

Abstract
Somatic copy-number alterations (SCNA) are a hallmark of many cancer types, but the mechanistic basis underlying their genome-wide patterns remains incompletely understood. Here we integrate data on DNA replication timing, long-range interactions between genomic material, and 331,724 SCNAs from 2,792 cancer samples classified into 26 cancer types. We report that genomic regions of similar replication timing are clustered spatially in the nucleus, that the two boundaries of SCNAs tend to be found in such regions, and that regions replicated early and late display distinct patterns of frequencies of SCNA boundaries, SCNA size and a preference for deletions over insertions. We show that long-range interaction and replication timing data alone can identify a significant proportion of SCNAs in an independent test data set. We propose a model for the generation of SCNAs in cancer, suggesting that data on spatial proximity of regions replicating at the same time can be used to predict the mutational landscapes of cancer genomes.
Cancer genomes display complex mutational landscapes including amplification, deletion and rearrangement of genetic material1. Many genomic alterations arise as a result of DNA damage or erroneous replication, whereas others occur because of replication-independent events (e.g., exchange reactions between sister chromatids). Eukaryotic DNA replication is spatiotemporally segregated: some regions are replicated early, whereas others are replicated late during S phase2–5. The proposed fractal organization of the genome6 brings together distant genomic regions of similar replication timing to form replication factories (Fig. 1a), where DNA synthesis takes place in multiple DNA regions simultaneously7,8. During replication, single-stranded DNA and DNA double-stranded ends can arise9,10, and interaction between physically proximal segments increases the risk of genetic alterations11 through mechanisms such as microhomology-mediated break-induced replication12. Therefore, patterns of nuclear organization and co-localization of replicating DNA strands may contribute to a mechanistic explanation of the genome-wide frequency and size distribution of genomic alterations in cancer. Indeed, the nuclear proximity of BCR and ABL13, which are also replicated at the same time during S phase, is causally linked to the formation of the BCR-ABL fusion oncogene driving leukemiagenesis. Although nuclear co-localization of chromosomal domains has been proposed to play a key role in generating translocation events14,15, the contributions of nuclear organization and replication timing to the genome-wide patterns of genomic alterations in cancer have not been systematically addressed. Here we propose that DNA replication timing, together with the long-range interaction patterns of the genome, is a predictor of the mutational landscapes of cancer genomes.
Figure 1.

Long-range DNA interactions and the distribution of SNCAs with regard to replication timing zones. (a) Organization of genomic DNA with long-range interactions between distant replication timing zones can increase the risk of interference between adjacent replication forks, leading to genomic alterations. (b–e) The two boundaries of SCNAs are significantly more likely to reside in genomic regions with the same replication timing than that expected by chance for all SCNAs (b), somatic copy number amplifications (SCNA-Amplifications) (c), somatic copy number deletions (SCNA-Deletions) (d) and SCNAs that overlap with known cancer genes listed in the Cancer Gene Census (SCNACGC) (e). Supplementary Module 3 provides separate analyses of SCNA amplifications and deletions as identified by GISTIC. The absolute number (n) of observed and expected cases is provided below each bar. *, P = 1.21 × 10–5; **, P = 1.98 × 10–5; ***, P = 3.76 × 10–6.
RESULTS
Data sets analyzed
We integrated three data sets on the boundaries of 331,724 SCNAs from 2,792 cancer samples classified into 26 cancer types1, genome-wide DNA replication timing4 and long-range DNA interactions6 (Online Methods). In brief, we used data1 on SCNAs in cancer genomes identified using Affymetrix single-nucleotide polymorphism (SNP)-arrays, which yielded copy number ratios derived from tumor samples and matched normal samples. We used replication timing data4 measured using a massively parallel sequencing-based technique across multiple human cell types. In this data set, the replication timing of genomic regions was categorized as ‘constant early’, ‘constant mid’, ‘constant late’ and ‘variable’. Finally, we used intra- and interchromosomal DNA interaction patterns6 in the human genome obtained using HiC, a method that probes the three-dimensional (3D) architecture of whole genomes by coupling proximity-based ligation with massively parallel sequencing. In HiC, two genomic regions represented by one or more sequence reads are likely proximal within the nucleus.
An initial analysis revealed that, in general, regions of similar replication timing cluster together spatially (Supplementary Module 1), supporting the concept of replication factories7,8. We focused on genomic regions that exhibited the same replication timing (constant early, mid or late) across several human cell types4, and we excluded SCNAs that had boundaries close to centromeres, telomeres or on sex chromosomes (Online Methods). We confirmed our results using multiple independent data sets and analyses; a summary of all data sets used1,4–6,16–18 is shown in Table 1. Our analyses revealed three key observations.
Table 1.
Summary of the data sets used in this study
| Feature | Cell type | Data sets |
|---|---|---|
| Somatic copy number alterations in cancer | Multiple cancer types | Ref. 1 |
| Glioblastoma | TCGA glioblastoma project17 | |
| Ovarian cancer | TCGA ovarian cancer project18 | |
| Long-range DNA interactions | Two tissue types | Ref. 6 |
| DNA replication timing | Multiple cell types | Ref. 4 |
| Lymphoblastoid cell line | Ref. 5 | |
| Cancer genes | Multiple cancer types | Cancer Gene Census project16 |
Determinants of cancer SCNAs
Our first observation was that cancer SCNAs arise preferentially in genomic regions that both have the same replication timing and share long-range interactions in the nucleus. We first determined that the two boundaries of SCNAs are significantly more likely to reside in genomic regions with the same replication timing than expected by chance (Fisher's exact test, P = 1.21 × 10–5; Fig. 1b). This pattern is consistent when considering only amplifications (Fig. 1c), deletions (Fig. 1d), SCNAs involving cancer genes (Fig. 1e and Supplementary Module 2) and also when using an alternative data set for genomic alterations in glioblastoma17 and for replication timing5 (Supplementary Module 2). We also obtained consistent results after adjusting for local GC content, chromatin status and lamin-associated domains (Supplementary Module 3). In general, SCNAs in our data set are large (median size 4.5 Mb) and can cover multiple replication timing zones each; therefore, most SCNAs likely arise owing to interactions between single-stranded DNAs or double-stranded ends from two replication timing zones that are far apart on linear DNA. Next, we integrated HiC data and found that for a majority of cases, the two boundaries of SCNAs were in close spatial proximity within the nucleus. This pattern was significantly unlikely to occur by chance (permutation test, P < 1 × 10–3; Supplementary Module 3).
About 32% of SCNAs have boundaries residing in genomic regions with different replication timing (SCNAdRT), and the mechanism of their generation may be different from that above. At ribosomal gene loci, for instance, transcription causes a local disruption of cohesin binding, leading to copy number changes19. We found that three-quarters of SCNAdRT overlapped at least at one boundary with expressed genes, and many shared long-range interactions between the genomic regions in which the boundaries reside (Supplementary Module 4). For those cases, transcription-coupled DNA damage and strand break may play a mutagenic role11,12,20.
SCNA distribution depends on replication timing of boundaries
Our second observation was that the prevalence of SCNA boundaries varied between early-, mid- and late- replication timing zones (Fig. 2a). Early-replicating regions were significantly depleted of SCNA boundaries, whereas late-replicating regions were enriched (Cochran-Armitage test, P < 2.2 × 10–16). This finding is consistent with recent observations that the germ line mutation rate is higher in late-replicating regions21. When we examined amplifications and deletions separately, we found that the above trend holds for deletion boundaries (Fig. 2b; Cochran-Armitage test, P < 2.2 × 10–16), whereas amplification boundaries were slightly enriched in early-replicating regions (Fig. 2c; Cochran-Armitage test, P = 1.59 × 10–4). In general, there is a preference for deletions over amplifications within late replicated regions as compared with early replicated regions (Fig. 2d; Mann Whitney test, P < 2.2 × 10–16). We obtained similar results after binning log2 ratios of cancer samples into amplifications, deletions and normal DNA copy number (Supplementary Module 5), and also using alternative data sets for genomic alterations in glioblastoma17 and replication timing5 (Supplementary Module 5). As genome-wide patterns of both replication timing and SCNAs are correlated with other genomic features, we tested for associations of these patterns with exon density, conserved elements, fragile sites, repeat elements, GC content, chromatin status and lamin-associated domains. We identified a significant association between the number of SCNA boundaries and replication timing zones even after controlling for these factors (P < 0.05 in all cases, Supplementary Module 6), indicating that our findings were probably not due to these covariates. In the absence of the availability of appropriate data sets to test for associations with other hidden covariates, we cannot exclude the possibility that the generation of SCNAs may be causally related to other factors.
Figure 2.

SCNA frequencies vary between different replication timing zones. (a–c) Observed (black) and expected (gray) proportions of SCNA boundaries in early-, mid- and late-replication timing zones. The absolute number (n) of observed and expected cases is shown inside each bar. Similar graphs are shown for somatic copy number amplifications (SCNA-Amplifications) (b) and somatic copy number deletions (SCNA-Deletions) (c). The triangle reflects the direction of enrichment. (d) SNP-chip log2 ratios indicating SCNAs with boundaries in genomic regions within early- and late-replication timing zones. The dashed line, drawn along the median of late-replication timing data points, serves to highlight the difference with early-replication timing data points. (e) SNP-chip log2 ratios indicating SCNAs with boundaries in genomic regions within early- and late-replication timing zones, which overlap with known cancer genes listed in the Cancer Gene Census (SCNACGC). Supplementary Module 6 provides the contingency tables for SCNA amplifications and deletions as identified by GISTIC. The dashed line serves the same purpose as in d. (f) Distribution frequencies of SCNA boundaries near early-, mid- and late-replication transition zones, after dividing the transition zones into 10-kb nonoverlapping windows. (g) Distribution of frequencies of SCNACGC boundaries near early-, mid- and late-replication transition zones, after dividing the transition zones into 10-kb nonoverlapping windows. Replication transition is continuous. *, P < 2.21 × 10–16; **, P = 1.59 × 10–4; ***, P < 2.2 × 10–16; ****, P = 1.42 × 10–8.
When analyzing a set of 18 well-characterized, cancer-associated genes such as MYC, APC and TP53, we found that a majority resides in early replicated regions, and that SCNAs that overlap with these genes often have boundaries in early replicated regions (Table 2); this finding is consistent with previous observations that gene-rich regions are generally replicated early2,4. We then investigated SCNAs overlapping with all cancer genes as classified by the Cancer Gene Census16. We found that, indeed, early replicated regions are enriched for boundaries of these SCNAs, which is notable because these SCNAs are often large (median size 4.5 Mb) and each cover multiple replication timing zones. Again, late replicated regions display a significant enrichment for deletions over amplifications (Fig. 2e; Mann-Whitney test, P = 1.42 × 10–8). We obtained similar results using SCNAs that overlap with peak regions as identified by the GISTIC algorithm1, which are enriched in oncogenes and tumor suppressor genes (Supplementary Module 5). Thus, boundaries of genomic alterations associated with cancer genes have different replication timing as compared to other genomic alterations. Note that although the trends we observed in Figure 2a–e have small effect sizes, they were statistically significant, were reconfirmed using alternative data sets and were consistent with findings observed in germ-line evolution21.
Table 2.
Replication timing for a list of curated cancer genes that are frequently amplified or deleted in many cancer types (as listed in the Cancer Gene Census)
| Gene | Type | Locus | Replication timing | Common SCNA event | Replication timing of the SCNA boundaries | Frequencies (early: late replication timing) |
|---|---|---|---|---|---|---|
| MYC | Oncogene | 8q24.21 | Constant early | Amplification |
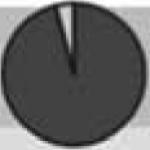
|
(23:1) |
| ERBB2 | Oncogene | 17q12 | Constant early | Amplification |

|
(66:3) |
| CDK4 | Oncogene | 12q14.1 | Constant early | Amplification |

|
(8:2) |
| MDM2 | Oncogene | 12q15 | Constant early | Amplification |

|
(16:1) |
| FGFR1 | Oncogene | 8p12 | Constant early | Amplification |

|
(31:24) |
| CRKL | Oncogene | 22q11.21 | Constant early | Amplification |

|
(28:2) |
| PRKCI | Oncogene | 3q26.2 | Constant early | Amplification |

|
(1:8) |
| CDK6 | Oncogene | 7q21.3 | Constant early | Amplification |

|
(13:1) |
| MET | Oncogene | 7q31.2 | Constant early | Amplification |

|
(1:0) |
| JUN | Oncogene | 1p32.1 | Constant early | Amplification |

|
(6:0) |
| BIRC2 | Oncogene | 11q22.2 | Constant early | Amplification |

|
(1:3) |
| MDM4 | Oncogene | 1q32.1 | Constant early | Amplification |

|
(20:1) |
| ETV6 | Tumor suppressor | 12p13.2 | Constant early | Deletion |

|
(42:0) |
| NF1 | Tumor suppressor | 17q11.2 | Constant early | Deletion |

|
(47:4) |
| ATM | Tumor suppressor | 11q23.1 | Constant early | Deletion |

|
(1:7) |
| PAX5 | Tumor suppressor | 9p13.2 | Constant early | Deletion |

|
(66:1) |
| TP53 | Tumor suppressor | 17p13.1 | Constant early | Deletion |

|
(19:1) |
| APC | Tumor suppressor | 5q21.1 | Constant early | Deletion |

|
(3:16) |
In the pie charts, dark and light gray indicates the proportion of SCNA boundaries in early- and late-replication timing regions.
It was recently reported22 that genomic instability is increased near replication timing transition zones at chr11q and chr21q; these regions harbor several cancer genes. To investigate whether this observation represents a genome-wide phenomenon, we analyzed the patterns of SCNA boundaries near early-, mid- and late-replication timing transition regions. When focusing on 200-kb windows centering on the mid-replicating regions that are flanked by early- and late-replicating regions, we found that the number of SCNA boundaries indeed displays a spike in the vicinity of mid-replicating regions (Fig. 2f), suggesting that replication transition zones harbor many SCNA boundaries. Analyzing the set of SCNAs that overlap with cancer genes from the Cancer Gene Census, we observed that many cancer genes occur near replication timing transition zones and that the frequency of their SCNA boundaries increases near replication transition zones (Fig. 2g). Our findings suggest that increased DNA damage occurring near replication transition regions22 is a genome-wide phenomenon.
In contrast to the boundaries of SCNAs, the genetic material within SCNAs is significantly enriched for early-replicating regions instead of late-replicating regions (Cochran-Armitage test, P < 2.2 × 10–16); this pattern is consistent for amplifications, deletions and regions harboring cancer genes. Although mutagenic processes predominantly operate on DNA breakpoints, cellular fitness depends on the functional implications of SCNAs during tumorigenesis; given that early-replicating regions are gene-dense and many of them also harbor cancer genes (Table 2), functional consequences and natural selection during cancer progression may also play a role in establishing this trend.
Replication timing and spatial proximity predict cancer SCNAs
Our third observation was that a significant proportion of SCNAs in cancer genomes can be identified using replication timing and long-range interaction data alone. We first determined that the probability of two genomic regions harboring SCNA boundaries depends on the extent of long-range interactions between them. Briefly, we divided the genome into nonoverlapping windows of 1 Mb size, and for pairs of regions that were at least 5 Mb apart on linear DNA, we investigated the numbers of HiC reads and SCNA boundaries between them. We found that noninteracting pairs of regions are substantially less likely to harbor SCNA boundaries than those pairs of regions that share at least one HiC read (Fig. 3a). Furthermore, the proportion of interacting pairs of regions that harbor one or more SCNAs increases with an increasing number of HiC reads connecting them. We obtained consistent results when focusing on the SCNAs that overlap with known cancer genes as listed in the Cancer Gene Census (Fig. 3b) and when analyzing amplifications and deletions separately (Supplementary Module 7). In a complementary analysis, we found that over >37% of large SCNAs (>5 Mb) have >10 HiC reads connecting the two boundary regions, whereas only 1.9% have no HiC reads between them (Fig. 3c). Furthermore, we determined that the length distributions of SCNAs depend on the replication timing of their boundaries and can be predicted from long-range interaction data between those regions (Supplementary Module 7). Using BCL6 as an example, we showed that (i) the majority of SCNAs spanning BCL6 have HiC reads between the pairs of boundaries, and (ii) pairs of regions not connected by HiC data are unlikely to harbor pairs of SCNA boundaries (Fig. 3d). Thus, using a learning data set of 331,724 SCNAs from 2,792 cancer samples, we established that replication timing and long-range interactions were associated with the location and size distribution of SCNAs in cancer.
Figure 3.

Genome-wide distributions of long-range interactions and SCNAs. (a) Pairs of genomic regions that were at least 5 Mb apart on the same chromosome were classified into four categories depending on whether they share no HiC reads, ≥1, >5 or >10 HiC reads. We estimated the proportion of region pairs that also harbor the two boundaries of one or more SCNAs. (b) Same as a, but only considering those SCNAs that overlap with known cancer genes. Supplementary Module 7 provides the information for SCNA amplifications and deletions as identified by GISTIC. (c) We classified large SCNAs (>5 Mb, black), and those that overlap with known cancer genes (gray) into four groups depending on the number of HiC reads that link the two boundary regions of the SCNAs. (d) Distributions of SCNAs and long-range interactions around BCL6, a cancer gene on human chromosome 3. The HiC reads that link two boundary regions of SCNAs are shown in red. (e) Pie chart showing the proportion of ovarian cancer SCNAs whose boundaries were predicted using replication timing and HiC data alone.
We then used a test data set of ovarian cancer copy number alterations from The Cancer Genome Atlas to test whether the pairs of boundaries of a proportion of genomic alterations can be identified using long-range interaction between regions of similar replication timing (Supplementary Module 8). Using this approach, we identified 78,412 pairs of genomic blocks of size 500 kb that were at least 5 Mb apart on linear DNA, had the same replication timing and shared at least one HiC read supporting long-range interactions between them. Overlaying ovarian cancer copy number data onto these blocks, we correctly identified the boundaries of 47% (317/675) of all large (>5 Mb) ovarian SCNAs with a resolution of 500 kb at the two boundaries (Fig. 3e). This finding was significantly higher than that expected by chance (Fisher's test, P < 1 × 10–20, Supplementary Module 8). Both the false-negative rate23 (1 – 317/675) and false-positive rate23 (1 – 317/78,412) were large; false negatives are partly attributed to the fact that not all genomic alterations arise from replication-coupled mechanisms (e.g., interaction between two replicating DNA segments). False positives, in contrast, can arise because the generation of SCNAs in cancer genomes is a stochastic event and not all theoretically possible SCNAs arise in any given tumor (the cancer genome is not saturated with genomic alterations). Such situations have been dubbed the false-positive paradox24—that is, false-positive results are more probable than true-positive results when the overall population has a low incidence of events (e.g., copy number alterations). Moreover, some genomic alterations may confer a fitness disadvantage to the cell so that these alterations may be lost from the population. Thus, any individual tumor contains a random set of SCNAs whose number and identity are determined by mutation and selection (Supplementary Module 8). The proportion of ovarian SCNAs detected using replication timing and HiC data alone depended on the choice of spatial resolution and the number of HiC reads (Supplementary Module 8), but for any parameter choice, the proportion remained significantly higher than that expected by chance (Fisher's test, P < 1 × 10–20). Finally, we found that pairs of genomic regions not connected by HiC reads and with different replication timing were significantly unlikely to form SCNAs. We also obtained similar results by cross-validation analyses using alternative data sets1,4,6 (Supplementary Module 8). These findings suggest that data on long-range interactions between distant genomic regions replicating at the same time can be used to predict the mutational landscapes of cancer genomes.
In summary, we report that (i) SCNA boundaries in cancer genomes predominantly resided in genomic regions with the same replication timing that share long-range interactions in the nucleus; (ii) the frequencies of SCNA boundaries differed between replication timing zones; and (iii) the patterns of replication timing and long-range interactions between distant genomic regions can help predict the landscape of SCNAs. We obtained consistent results using a filtered list of SCNAs excluding complex SCNAs that involve multiple alterations (Supplementary Module 9) and using permutation analyses to complement our statistical approaches (Supplementary Module 10). Moreover, even though epigenetic states, GC content and other factors affect DNA replication timing2,3,15 and long-range interactions6, our control calculations suggest that our conclusions hold true even after controlling for these factors (Supplementary Modules 2,6,9). Note, however, that these findings are not based on data of genomic alterations, replication timing and long-range interactions obtained from the same samples.
DISCUSSION
Our findings offer insights into a possible mechanism underlying genome-wide mutational landscapes in cancer. The 3D organization of the genome6 brings together distant genomic regions of similar replication timing to form replication factories, where DNA is replicated in multiple regions simultaneously7,8. Single-stranded DNA (ssDNA) and spontaneous DNA breaks frequently arise during replication9,10, which often lead to single-strand ends initiating processes such as fork stalling and template switching (FosTes)25 or erroneous microhomology-mediated break-induced recombination12. Such recombination can repair double-strand ends when stretches of single-stranded DNA are available in proximity in 3D and share microhomology with the 3′ single-stranded end from the collapsed replication fork. Single-stranded DNA occurs in replication forks, stalled transcription complexes, at DNA secondary structures and in other situations such as in promoter regions and replication origins12. In fact, DNA secondary structures such as G-quadruplexes were shown to promote genomic instability in cancer26,27. The prevalence of these mechanisms during different stages of S phase may play a role in the occurrence of amplifications and deletions. In addition, these mechanisms may work in concert with other processes to drive the generation of alterations involving genomic regions that reside in close proximity within the 3D structure of the nucleus11,20. Finally, the number of SCNA boundaries varies between different replication timing zones; the effect sizes are small but are statistically significant, reconfirmed using alternative data sets and consistent with findings observed in germ-line evolution21. The preference for deletions over amplifications in late replication timing zones may be, at least partly, due to a depletion of the total dNTP pool toward late replication, which was demonstrated to affect DNA excision repair28, trigger deletions29 and potentially escape S phase checkpoints30. These features may work alongside other genomic and epigenomic factors as well as natural selection operating on cancer cells to establish the observed trend.
Because replication timing and nuclear organization are, to some extent, epigenetically regulated2,3,15 and may thus vary between tissue types4,6, our proposed model has the potential to help explain tissue- and cancer type-specific mutational landscapes. Although we focused on solid tumors, our findings may also be applicable to other cancer types such as leukemias. The knowledge that genomic alterations can occur between pairs of interacting genomic regions may help design cancer genome profiling studies toward the identification of novel low-frequency translocation, amplification and deletion events from primary samples, which remains a challenge using current techniques31. In summary, our findings elucidate the roles of DNA replication timing and higher order genomic organization in shaping the mutational landscapes of cancer genomes, and suggest a model for genomic instability in cancer.
ONLINE METHODS
We obtained data on boundaries of 331,724 somatic copy number alterations (SCNAs) from 2,792 cancer samples classified into 26 cancer subtypes1. We obtained genome-wide DNA replication timing data from reference 4, which was generated using a massively parallel sequencing-based technique (Repli-seq). Integrating replication timing information from multiple cell types, the authors categorized genomic regions as constant early, constant mid, constant late and variable across these cell types. We focused on those genomic regions that have constant replication timing across all cell types, that is, that are constant early, constant mid or constant late. We also obtained long-range DNA interaction data from reference 6; these authors employed a massively parallel sequencing technique (HiC) to investigate intra- and inter-chromosomal DNA interaction patterns in the human genome by coupling proximity-based ligation with sequencing. They designated two genomic regions that are represented by one or more HiC sequence reads to be likely in proximity of each other within the nucleus; the number of HiC reads between genomic regions reflects the extent or strength of interaction. The strength of long-range interactions, measured by the number of HiC reads between two genomic regions at a resolution of 1 Mb, was very similar between GM06990 and K562 cell lines (Pearson correlation coefficient: 0.803). This association was even stronger when we focused on interactions between constant replicating regions (Pearson correlation coefficient: >0.82).
Our analyses were done by overlaying SCNA, DNA replication timing and long-range DNA interaction data on the human genome build hg18. Only auto-somes were analyzed for technical reasons such as the gender of the patients from whom the cancer samples were obtained. Furthermore, we excluded SCNAs with boundaries residing within 1 Mb regions from the telomeres and centromeres. This choice was made as many SCNAs arise due to gain or loss of chromosomal arms and telomeric instability. These alterations are driven by distinct molecular mechanisms such as the breakage-fusion-bridge cycle11; moreover, telomeric and centromeric regions are rich in repeats and hence difficult to sequence and assemble, and often have low coverage in high-throughput sequencing data32. The median length of the SCNAs in the filtered data set was ~4.5 Mb (deletions, 5.5 Mb; amplifications, 3.8 Mb). Because some genomic regions display complex patterns of genomic alterations, that is, multiple insertions, deletions or rearrangements in the same locus in a sample, we repeated our analyses excluding complex SCNAs and also those with a low signal to noise ratio, and obtained consistent results (Supplementary Module 9). We also found that mapability of short read sequences is unlikely a concern when using replication timing and HiC data sets (Supplementary Module 9).
We obtained a curated list of cancer genes frequently amplified or deleted in different cancer types from the Cancer Gene Census (December 2010 release)16. We also obtained a list of genomic regions significantly amplified or deleted in the cancer samples as detected by GISTIC (termed “GISTIC peak regions”) from reference 1. Many of these regions overlap with known cancer genes based on the Cancer Gene Census.
All key analyses were repeated using alternative data sets for DNA replication timing5 and SCNAs in glioblastoma (TCGA batch 1)17. We also performed cross-validation analyses by overlaying SCNA data sets from reference 1, replication timing data sets from reference 4, and HiC data from reference 6, as described in Supplementary Module 8, and permutation analyses as described in Supplementary Module 10. Moreover, we performed a detailed analysis using newly published TCGA ovarian cancer data (http://www.cancergenome. nih.gov/), as shown in Figure 3 and Supplementary Module 8. We obtained consistent results when using those alternative data sets and analysis strategies. Statistical analyses were performed using R.
Supplementary Material
AcknowleDgMentS
We would like to thank K. Polyak, A. Melnick, K.J. Patel, A. Chakravarti and R. Beroukhim for comments and discussions. S.D. is a recipient of Human Frontier Science Program long-term fellowship and is a Research Fellow at King's College, Cambridge. This work was funded by the National Cancer Institute's initiative to found Physical Science-Oncology Centers (U54CA143798).
Footnotes
AUTHOR CONTRIBUTIONS
S.D. and F.M. designed the experiments and wrote the paper. S.D. performed the analysis.
Supplementary information is available on the Nature Biotechnology website.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Beroukhim R, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu. Rev. Biochem. 2002;71:333–374. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- 3.Gilbert DM. Evaluating genome-scale approaches to eukaryotic DNA replication. Nat. Rev. Genet. 2010;11:673–684. doi: 10.1038/nrg2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hansen RS, et al. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl. Acad. Sci. USA. 2010;107:139–144. doi: 10.1073/pnas.0912402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woodfine K, et al. Replication timing of human chromosome 6. Cell Cycle. 2005;4:172–176. doi: 10.4161/cc.4.1.1350. [DOI] [PubMed] [Google Scholar]
- 6.Lieberman-Aiden E, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meister P, Taddei A, Gasser SM. In and out of the replication factory. Cell. 2006;125:1233–1235. doi: 10.1016/j.cell.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 8.Ryba T, et al. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010;20:761–770. doi: 10.1101/gr.099655.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saleh-Gohari N, et al. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol. Cell. Biol. 2005;25:7158–7169. doi: 10.1128/MCB.25.16.7158-7169.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004;118:699–713. doi: 10.1016/j.cell.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 11.Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nat. Rev. Genet. 2009;10:551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lukasova E, et al. Localisation and distance between ABL and BCR genes in interphase nuclei of bone marrow cells of control donors and patients with chronic myeloid leukaemia. Hum. Genet. 1997;100:525–535. doi: 10.1007/s004390050547. [DOI] [PubMed] [Google Scholar]
- 14.Wijchers PJ, de Laat W. Genome organization influences partner selection for chromosomal rearrangements. Trends Genet. 2011;27:63–71. doi: 10.1016/j.tig.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 15.Misteli T, Soutoglou E. The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat. Rev. Mol. Cell Biol. 2009;10:243–254. doi: 10.1038/nrm2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Futreal PA, et al. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.TCGA Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.TCGA Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi T, Ganley AR. Recombination regulation by transcription-induced cohesin dissociation in rDNA repeats. Science. 2005;309:1581–1584. doi: 10.1126/science.1116102. [DOI] [PubMed] [Google Scholar]
- 20.Zhang F, Gu W, Hurles ME, Lupski JR. Copy number variation in human health, disease, and evolution. Annu. Rev. Genomics Hum. Genet. 2009;10:451–481. doi: 10.1146/annurev.genom.9.081307.164217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stamatoyannopoulos JA, et al. Human mutation rate associated with DNA replication timing. Nat. Genet. 2009;41:393–395. doi: 10.1038/ng.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watanabe Y, et al. Chromosome-wide assessment of replication timing for human chromosomes 11q and 21q: disease-related genes in timing-switch regions. Hum. Mol. Genet. 2002;11:13–21. doi: 10.1093/hmg/11.1.13. [DOI] [PubMed] [Google Scholar]
- 23.Fisher RA. The Design of Experiments. edn. 8 Hafner; Edinburgh: 1966. [Google Scholar]
- 24.Rheinfurth MH, Howell LW. Probability and Statistics in Aerospace Engineering. NASA. 1998 [Google Scholar]
- 25.Slack A, Thornton PC, Magner DB, Rosenberg SM, Hastings PJ. On the mechanism of gene amplification induced under stress in Escherichia coli. PLoS Genet. 2006;2:e48. doi: 10.1371/journal.pgen.0020048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De S, Michor F. DNA secondary structures and epigenetic determinants of cancer genome evolution. Nat. Struct. Mol. Biol. 2011;18:950–955. doi: 10.1038/nsmb.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao J, Bacolla A, Wang G, Vasquez KM. Non-B DNA structure-induced genetic instability and evolution. Cell. Mol. Life Sci. 2010;67:43–62. doi: 10.1007/s00018-009-0131-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Snyder RD. Consequences of the depletion of cellular deoxynucleoside triphosphate pools on the excision-repair process in cultured human fibroblasts. Mutat. Res. 1988;200:193–199. doi: 10.1016/0027-5107(88)90082-6. [DOI] [PubMed] [Google Scholar]
- 29.Song S, Wheeler LJ, Mathews CK. Deoxyribonucleotide pool imbalance stimulates deletions in HeLa cell mitochondrial DNA. J. Biol. Chem. 2003;278:43893–43896. doi: 10.1074/jbc.C300401200. [DOI] [PubMed] [Google Scholar]
- 30.Kumar D, Viberg J, Nilsson AK, Chabes A. Highly mutagenic and severely imbalanced dNTP pools can escape detection by the S-phase checkpoint. Nucleic Acids Res. 2010;38:3975–3983. doi: 10.1093/nar/gkq128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat. Rev. Genet. 2010;11:685–696. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 32.Koboldt DC, Ding L, Mardis ER, Wilson RK. Challenges of sequencing human genomes. Brief. Bioinform. 2010;11:484–498. doi: 10.1093/bib/bbq016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.