

Abstract
Vascular endothelial cells (ECs) lining the blood vessels are the preferred primary targets of pathogenic Rickettsia species in the host. In response to oxidative stress triggered by infection, ECs launch defense mechanisms such as expression of heme oxygense-1 (HO-1). Previous evidence from an established animal model of Rocky Mountain spotted fever also suggests selective modulation of anti-oxidant enzyme activities in the target host tissues. In this study, we have examined the expression profiles of HO-1 and COX-2 in different tissues during R. conorii infection of susceptible C3H/HeN mice. RNA hybridization with murine HO-1 and COX-2-specific complementary DNA probes revealed increased HO-1 expression in the liver and brain of mice infected with three different doses of R. conorii ranging from 2.25×103 to 2.25×105 pfu, relatively non-remarkable changes in the lungs, and a trend for down-regulation in the spleen. The most prominent HO-1 response was evident in the liver with ~4-fold increase on day 4 post-infection, followed by a decline on day 7. HO-1 expression in the brain, however, peaked with significantly higher levels on day 7. Following infection with both sub-lethal as well as lethal doses of infection, the transcript encoding COX-2 also displayed a pattern of increased expression in the liver and brain. Although immunohistochemical staining revealed increased abundance of HO-1 protein in the liver of infected mice, adjoining serial sections did not exhibit positive staining for COX-2 in infected tissues. The levels of monocyte chemoattracttant protein-1 (MCP-1) and keratinocyte-derived cytokine (KC) were significantly higher in the sera of infected mice and corresponded with the onset and severity of the disease. Treatment of infected animals with antioxidants α-lipoic acid and N-acetylcysteine and HO inhibitor stannous protoporphyrin (SnPPIX) showed only selective beneficial effects on HO-1 and COX-2 expression in the liver and spleen and serum levels of KC and MCP-1. R. conorii infection of susceptible mice, therefore, results in selective regulation of the expression of HO-1 and COX-2 in a manner dependent on the target host tissue’s cellular environment and the propensity of infection with rickettsiae.
1. Introduction
Transmitted in nature by the bite of an infected tick, caused by Rickettsia conorii, and typically characterized by an eschar or ‘tache-noire’ at the tick attachment site, Mediterranean spotted fever (MSF) is a human rickettsial disease, the prognosis of which can range from satisfactory to serious complications depending on the virulence of the pathogen [1,2]. Rickettsiae are obligate intracellular, gram-negative bacteria, which display unique tropism for microvascular endothelium in their mammalian hosts, resulting in disseminated endothelial infection, altered barrier function and inflammation of the vasculature, and compromised vascular permeability, collectively referred to as ‘rickettsial vasculitis’ [3,4]. Published findings from in vitro infection of human umbilical vein-derived endothelial cells (HUVECs) with spotter fever group (SFG) rickettsiae have yielded evidence for the ‘host cell activation’, characterized by increased expression of surface adhesion molecules (intercellular adhesion molecule-1 and E-selectin), cytokines (IL-1α), and chemokines (interleukin-8 and monocyte chemoattractant protein-1) and mediated, for the most part, by the activation of nuclear transcription factor-κB (NF-κB) and p38 stress-activated protein kinase [5-10]. Compelling evidence also documents that endothelial cells infected in vitro with R. rickettsii, the prototypical SFG species and the causative agent of Rocky Mountain spotted fever (RMSF), experience considerable oxidative stress due to increased production and accumulation of superoxide radical and hydrogen peroxide [11,12], and consequently activate important cellular anti-oxidant defense mechanisms such as increased expression of the inducible isozyme of heme oxygenase, HO-1 [13]. An important and physiologically relevant regulatory function of HO-1 in the vasculature is to regulate the activity of the cyclooxygenase (COX) system [14], which is responsible for the generation of a number of vasoactive substances including prostaglandins. Among the well-characterized COX isoforms, COX-1 primarily performs housekeeping functions, whereas COX-2 is tremendously sensitive to a variety of physiologic and pathologic stimuli and exhibits increased expression after endothelial cell infection with SFG rickettsiae in vitro and ex vivo [15]. Our recent studies, however, further suggest that there exist differences in the COX-2 response of different types of host endothelial cells to R. rickettsii infection. Whereas infection of primary cultures of macrovascular HUVECs (derived from a large vessel) results in an apparent biphasic pattern of induced expression, microvascular EC (HMECs) are largely refractory to prominent changes in the regulation of COX-2 expression at the levels of both mRNA and protein [15-17]. On the other hand, induction of HO-1 in both macro- as well as microvascular endothelium is relatively identical [17].
Using an established mouse model of RMSF based on the susceptibility of C3H/HeN mice to intravenously administered R. conorii [18,19], our laboratory has also reported on the selective and differential modulation of key antioxidant enzymes of the glutathione redox cycle, namely glutathione peroxidase, glutathione reductase, and glucose-6-phosphate dehydrogenase, and superoxide scavenging system, namely superoxide dismutase in the target host tissues, including brain, lungs, and testes, during in vivo rickettsiosis [20]. Since cumulative evidence from in vitro and ex vivo models of infection implicates HO-1, COX-2, and chemokines as important regulators of host defense/inflammatory networks and determinants of pathogenetic mechanisms during Rickettsia infection, we hypothesized that their activation may differ in various target organs and in response to varying doses and outcomes of infection. Thus, the aims of the present study were to investigate and compare the transcriptional activation of HO-1 and COX-2 in different target tissues in the host as well as systemic secretion of chemokines KC (the murine homolog of IL-8; keratinocyte factor) and MCP-1 in C3H/HeN mice infected with the sub-lethal and lethal doses of R. conorii in the presence and absence of treatment with compounds known to protect against oxidative stress.
2. Materials and methods
2.1. Cultivation and enumeration of Rickettsia organisms
Rickettsia conorii (Malish 7 strain), a human isolate from South Africa with unknown passage history was obtained from Dr G. Dasch (Centers for Disease Control and Prevention, Atlanta, GA). The organisms were propagated in Vero C1008 cells (American Type Culture Collection, Rockville, MD) and then purified from heavily-infected host cells as described previously [20]. The titers of viable rickettsiae in such preparations were determined by plaque titration assay on Vero cell monolayers as the number of plaque forming units (pfu) per ml. The results of plaque assay were subsequently confirmed by a TaqMan® quantitative-PCR procedure using primer pair Rp877p-Rp1258n for rickettsial citrate synthase (gltA) gene and an internal probe with 100% homology to R. conorii [21]. For infection, a purified stock of R. conorii was appropriately diluted in sterile normal saline prior to administration into mice via tail vein injection.
2.2. Mouse model of infection
Six to eight weeks old, male C3H/HeN mice (Harlan Sprague Dawley Inc., Indianapolis, IN) were infected with viable R. conorii by intravenous injection in the tail vein. Three different doses with a log difference, i.e. 2.25×103, 2.25×104, and 2.25×105 pfu per mouse, in a total volume 200μl were used representing low, medium (sub-lethal), and high (lethal) levels of infection. For each experiment, a group of animals receiving only intravenous injection of saline was included to serve as uninfected controls. At day 4, 7, or 10 post-injection, mice were anaesthetized using 45 mg/kg Phenobarbital sodium intraperitoneally and blood samples were collected by direct cardiac puncture. Organs were removed under sterile conditions for further processing for RNA isolation and immunohistochemical studies.
2.3. Treatment with the modulators of oxidative stress
In some experiments, mice were treated with either α-lipoic acid [(α-LA or DL-6,8-thioctic acid); from Sigma Chemical Company, St. Louis, MO] or N-acetyl-L cysteine (NAC; from Sigma), both of which are potent antioxidants known to stimulate the biosynthesis and intracellular accumulation of reduced glutathione (GSH). In addition, Stannous (IV) Protoporphyrin IX dichloride (SnPPIX; from Frontier Scientific Inc., Logan, UT), a potent inhibitor of heme oxygenase, was also administered to a cohort of animals. Briefly, α-LA was dissolved in sterile normal saline and the pH was then adjusted to 7.4. α-LA was administered as a once daily dose of 150 mg/kg body weight by oral gavage for three days prior to and during the course of infection. SnPPIX was dissolved in 0.1 N sodium hydroxide followed by adjustment of the solution’s pH to 7.0 and was administered intraperitoneally at 100 μmol/kg body weight. NAC, dissolved in sterile PBS, was administered at 50 mg/kg body weight via intra-peritoneal injection. Both SnPP and NAC were given to mice as once daily dose 24 h prior to and during the course of infection. The groups of control animals were handled simultaneously, but were administered with the corresponding volumes of sterile PBS.
2.4. Measurement of chemokine secretion
Serum samples from R. conorii-infected mice and simultaneously processed control (uninfected) animals were prepared by centrifugation of the whole blood at 10,000g for 5 minutes to remove the erythrocytes and kept frozen at -80°C until further analysis. The amounts of MCP-1 and KC were determined by enzyme-linked immunosorbent assay (ELISA) kits following the instructions provided by the manufacturer (R&D systems Inc., Minneapolis, MN). The detection ranges of both MCP-1 and KC for the assay systems used were from 2 to 1000 pg/ml. At least two dilutions of each sample were assayed in duplicate and only the OD values corresponding to the linear range of the standard curve were used in calculations to determine the chemokine concentration.
2.5. Northern blot analysis
At the time of sacrifice, sections of organs (lungs, liver, spleen, and brain) were collected aseptically and quickly immersed in an RNA later™ stabilization solution (Ambion Applied Biosystems, Foster City, CA) until subjected to isolation of total RNA using a TRI-reagent™ protocol according to the manufacturer’s manual. Equal amounts of RNA (6-10 μg) from infected mouse tissues and corresponding controls were subjected to Northern blot analysis using radioactively labeled cDNA probes as described earlier [13,16,17]. Murine HO-1 probe was synthesized by PCR amplification of a 155 bp fragment using “Super Array” primer set (Frederick, MD). Mouse-specific COX-2 probe and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) probe were obtained from Cayman Chemical Company, Ann Arbor, MI, and US Biological Company, Swampscott, MA, respectively. The differences in the loading of individual samples on different gel lanes were corrected by stripping and re-probing of the blotted membranes for the housekeeping gene GAPDH.
2.6. Immunohistochemical staining
Tissues fixed in 10% (v/v) formalin at the time of animals’ sacrifice were embedded in paraffin and sectioned at 5μm on a microtome. Distribution of rickettsiae in target host tissues was evaluated using rabbit polyclonal anti-R. rickettsii serum (provided by Dr. Ted Hackstadt, NIH/NIAID Rocky Mountain Laboratories, Hamilton, MT). Anti-murine HO-1 and COX-2 rabbit polyclonal antibodies were purchased from Assay Designs, Ann Arbor, MI, and Cayman Chemical Company, Ann Arbor, MI, respectively. Immunohistochemical staining was performed according to our previously established protocols and procedures [20].
2.7. Densitometric and statistical analysis
The radiograms with optimum exposure were scanned in the grayscale mode using a HP ScanJet 6300C scanner at a resolution of 600 dpi for quantitative analysis of data from Northern blots. Volume analysis was performed using ImageQuant software, version 3.3 and band intensities were determined as densitometric units. In order to compare between experimental conditions, the normalized band intensity for uninfected control in each experiment was assigned a value of 1. All data were calculated as the mean ± standard error and a minimum of three animals were included for each experimental condition. The comparisons between the study and the control groups were performed using Student’s t test and the p values of ≤0.05 were considered to be statistically significant.
3. Results
3.1. HO-1 mRNA expression in tissues of mice infected with different doses of R. conorii
Oxidative stress has been implicated as an important contributor to the pathogenesis associated with spotted fever rickettsiae in both in vitro and in vivo models of infection [11,12,20]. However, the involvement of HO-1 as an anti-oxidant host protective mechanism has only been established for in vitro responses of endothelial cells of different origin to R. rickettsii [13,17]. To fill this critical gap in the knowledge, we first investigated HO-1 mRNA expression in different tissues (brain, liver, lungs, and spleen) after infection with three different doses of R. conorii ranging from 2.25×103 to 2.25×105 pfu per mouse. Northern blot analysis indicated significantly increased expression of HO-1 transcript in the liver of mice on day 4 post-infection, the extent of which was relatively similar (an average of about 4-fold) for all three doses of infection mentioned above. At a later time (day 7), the levels of HO-1 expression showed a slight decrease, but were still significantly higher than the basal level in uninfected controls (Figure 1A). In contrast, the spleens from mice infected with the low and medium doses of R. conorii had significantly lower HO-1 expression than in controls, whereas those from the high dose of 2.25×105 pfu, causing 100% mortality after day 4, did not yield a significant difference from the basal levels of expression (Figure 1B). Although a pattern of gradual increase in HO-1 expression in the brains of infected mice on day 4 was noticeable, statistically significant induction of ~2.5-fold on day 7 was clearly evident in mice receiving either low or medium (sub-lethal) doses of infection (Figure 1C). Finally, lungs of infected mice showed only slightly higher HO-1 expression on day 4 for mice infected with the low dose and on day 7 for mice infected with the medium dose. The lethal dose of infection, in contrast, did not yield any statistically significant changes in pulmonary HO-1 expression (Figure 1D). Thus, although administration of low and medium doses of R. conorii to susceptible mice stimulates HO-1 expression in the liver and brain, pulmonary HO-1 remains relatively unaffected while splenic HO-1 reveals a pattern of suppression. Also, with the exception of hepatic HO-1 expression, lethal dose of infection appears to disable the host’s ability to launch an anti-oxidant response via induction of HO-1.
Figure 1.
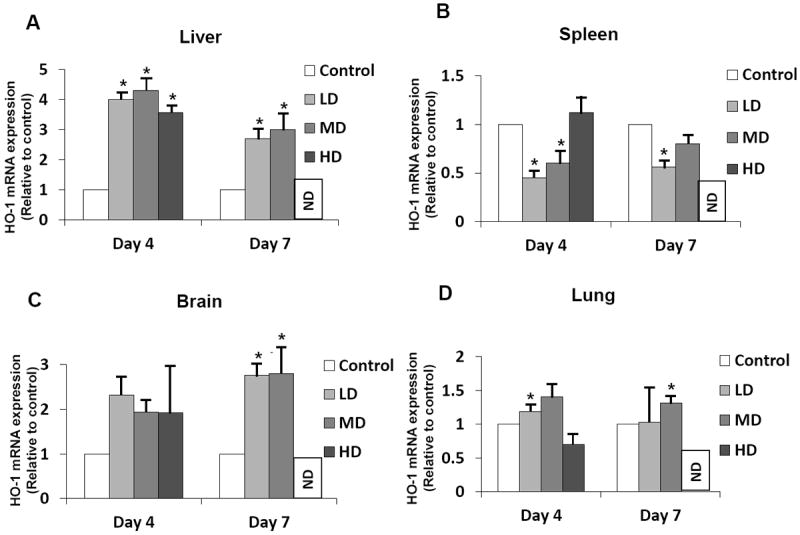
Comparative evaluation of heme oxygenase-1 (HO-1) mRNA expression during in vivo R. conorii infection. C3H/HeN mice infected with 2.25×103 (Low dose: LD), 2.25×104 (Median dose: MD), and 2.25×105 (High dose: HD) plaque forming units of R. conorii via intravenous administration were sacrificed on day 4 and day 7 post-infection (with the exception of animals infected with the high dose, which usually die prior to day 7) to collect tissue samples in a preservative RNA later™ solution and storage at -20°C. RNA from the liver, spleen, brain, and lung was isolated using Tri-reagent® protocol and subjected to northern blotting with a murine HO-1-specific cDNA probe labeled using 32P-[αCTP]. The blots were then stripped and probed with GAPDH to normalize for variations in sample loading on different lanes. For the ease of comparison among experimental conditions, the basal level of HO-1 expression in tissues from uninfected controls was assigned a value of 1. Histograms represent the average values from a minimum of three independent observations. Error bars represent standard error of the mean and * denotes significant change (p≤0.05) in comparison to the basal expression in controls. ND = not done.
3.2. Transcriptional activation of COX-2 in tissues of mice infected with different doses of R. conorii
Because published evidence implicates regulation of cyclooxygenase by heme-HO system in microvascular endothelial cells, we next determined the steady-state levels of COX-2 mRNA in different tissues of mice infected with different doses of R. conorii employing Northern blot analysis with a murine COX-2-specific cDNA probe. In animals receiving the low dose of R. conorii, the levels of hepatic COX-2 expression displayed only slight increase at day 4 as well as day 7 post-infection, but statistically significant yet modest increase was evident on day 4 in the cohort of mice infected with the high, lethal dose of R. conorii. Corresponding to the response to both low and high doses of infection, the animals infected with the medium dose also displayed only modest change in the steady-state levels of COX-2 mRNA in the liver (Figure 2A). In the spleen, however, there was significant decrease in the COX-2 mRNA expression on day 4 with low and high doses of infection, and on day 7 with the medium dose of R. conorii, which was consistent with the down-regulation of HO-1 activity (Figure 2B). In brain, low dose of infection did not cause any change, whereas medium and high doses produced significant increase of COX-2 expression (Figure 2C). Also, lungs of infected animals did not demonstrate any significant changes in COX-2 transcription regardless of the dose and time of infection (Figure 2D).
Figure 2.
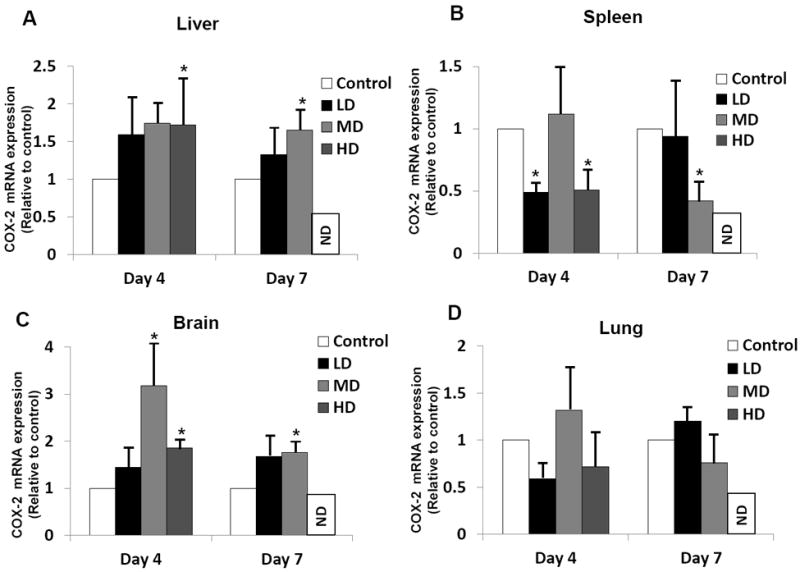
Comparative analysis of cyclooxygenase-2 (COX-2) mRNA expression during in vivo R. conorii infection. C3H/HeN mice infected with 2.25×103 (Low dose: LD), 2.25×104 (Median dose: MD), and 2.25×105 (High dose: HD) plaque forming units of R. conorii via intravenous inoculation were sacrificed on day 4 and day 7 post-infection (with the exception of animals receiving high dose, which usually die prior to day 7) to collect tissue samples in a preservative RNA later™ solution and storage at -20°C. RNA from the liver, spleen, brain, and lung was isolated using Tri-reagent® protocol and subjected to northern blotting with a murine COX-2-specific cDNA probe labeled using 32P-[αCTP]. The blots were then stripped and probed with GAPDH to normalize for variations in sample loading on different lanes. For the ease of comparison among experimental conditions, the basal level of COX-2 expression in tissues from uninfected controls was assigned a value of 1. Histograms represent the average values from a minimum of three independent observations. Error bars represent standard error of the mean and * denotes significant change (p≤0.05) in comparison to the basal expression in controls. ND = not done.
3.3. Immunohistochemical evaluation of HO-1 and COX-2 expression in infected tissues
To further determine whether or not changes in HO-1 and COX-2 mRNA expression translate into increased protein synthesis in target organ systems, we next performed immunohistochemical staining of serial tissue sections using antibodies capable of specifically detecting spotted fever group rickettsiae, HO-1, and COX-2. As shown in Figure 3, negligible to minimal levels of background staining was seen in the liver of uninfected controls (panel A), and distinct foci of positive staining for rickettsial antigen confirming the presence of R. conorii were evident in the liver (panel B). As reported previously [20], rickettsiae in the liver were apparently localized to either sinusoidal endothelial cells lining vessels or to the loci of granulomatous inflammation. The livers from uninfected mice had very low background staining for both HO-1 and COX-2 (Panels C and E). In the liver of infected mice, on the other hand, endothelial cells lining the sinusoidal vessels as well as hepatocytes were distinctly positive for HO-1 protein (Panel D), but there were no apparent changes in the expression of COX-2 (Panel F).
Figure 3.
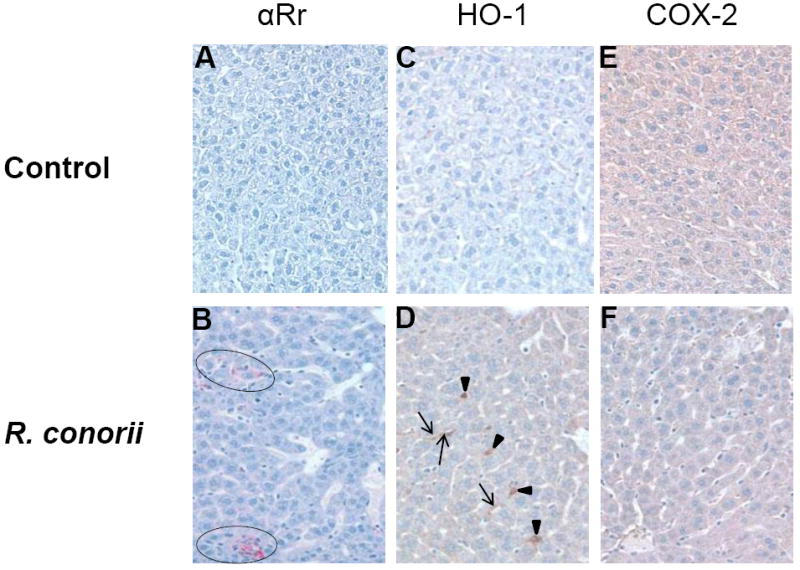
Immunohistochemical analysis of HO-1, COX-2, and the presence of rickettsiae in the hepatic tissue of mice. Liver sections from uninfected and R. conorii-infected mice were subjected to staining using anti-HO-1, anti-COX-2, and anti-Rickettsia antibodies followed by counterstaining with hematoxylin as described in Materials and Methods. Panels A, C, and E show negative staining for R. conorii, HO-1, and COX-2 in the liver of an uninfected mouse (control). Panels B, D, and F represent photomicrographs of liver sections obtained on day 4 from a mouse infected with 2.25×105 plaque forming units of R. conorii. Intense inflammatory infiltrates with positive staining for R. conorii (circled) were present in the liver (Panel B). As shown in Panel D, expression of HO-1 in the endothelium lining of hepatic sinusoids (indicated by arrows) and hepatocytes (arrowheads) was also clearly evident, yet there was no evidence of positive staining with anti-COX2 antibody (Panel F). Original magnification ×200.
3.4. Effects of oxidant stress modulators on HO-1 and COX-2 activation in target tissues during R. conorii infection
Increased generation and propagation of reactive oxygen species is one of the major mechanisms underlying vascular stress and dysfunction [11,12]. Consequently, known antioxidants and inducers of HO activity may exert their beneficial effects by preventing oxidant-mediated vascular distress and damage. In this context, we sought to determine the effects of potent antioxidants αLA and NAC and an HO inhibitor SnPPIX on HO-1 and COX-2 expression during R. conorii infection in vivo. HO-1 expression exhibited a trend for increased mRNA expression in the liver of infected animals treated with NAC, in comparison to those infected but not treated, but the change was not statistically significant (Figure 4A). In animals receiving oral administration of α-LA during the infection, HO-1 expression was significantly increased in the spleen (Figure 4B). In contrast, α-LA treatment led to suppression of infection-induced HO-1 expression response in the liver and the effect was statistically significant when compared to the corresponding controls comprised of infected mice receiving no treatment. Further, COX-2 transcriptional activation in both the liver and the spleen was up-regulated in animals treated with SnPPIX versus infection alone (Figures 4C and 4D). In addition, administration of NAC during infection also caused suppression of splenic COX-2 expression, an effect which was found to be statistically significant. The brain and lungs of infected animals administered with either of the compounds, i.e. α-LA, NAC, or SnPPIX, did not exhibit any significant differences in the transcriptional activation of HO-1 and COX-2 in comparison with the tissues from the corresponding cohort of infected, but untreated animals (data not shown). Thus, treatment of mice with the modulators of oxidative stress during the course of infection revealed only selective effects on the status of HO-1 and COX-2 expression in the target host tissues examined.
Figure 4.
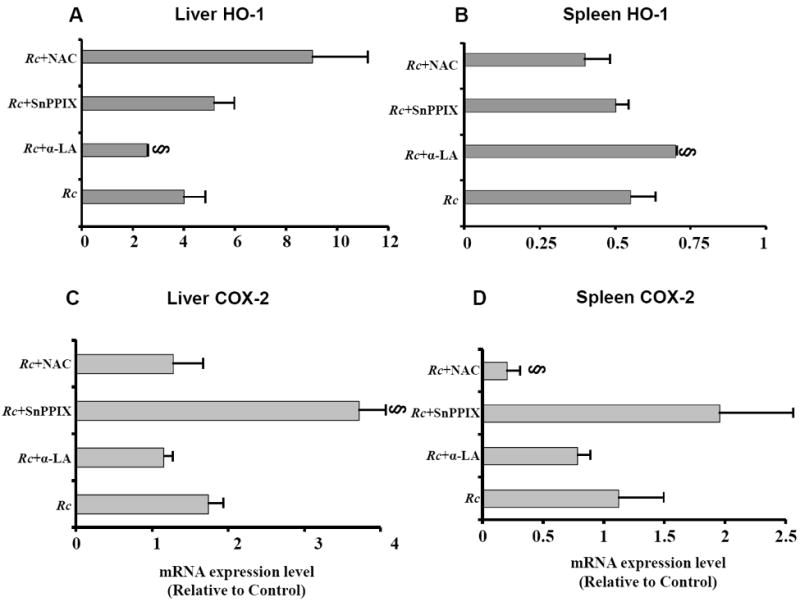
Effect of anti-oxidant compounds α-lipoic acid (α-LA) and N-acetylcysteine (NAC), and heme oxygenase inhibitor stannous protoporphyrin IX hydrochloride (SnPPIX) on the expression of hepatic and splenic HO-1 and COX-2 during R. conorii infection. Mice infected with 2.25×104 plaque forming units of R. conorii received the treatments or vehicle controls as detailed in the Materials and Methods. RNA from the liver and spleen was isolated using Tri-reagent® protocol and subjected to northern blotting with murine HO-1 and COX-2-specific cDNA probes labeled using 32P-[αCTP]. The blots were then stripped and probed with GAPDH to normalize for any variations in sample loading on different lanes. The data in the bar diagrams are presented as the mean ± SEM from three independent animals and represent changes in relation to the uninfected controls, which were given a value of 1. The symbol § signifies a statistically significant change in comparison to the infected animals receiving no treatment (labeled as Rc).
3.5. Secretion of chemokines during R. conorii infection of C3H/HeN mice and effects of the modulators of oxidative stress
R. conorii infection of host cells in vitro induces a pro-adhesive and pro-inflammatory phenotype characterized by increased expression of adhesion molecules and release of cytokines and chemokines including IL-8 and MCP-1 [8,21]. The levels of both MCP-1 and KC (murine analog of IL-8) in the serum samples from mice infected with all three doses of R. conorii were found to be dramatically increased on day 2 and day 4 post-infection, attaining the maximum level on day 4 for mice infected with the low and medium doses of R. conorii and on day 2 for the lethal dose of infection (Figures 5A and 5B). On day 7, the levels of both chemokines were comparatively lower than at earlier times. Specifically, the serum concentrations of MCP-1 on day 7 were significantly lower than their peak levels on day 4 post-infection (Figure 5A). Thus, it is evident that enhanced MCP-1 secretion is dependent on the infectious dose of R. conorii and correlates with the course of infection. Intriguingly, the serum KC levels in mice infected with the low and medium doses of R. conorii on both day 2 and day 4 were significantly higher in comparison to the uninfected controls, but animals receiving high dose displayed a relatively less robust KC response (Figure 5B). Again, modulators of oxidant stress had only selective effects on the secretion of chemokines during R. conorii infection. The serum MCP-1 levels in infected mice treated with SnPPIX and NAC were significantly decreased in comparison with untreated animals infected with medium dose for 4 days (Figure 5C). The levels of KC under similar experimental conditions were also decreased in mice treated with NAC (Figure 5D). Thus, treatment with NAC resulted in at least partial attenuation of chemokine secretion during infection with a sub-lethal dose; SnPPIX only selectively affected the levels of MCP-1; and α-LA had no significant effect. Intriguingly, the weight loss data from infected animals with all of these treatments did not yield any significant differences when compared with the group of animals that were infected but then left untreated (not shown).
Figure 5.
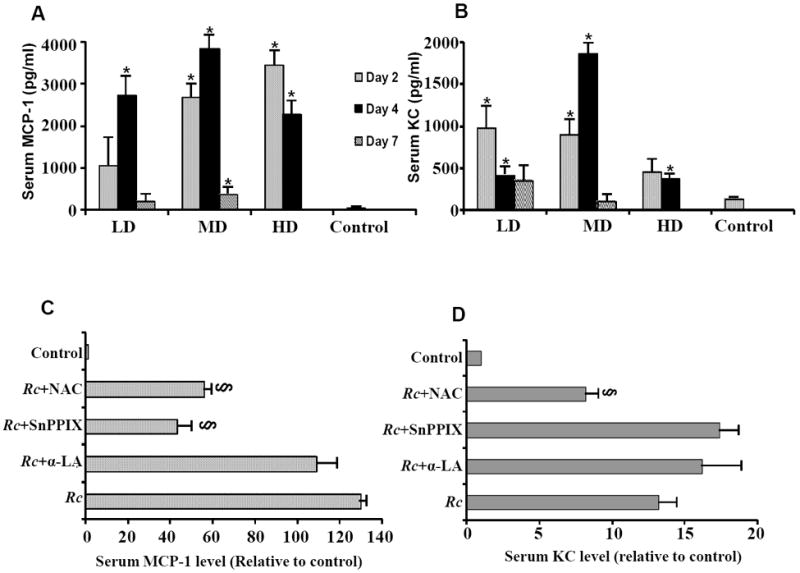
Serum levels of chemokines monocyte chemoattractant protein-1 (MCP-1) and keratinocyte-derived cytokine (KC: functional homolog of IL-8) during R. conorii infection of susceptible mice. C3H/HeN mice infected with 2.25×103 (Low dose: LD), 2.25×104 (Median dose: MD), and 2.25×105 (High dose: HD) plaque forming units of R. conorii via intravenous inoculation were sacrificed on day 2, day 4 and day 7 post-infection (with the exception of high dose animals, which usually die prior to day 7) to collect serum samples that were kept frozen at -20°C until ELISA measurements. The data in vertical bars (panels A and B) are presented as the mean ± SEM of absolute chemokine concentrations from at least three independent observations for each experimental condition. The asterisk (*) denotes significantly higher levels in comparison to the baseline controls. The effects of α-lipoic acid (α-LA), N-acetylcysteine (NAC), and stannous protoporphyrin IX hydrochloride (SnPPIX) on MCP-1 and KC levels are summarized in Panels C and D, respectively. Please note that the values in horizontal bars represent fold-changes (mean ± SEM of three separate observations) in comparison to serum MCP-1 or KC levels in uninfected controls, which were assigned a value of 1. The symbol § signifies statistically significant difference in relation to the animals infected with R. conorii, but no treatment (labeled as Rc).
4. Discussion
Rickettsioses now represent important reemerging infectious diseases with increasing global burden of morbidity and mortality, the pathology of which is generally attributable to disseminated infection of vascular endothelium and systemic inflammatory responses [19,22]. Because infection of cultured endothelial cells with R. rickettsii results in increased generation of harmful reactive oxygen species, oxidant-induced damage and dysfunction have been projected as one of the critical mechanisms underlying cellular injury and pathogenesis [4,11,12]. We have previously reported on the modulation of enzyme systems playing an important role in glutathione redox mechanisms and reducing equivalents (NADH, NADPH) generating pathways in an established mouse model of RMSF [20]. Here, we demonstrate selective regulation of HO-1, a cytoprotective enzyme known for its involvement in cellular defense against oxidative stress and inflammatory insults [23]. R. conorii infection induced HO-1 mRNA in the liver and brain, albeit the peak response in the latter occurred later, likely corresponding to the levels of pathogen burden in target tissues during the course of infection. Although lack of evidence for the detection of rickettsiae in the brain is a limitation of the present study, published quantitative PCR data demonstrate a progressive increase in rickettsial load peaking at day 5 in the liver, lungs, and brain of mice infected with both lethal and sub-lethal doses of R. conorii and as expected, higher (~10-fold) pathogen burden with the lethal dose [24]. In addition, we were also able to recover viable, plaque-forming rickettsiae from the brains of infected mice, the titers of which were proportional to the dose and duration of infection [20]. Increased HO-1 expression, however, did not represent a generalized response to infection as the transcript levels in the spleen and lungs were either suppressed or only marginally affected. Lungs are highly vascularized tissues with significant accumulation of rickettsiae during in vivo infection [20,24]. Although human pulmonary microvascular and artery ECs respond to R. rickettsii by stimulating HO-1 response in vitro [17], blunted induction of HO-1 in vivo likely reflects compromised ability of the host to prevent pulmonary insults owing to oxidative stress. Inability to induce or insufficient HO-1 expression have been associated with a number of pulmonary ailments, including interstitial pneumonia, pulmonary edema, and adult respiratory distress syndrome, which constitute major complications of severe and fatal outcomes of rickettsioses in humans [25-27]. Conversely, sustained expression of HO-1 in the lung protects against deleterious effects of hypoxia and particularly against pulmonary inflammation, hypertension, and vessel wall hypertrophy [28]. On the other hand, HO-1 can also exert pro-oxidant effects in certain situations by virtue of releasing more free iron, as has been demonstrated by up-regulation of HO-1 in the spleen of animals treated with hemolytic agents, including aniline [29]. Thus, suppression of splenic HO-1 expression during infection may represent a protective response to curtail the free iron-mediated oxidant stress for the benefit of the host.
The understanding of the roles of HO-1 and its reaction products in microbial host defense mechanisms is now beginning to expand [30], yet there are relatively few reports pertaining to its significance in animal models of infection closely mimicking human disease. The induction of HO-1 is an important cytoprotective mechanism in sepsis [30,31]. Recent studies have shown increased expression of HO-1 in macrophages infected with Mycobacterium tuberculosis and regulation of bacterial ‘dormancy regulon’ by carbon monoxide, a by-product of HO activity [32]; induced expression in the liver and potent cytoprotective function in a mouse model of salmonellosis [33]; and down-regulation of placental HO-1 by Listeria monocytogenes leading to infectious abortion [34]. In contrast, abortion due to Brucella abortus infection in pregnant mice is attributed to HO-1 expression in the placenta [35] and HO-1 induction in an experimental model of pneumocoocal meningitis results in iron-mediated oxidative damage in the brain [36]. Interestingly, in a mouse model of secondary bacterial pneumonia due to Streptococcus pneumoniae after initial influenza infection, the expression of HO-1 was significantly higher in the lungs, but not in other tissues [37]. Our findings demonstrate selective HO-1 induction in the liver and brain, only subtle and mostly insignificant changes in the lungs, and apparent suppression in the spleen during R. conorii infection.
In endothelial cells, the heme-HO system functions as a negative regulator of COX-2 expression, influencing the production of vasoactive prostanoids [14,38]. In response to lipopolysaccharide, however, COX-2 can either be positively or negatively affected by HO-1 depending on the cell-type [39,40]. In the mouse model of RMSF, both HO-1 and COX-2 exhibit an apparently similar pattern of regulation as evidenced by highest increase in COX-2 expression in the liver, a relatively modest up-regulation in the brain, suppression in the spleen, and lack of significant responses in the lungs. This correlates with our recent in vitro findings suggesting that primary human pulmonary endothelial cells of microvascular origin do not express COX-2 during in vitro infection with R. rickettsii and display a pattern of differential HO-1 and COX-2 activation in tissue-specific endothelial cells [17]. Accordingly, we infer that the expression of transcripts for both HO-1 and COX-2 displays selective modulation during R. conorii infection, likely depending on the cellular environment of the tissues harboring the pathogen. Derived from arachidonic acid metabolism by the COX enzyme system, prostaglandins are considered to be important lipid mediators of acute inflammatory response and alterations in vascular permeability. COX2-deficient mice have been shown to display resistance against detrimental effects of endotoxemia [41]. On the other hand, a recent study has implicated COX-2 in the production of an antimicrobial peptide human-β-defensin and killing of Staphylococcous aureus [42]. During rickettsioses in vivo, vascular inflammation is a primordial response that may protect against infection-induced injury by restoring damaged tissue to its normal physiology. In this context, whether or not induced COX-2 expression and/or activity in the target host tissues on a selective basis serves to initiate controlled inflammation and host defense or contributes to the disease pathogenesis remains to be investigated further. Such studies will allow us to address an important limitation of this study that despite increased mRNA in the liver, we were unable to detect COX-2 protein through immunohistochemical analysis. Nevertheless, our current data indicate that R. conorii infection affects the transcriptional expression of HO-1 and COX-2 in various organ systems of C3H/HeN mice, suggesting the suitability of this established in vivo model of rickettsiosis to further delineate the potential roles of these important regulatory enzymatic mechanisms in the pathogenesis of rickettsial diseases.
There is now ample evidence suggesting expression and secretion of cytokines and chemokines by host cells infected in vitro with R. rickettsii and R. conorii. Capable of attracting inflammatory cells to the foci of infection and fine-tuning immune responses via regulation of others, such mediators include, but are likely not limited to, IL-1α (but not IL-1β), IL-8, and MCP-1 [6,7,9,10]. Of note, certain inflammatory chemokines known to specifically target activated T cells through CXCR3 receptor, namely CXCL9 (Mig) and CXCL10 (IP-10), were expressed in the brain, liver, and lungs of infected mice, but were not detectable in mouse endothelial cells infected in vitro with R. conorii[24]. Similarly, fractalkine (CXCL1) is expressed in all of the investigated organ systems in the mouse model of RMSF [43], but the status of its production by infected endothelium remains to be determined. The present study indicates increased circulating levels of KC and MCP-1 in the blood of mice infected with R. conorii. While it is apparent from the presented data that serum concentrations of chemokines reflect the onset and severity of disease, it remains to be determined whether or not they are also useful indicators of recovery from the infection. Although correlation between in vitro and in vivo findings would implicate endothelial cells as the major source of these potent chemoattractants for macrophages and neutrophils [6,9,10], the possibility of contributions from other host cells that may be infected in vivo can’t be completely ruled out. Despite this caveat, our results are in agreement with a recent comparative analysis of patients suffering from MSF and African tick bite fever (ATBF) due to R. africae, attributing higher degree of inflammation during MSF to markedly elevated IL-8 and soluble adhesion molecules, when compared with either negligible or only a modest increase during relatively milder ATBF [44].
Although multiple lines of evidence point towards a protective role for the potent anti-oxidant α-LA against R. rickettsii-induced cellular damage by increasing the levels of thiols and glutathione peroxidase and diminishing the peroxide content of ECs in vitro and in the mouse model of RMSF used in the present study [20,45,46], we did not observe a significant effect of α-LA on both the regulation of HO-1 and COX-2 and the chemokine levels during infection. This supports the previous suggestion of complex interactions with the cellular metabolic machinery, a component of which likely is differential regulation of anti-oxidant enzyme systems. Instead, administration of NAC, which also possesses a strong anti-oxidant activity, was able to further induce the hepatic HO-1 expression and suppress the levels of circulating chemokines in R. conorii-infected mice. Such an effect is consistent with the cytoprotective effects of NAC against rhinovirus-induced nuclear factor κB activation and IL-8 secretion by respiratory epithelial cells in vitro [47], in a rat model of neurophilic lung inflammation and chemoattractant expression in vivo [48], and clinically in patients with sepsis [49]. Furthermore, as has been reported for the predominantly negative crosstalk between HO-1 and COX-2 [14,37], inhibition of HO-1 by SnPPIX led to increased COX-2 expression in the liver and spleen. Selective down-regulation of only MCP-1 by SnPPIX is also rather intriguing, the potential explanation for which may be that infection-induced HO-1 may also play a cytoprotective role by inducing the synthesis of MCP-1. It is important to consider that both HO-1 and MCP-1 have now been documented as oxidant response mediators, the regulation of which during oxidative stress is also dependent on the cell-type under investigation. Although the effects of COX-2-specific inhibition on serum chemokines were not investigated in our study, administration of celecoxib to mice infected with influenza virus had no significant affect on the levels of serum MCP-1 or KC [50], indicating the likelihood of potential contributions from other, as yet unidentified, mechanisms in the production of KC during R. conorii infection.
In conclusion, R. conorii infection of susceptible mice results in increased serum levels of MCP-1 and KC; selective regulation of the expression of HO-1 and COX-2 in apparent correlation with the target host tissue’s cellular environment and the propensity of infection; and selective beneficial effects of antioxidants α-lipoic acid/N-acetylcysteine and HO inhibitor (SnPPIX) on HO-1 and COX-2 expression in the liver and spleen and serum levels of the chemokine MCP-1.
Acknowledgments
We thank Julia Ablaeva and Semion Kiriakidi for excellent technical assistance throughout the course of this study. This research was supported in part by USPHS grants AI067613 and AI076697 from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health, Bethesda, MD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rovery C, Raoult D. Mediterranean spotted fever. Infect Dis Clin North Am. 2008;22:515–30. doi: 10.1016/j.idc.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 2.Sousa R, França A, Dória Nòbrega S, Belo A, Amaro M, Abreu T, Poças J, Proença P, Vaz J, Torgal J, Bacellar F, Ismail N, Walker DH. Host- and microbe-related risk factors for and pathophysiology of fatal Rickettsia conorii infection in Portuguese patients. J Infect Dis. 2008;198:576–85. doi: 10.1086/590211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hackstadt T. The biology of rickettsiae. Infect Agents Dis. 1996;5:127–43. [PubMed] [Google Scholar]
- 4.Sahni SK, Rydkina E. Host-cell interactions with pathogenic Rickettsia species. Future Microbiol. 2009;4:323–39. doi: 10.2217/FMB.09.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sporn LA, Lawrence SO, Silverman DJ, Marder VJ. E-selectin-dependent neutrophil adhesion to Rickettsia rickettsii-infected endothelial cells. Blood. 1993;81:2406–12. [PubMed] [Google Scholar]
- 6.Kaplanski GN, Teysseire N, Farnarier C, Kaplanski S, Lissitzky J-C, Durand J-M, Soubeyrand J, Dinarello CA, Bongrand P. IL-6 and IL-8 production from cultured human endothelial cells stimulated by infection with Rickettsia conorii via a cell-associated IL-1α-dependent pathway. J Clin Invest. 1995;96:2839–44. doi: 10.1172/JCI118354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sporn LA, Marder VJ. Interleukin-1α production during Rickettsia rickettsii infection of cultured endothelial cells: Potential role in autocrine cell stimulation. Infect Immun. 1996;64:1609–13. doi: 10.1128/iai.64.5.1609-1613.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dignat-George F, Teysseire N, Mutin M, Bardin N, Lesaule G, Raoult D, Sampol J. Rickettsia conorii infection enhances vascular cell adhesion molecule-1 and intercellular adhesion molecule-1-dependent mononuclear cell adherence to endothelial cells. J Infect Dis. 1997;175:1142–52. doi: 10.1086/520353. [DOI] [PubMed] [Google Scholar]
- 9.Clifton DR, Rydkina E, Huyck H, Pryhuber G, Freeman RS, Silverman DJ, Sahni SK. Expression and secretion of chemotactic cytokines IL-8 and MCP-1 by human endothelial cells after Rickettsia rickettsii infection: regulation by nuclear transcription factor NF-κB. Int J Med Microbiol. 2005;295:267–78. doi: 10.1016/j.ijmm.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 10.Rydkina E, Silverman DJ, Sahni SK. Activation of p38 stress-activated protein kinase during Rickettsia rickettsii infection of human endothelial cells: role in the induction of chemokine response. Cell Microbiol. 2005;7:1519–30. doi: 10.1111/j.1462-5822.2005.00574.x. [DOI] [PubMed] [Google Scholar]
- 11.Eremeeva ME, Santucci LA, Popov VL, Walker DH, Silverman DJ. Rickettsia rickettsii infection of human endothelial cells: oxidative injury and reorganization of the cytoskeleton. In: Raoult D, Brouqui P, editors. Rickettsiae and rickettsial diseases at the turn of the third millennium. Elsevier Press; Paris: 1999. pp. 128–44. [Google Scholar]
- 12.Eremeeva ME, Dasch GA, Silverman DJ. Quantitative analyses of variations in the injury of endothelial cells elicited by 11 isolates of Rickettsia rickettsii. Clin Diagn Lab Immunol. 2001;8:788–96. doi: 10.1128/CDLI.8.4.788-796.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rydkina E, Sahni A, Silverman DJ, Sahni SK. Rickettsia rickettsii infection of cultured human endothelial cells induces heme oxygenase 1 expression. Infect Immun. 2002;70:4045–52. doi: 10.1128/IAI.70.8.4045-4052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haider A, Olszanecki R, Gryglewski R, Schwartzman ML, Lianos E, Kappas A, Nasjletti A, Abraham NG. Regulation of cyclooxygenase by the heme-heme oxygenase system in microvessel endothelial cells. J Pharmacol Exp Therap. 2002;300:188–94. doi: 10.1124/jpet.300.1.188. [DOI] [PubMed] [Google Scholar]
- 15.Rydkina E, Sahni A, Baggs RB, Silverman DJ, Sahni SK. Infection of human endothelial cells with spotted fever group rickettsiae stimulates cyclooxygenase-2 expression and release of prostaglandins. Infect Immun. 2006;74:5067–74. doi: 10.1128/IAI.00182-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rydkina E, Turpin LC, Silverman DJ, Sahni SK. Rickettsia rickettsii infection of human pulmonary microvascular endothelial cells: modulation of cyclooxygenase-2 expression. Clin Microbiol Infect. 2009;15(Supplement 2):300–02. doi: 10.1111/j.1469-0691.2008.02247.x. [DOI] [PubMed] [Google Scholar]
- 17.Rydkina E, Turpin LC, Sahni SK. Rickettsia rickettsii infection of human macrovascular and microvascular endothelial cells reveals activation of both common and cell type-specific host response mechanisms. Infect Immun. 2010;78:2599–606. doi: 10.1128/IAI.01335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walker DH, Popov VL, Wen J, Feng HM. Rickettsia conorii infection of C3H/HeN mice. A model of endothelial-target rickettsiosis. Lab Invest. 1994;70:358–68. [PubMed] [Google Scholar]
- 19.Walker DH, Ismail N. Emerging and re-emerging rickettsioses: endothelial cell infection and early disease events. Nat Rev Microbiol. 2008;6:375–86. doi: 10.1038/nrmicro1866. [DOI] [PubMed] [Google Scholar]
- 20.Rydkina E, Sahni SK, Santucci LA, Turpin LC, Baggs RB, Silverman DJ. Selective modulation of antioxidant enzyme activities in host tissues during Rickettsia conorii infection. Microb Pathog. 2004;36:293–301. doi: 10.1016/j.micpath.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 21.Rydkina E, Sahni A, Silverman DJ, Sahni SK. Comparative analysis of host cell signaling mechanisms activated in response to infection with Rickettsia conorii and Rickettsia typhi. J Med Microbiol. 2007;56:896–906. doi: 10.1099/jmm.0.47050-0. [DOI] [PubMed] [Google Scholar]
- 22.Nicholson WL, Allen KE, McQuiston JH, Breitschwerdt EB, Little SE. The increasing recognition of rickettsial pathogens in dogs and people. Trends Parasitol. 2010;26:205–12. doi: 10.1016/j.pt.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 23.Durante W. Targeting heme oxygenase-1 in vascular disease. Curr Drug Targets. 2010;11:1504–16. doi: 10.2174/1389450111009011504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valbuena G, Bradford W, Walker DH. Expression analysis of the T-cell-targeting chemokines CXCL9 and CXCL10 in mice and humans with endothelial infections caused by rickettsiae of the spotted fever group. Am J Pathol. 2003;163:1357–69. doi: 10.1016/S0002-9440(10)63494-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mumby S, Upton RL, Chen Y, Stanford SJ, Quinlan GJ, Nicholson AG, Gutteridge JM, Lamb NJ, Evans TW. Lung heme oxygenase-1 is elevated in acute respiratory distress syndrome. Crit Care Med. 2004;32:1130–35. doi: 10.1097/01.ccm.0000124869.86399.f2. [DOI] [PubMed] [Google Scholar]
- 26.Sheu CC, Zhai R, Wang Z, Gong MN, Tejera P, Chen F, Su L, Thompson BT, Christiani DC. Heme oxygenase-1 microsatellite polymorphism and haplotypes are associated with the development of acute respiratory distress syndrome. Intensive Care Med. 2009;35:1343–51. doi: 10.1007/s00134-009-1504-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amaro M, Bacellar F, França A. Report of eight cases of fatal and severe Mediterranean spotted fever in Portugal. Ann NY Acad Sci. 2003;990:331–43. doi: 10.1111/j.1749-6632.2003.tb07384.x. [DOI] [PubMed] [Google Scholar]
- 28.Minamino T, Christou H, Hsieh CM, Liu Y, Dhawan V, Abraham NG, Perrella MA, Mitsialis SA, Kourembanas S. Targeted expression of heme oxygenase-1 prevents the pulmonary inflammatory and vascular responses to hypoxia. Proc Natl Acad Sci USA. 2001;98:8798–803. doi: 10.1073/pnas.161272598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Ma H, Boor PJ, Ramanujam VM, Ansari GA, Khan MF. Up-regulation of heme oxygenase-1 in rat spleen after aniline exposure. Free Radic Biol Med. 2010;48:513–8. doi: 10.1016/j.freeradbiomed.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung SW, Hall SR, Perrella MA. Role of haem oxygenase-1 in microbial host defence. Cell Microbiol. 2009;11:199–207. doi: 10.1111/j.1462-5822.2008.01261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chung SW, Liu X, Macias AA, Baron RM, Perrella MA. Heme oxygenase-1-derived carbon monoxide enhances the host defense response to microbial sepsis in mice. J Clin Invest. 2008;118:239–47. doi: 10.1172/JCI32730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiloh MU, Manzanillo P, Cox JS. Mycobacterium tuberculosis senses host-derived carbon monoxide during macrophage infection. Cell Host Microbe. 2008;3:323–30. doi: 10.1016/j.chom.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zaki MH, Fujii S, Okamoto T, Islam S, Khan S, Ahmed KA, Sawa T, Akaike T. Cytoprotective function of heme oxygenase 1 induced by a nitrated cyclic nucleotide formed during murine salmonellosis. J Immunol. 2009;182:3746–56. doi: 10.4049/jimmunol.0803363. [DOI] [PubMed] [Google Scholar]
- 34.Tachibana M, Hashino M, Nishida T, Shimizu T, Watarai M. Protective role of heme oxygenase-1 in Listeria monocytogenes-induced abortion. PLoS One. 2011;6:e25046. doi: 10.1371/journal.pone.0025046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tachibana M, Watanabe K, Yamasaki Y, Suzuki H, Watarai M. Expression of heme oxygenase-1 is associated with abortion caused by Brucella abortus infection in pregnant mice. Microb Pathog. 2008;45:105–9. doi: 10.1016/j.micpath.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 36.Ren H, Leib SL, Ferriero DM, Täuber MG, Christen S. Induction of haem oxygenase-1 causes cortical non-haem iron increase in experimental pneumococcal meningitis: evidence that concomitant ferritin up-regulation prevents iron-induced oxidative damage. J Neurochem. 2007;100:532–44. doi: 10.1111/j.1471-4159.2006.04230.x. [DOI] [PubMed] [Google Scholar]
- 37.Smith MW, Schmidt JE, Rehg JE, Orihuela CJ, McCullers JA. Induction of pro- and anti-inflammatory molecules in a mouse model of pneumococcal pneumonia after influenza. Comp Med. 2007;57:82–9. [PMC free article] [PubMed] [Google Scholar]
- 38.Li Volti G, Seta F, Schwartzman ML, Nasjletti A, Abraham NG. Heme oxygenase attenuates angiotensin II-mediated increase in cyclooxygenase-2 activity in human femoral endothelial cells. Hypertension. 2003;41:715–19. doi: 10.1161/01.HYP.0000049163.23426.66. [DOI] [PubMed] [Google Scholar]
- 39.Pi SH, Jeong GS, Oh HW, Kim YS, Pae HO, Chung HT, Lee SK, Kim EC. Heme oxygenase-1 mediates nicotine- and lipopolysaccharide-induced expression of cyclooxygenase-2 and inducible nitric oxide synthase in human periodontal ligament cells. J Periodontal Res. 2010;45:177–83. doi: 10.1111/j.1600-0765.2009.01215.x. [DOI] [PubMed] [Google Scholar]
- 40.Shih R-H, Yang C-M. Induction of heme oxygenase-1 attenuates lipopolysaccharide-induced cyclooxygenase-2 expression in mouse brain endothelial cells. J Neuroinflammation. 2010;7:86. doi: 10.1186/1742-2094-7-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ejima K, Perrella MA. Alteration in heme oxygenase-1 and nitric oxide synthase-2 gene expression during endotoxemia in cyclooxygenase-2-deficient mice. Antioxid Redox Signal. 2004;6:850–7. doi: 10.1089/ars.2004.6.850. [DOI] [PubMed] [Google Scholar]
- 42.Bernard JJ, Gallo RL. Cyclooxygenase-2 enhances antimicrobial peptide expression and killing of Staphylococcus aureus. J Immunol. 2010;185:6535–44. doi: 10.4049/jimmunol.1002009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valbuena G, Walker DH. Expression of CX3CL1 (fractalkine) in mice with endothelial-target rickettsial infection of the spotted-fever group. Virchows Arch. 2005;446:21–27. doi: 10.1007/s00428-004-1120-3. [DOI] [PubMed] [Google Scholar]
- 44.Damås JK, Davì G, Jensenius M, Santilli F, Otterdal K, Ueland T, Flo TH, Lien E, Espevik T, Frøland SS, Vitale G, Raoult D, Aukrust P. Relative chemokine and adhesion molecule expression in Mediterranean spotted fever and African tick bite fever. J Infect. 2009;58:68–75. doi: 10.1016/j.jinf.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 45.Eremeeva ME, Silverman DJ. Rickettsia rickettsii infection of the EA.hy 926 endothelial cell line: morphological response to infection and evidence for oxidative injury. Microbiology. 1998;144:2037–48. doi: 10.1099/00221287-144-8-2037. [DOI] [PubMed] [Google Scholar]
- 46.Eremeeva ME, Silverman DJ. Effects of the antioxidant α-lipoic acid on human umbilical vein endothelial cells infected with Rickettsia rickettsii. Infect Immun. 1998;66:2290–9. doi: 10.1128/iai.66.5.2290-2299.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Biagioli MC, Kaul P, Singh I, Turner RB. The role of oxidative stress in rhinovirus induced elaboration of IL-8 by respiratory epithelial cells. Free Radic Biol Med. 1999;26:454–62. doi: 10.1016/s0891-5849(98)00233-0. [DOI] [PubMed] [Google Scholar]
- 48.Blackwell TS, Blackwell TR, Holden EP, Christman BW, Christman JW. In vivo antioxidant treatment suppresses nuclear factor-κB activation and neutrophilic lung inflammation. J Immunol. 1996;157:1630–7. [PubMed] [Google Scholar]
- 49.Paterson RL, Galley HF, Webster NR. The effect of N-acetylcysteine on nuclear factor-kappa B activation, interleukin-6, interleukin-8, and intercellular adhesion molecule-1 expression in patients with sepsis. Crit Care Med. 2003;31:2574–8. doi: 10.1097/01.CCM.0000089945.69588.18. [DOI] [PubMed] [Google Scholar]
- 50.Carey MA, Bradbury JA, Rebolloso YD, Graves JP, Zeldin DC, Germolec DR. Pharmacologic inhibition of COX-1 and COX-2 in influenza A viral infection in mice. PLoS One. 2010;5:e11610. doi: 10.1371/journal.pone.0011610. [DOI] [PMC free article] [PubMed] [Google Scholar]