex interacts with the Ser5-phosphorylated RNA polymerase II C-terminal domain. Nat Struct Mol Biol. 2008;15:795–804. doi: 10.1038/nsmb.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nature methods. 2009;6:359–62. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 55.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.