

Abstract
Progesterone plays a key role in the development, differentiation and maintenance of female reproductive tissues and has multiple non-reproductive neural functions. Depending on the cell and tissue, the hormonal environment, growth conditions and the developmental stage, progesterone can either stimulate cell growth or inhibit it while promoting differentiation. Progesterone receptors (PRs) belong to the steroid hormone receptor superfamily of ligand-dependent transcription factors. PR proteins are subject to extensive post-translational modifications that include phosphorylation, acetylation, ubiquitination and SUMOylation. The interplay among these modifications is complex with alteration of the receptors by one factor influencing the impact of another. Control over these modifications is species-, tissue- and cell-specific. They in turn regulate multiple functions including PR stability, their subcellular localization, protein-protein interactions and transcriptional activity. These complexities may explain how tissue- and gene-specific differences in regulation are achieved in the same organism, by the same receptor protein and hormone. Here we review current knowledge of PR post-translational modifications and discuss how these may influence receptor function focusing on human breast cancer cells. There is much left to be learned. However, our understanding of this may help to identify therapeutic agents that target PR activity in tissue-specific, even gene-specific ways.
Keywords: Progesterone receptor, Post-translational Modification, SUMOylation, Phosphorylation, transcriptional activity, Breast cancer
1. Introduction
Progesterone has a remarkable repertoire of functions from normalizing blood sugar levels and facilitating thyroid hormone action to regulating menstrual cycles. The survival of the embryo in the uterus is absolutely dependent on this hormone. Progesterone plays a key role in normal development of the female reproductive tract including establishment of puberty, mammary gland development, and sexual behavior regulated by the brain. Progesterone and synthetic progestins are used clinically for contraception and menopausal hormone replacement therapy, and to treat a variety of medical conditions including abnormal uterine bleeding, amenorrhea, endometriosis, anorexia, traumatic brain injury and cancers. Depending on the cell, the hormonal microenvironment and the developmental state of tissues, progestins can stimulate growth or inhibit growth and promote differentiation. In the uterus for example its actions are proliferative in stroma but anti-proliferative in endometria; in the breast it is mainly differentiative [1]. In the uterus progesterone protects against the endometrial tumor-promoting effects of estrogens. In the breast, progestins are not protective; rather they raise the risk of breast cancers [2]. Such tissue-specific effects make it difficult to generalize about the actions of this complex hormone. Adding complexity is the fact that progesterone and synthetic progestins target at least 3 different progesterone receptors (PR). In human breast cancer cells these PR isoforms are ~94 kDa PR-A, ~110 kDa PR-B and ~60 kDa PR-C [3]. Each isoform subserves different functions. For example, in knockout-mice PR-B are necessary for regulating normal mammary gland alveologenesis while PR-A are required for uterine development [4–7]. Breast cancer patients with PR-A rich tumors have poorer disease-free survival rates than patients whose tumors overexpress PR-B [8]. In human breast cancer cell lines regulation of most genes is isoform specific, isoform ratios vary among models, and gene sets vary among tissues in vivo; all of which may also explain the tissue specificity of this hormone [9].
PRs are members of the steroid hormone receptor superfamily of ligand-dependent transcription factors. They are found in all vertebrates and are ancient molecules, anteceded only by estrogen receptors (ERs) [10]. Like other nuclear recept ors, PRs are multidomain proteins consisting of a central DNA-binding domain (DBD); large N-termini with a proximal activation function (AF-1) common to PR-A and PR-B; a distal AF-3 in the B-upstream segment (BUS) restricted to PR-B; and at their C-termini, a nuclear localization signal in the hinge region upstream of a ligand binding domain (LBD) (figure 1). The latter contains an AF-2 [3, 11–14]. The ligand-independent AF-1 mediates protein-protein interactions with general transcription factors and with coactivators or corepressors thereby up- or down-regulating the direction of transcription [15]. The DBD is highly homologous among the steroid receptors (except for ER) and is involved in DNA-binding, additional protein-protein interactions [16] and possibly receptor dimerization through contact with the LBD [17]. The LBD contacts chaperone proteins and binds transcription factors via a classical LxxLL motif [18]. Unlike AF-1, the AF-2 of PRs is ligand dependent [17]. PR-C lack the entire N-terminus and first zinc finger of the DBD. Thus they cannot bind DNA and may be transcriptionally inactive [3]. Nevertheless, PR-C are up-regulated in the endometrium at parturition, where these odd receptors suppress the actions of PR-A and PR-B by sequestering progesterone and signal the onset of labor [19].
Figure 1.
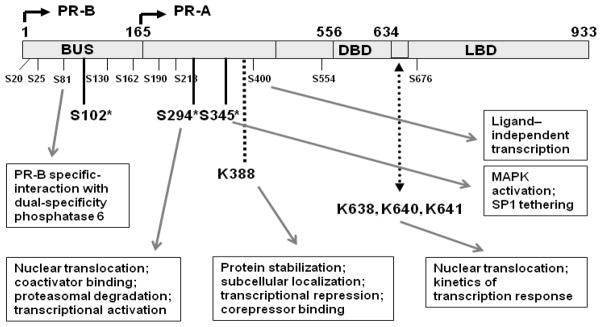
Post-translational modification of progesterone receptors. Schematic of PR-A and PR-B showing the location of basal and hormone dependent (*) serine (S) phosphorylation sites; the Lys (K)388 SUMO conjugation site within an IKEE motif; and an acetylation consensus KxKK site (amino acids 638-641). Site(s) for ubiquitination remain unclear. BUS, B-upstream segment; DBD, DNA binding domain; LBD, ligand binding domain. Also shown are putative functions of some sites.
PRs can be found in both the cytoplasm and nucleus of target cells. Classically, unliganded PRs are thought to reside mainly in cytoplasmic compartments as inactive proteins complexed to suppressor heat shock protein (hsp) 90 and hsp70, to immunophilins and to other factors. Upon ligand binding PRs are phosphorylated, the suppressor proteins dissociate, the receptors dimerize, translocate to the nucleus, and bind to target genes at specific palindromic progesterone-response elements (PREs) located in active chromatin poised for transcription. There they recruit coactivators or corepressors that further modify chromatin and facilitate receptor interactions with the general transcription apparatus [20]. Besides binding directly at PREs PR-A and PR-B also regulate transcription indirectly by being tethered to other DNA-bound transcription factors including SP-1, AP-1 or STAT5 [21–25].
Details of this “classical” model have recently come into question. For instance, it cannot explain data showing that PRs are localized to the nucleus in the absence of hormone; that dimerization may not be required for PR-mediated transcription [9]; and that PRs also act through cell membrane and cytoplasmic signaling pathways [9, 26]. Specifically with regard to dimerization, analytical ultracentrifugation sedimentation velocity and equilibrium analyses show that highly purified and functionally homogeneous PR-B undergo self-association with an interaction constant in the micromolar range [27]. Since endogenous cellular PR levels are in the nanomolar range this suggests that in the cell, receptors exist largely in monomeric states. The energetics of PR isoform binding to DNA have also been studied by quantitative DNaseI footprinting using a single palindromic PRE or two tandem PREs [28]. These data indicate that successive monomer binding to a palindromic PRE is thermodynamically favored over the binding of preformed PR dimers. Bain et al [25] conclude that PR monomer binding to arrays of tandem PRE half-sites in natural promoters is favorable, cooperative and robust, and leads to synergistic recruitment of coregulators such as steroid receptor coactivators (SRCs) [28]. This model is supported by studies showing that the majority of PREs on endogenous promoters tend to be half-sites rather than palindromes [9].
PR-dependent transcriptional activation or repression depends on the nature of the promoter being regulated and the cofactors recruited to the complex. Generally, coactivators such as SRCs are, as their name implies, transactivators because their histone acetylase activity opens chromatin and fosters basal transcription factor recruitment [29]. Corepressors on the other hand are histone deacetylases able to recruit other histone deacetylases to the DNA-bound receptors [30, 31]. PRs can also negatively regulate transcription either directly or indirectly by transrepression; interfering with or being interfered by, other DNA-bound factors. Thus PRs can transrepress ERs on a synthetic ER-dependent promoter [14] and on a natural promoter such as that of lipocalin2 [32]. On the other hand, the RelA (p65) subunit of NF-κB specifically inhibits transcription by liganded PR [33] resulting in gene repression.
Like the activity of many transcription factors, that of PR proteins is controlled further by post-translational modifications that include phosphorylation, ubiquitination, SUMOylation and acetylation (Figure 1). Among other things, these modifications driven by cell surface signaling pathways modify the stability, hormone sensitivity and nuclear localization [34–38] of both liganded and unliganded receptors. For example p38 and p42/44 mitogen activated protein kinase (MAPK) alter PR-A/PR-B ratios [39] thereby influencing breast cancer progression. In turn PRs modify cell signaling including those of MAPK and PI3K/AKT [40]. These effects are mediated through non-genomic mechanisms by a small fraction of classic liganded PRs that reside in the cytoplasm and associate with tyrosine kinases and other signaling factors [20, 41–44]. Progestins are thought to activate cytoplasmic signaling cascades especially of c-SRC, either by promoting interactions between PR and ER to activate c-Src and PI3K, or by direct interactions between PR and the SH3 domain of c-Src [45]. However, besides the classic PRs [46] alternative cell surface membrane PRs (mPR) may also play a role in non-genomic, especially rapid effects of progestins. Such a mechanism has been invoked for rapid progestin signaling associated with oocyte maturation and rapid signaling in the brain and breast cancers [44]. Because of the complexity by which progestins and PRs mediate their actions, the studies reviewed below focus mainly on the role of post-translational modifications as they impact classic PRs and their regulation of transcription.
2. Phosphorylation
Phosphorylation controlled by cell surface growth factors and cytoplasmic kinases regulates steroid receptor nuclear translocation, dimerization, DNA binding, coregulator interactions and ultimately, transcriptional activity [47, 48]. Phosphorylation is generally considered to be a positive regulator by integrating growth-factor initiated signaling with steroid hormone signaling. For instance, with regard to PRs, compared to their under-phosphorylated counterparts, phosphorylated PRs are ultrasensitive to sub-physiological levels (0.1 nM range) of progestins [49]. This may explain how growth factors like epidermal growth factor (EGF) potentiate proliferative effects of progesterone. In the case of co-stimulation by EGF plus progesterone, this synergism promotes ductal branching and lobuloalveolar development and diffrentiation of the mammary gland [50]. Similarly, progestins and EGFs act synergistically to upregulate mRNA or protein levels for a number of growth-regulatory genes [51] including cyclin D1 and MAPK-dependent cyclin E [52]. Cyclins, in turn, regulate progression of cells through the cell cycle by interacting with CDKs. Progestins activate cyclin dependent kinase 2 (CDK2) [53] and PRs are predominantly phosphorylated by CDK2 at proline-directed (S/TP) sites [54, 55] perhaps allowing for the coordinate regulation of PR action during cell cycle progression. In support of this idea, Narayanan and co-workers [56] report that PR activity is highest in S-phase, lower in G0/G1, and impaired during G2/M concomitant with PR dephosphorylation. Overexpression of either cyclin A or CDK2 enhances both PR and androgen receptor (AR) transcriptional activity. Cyclin A interacts with the N-terminus of PR, and CDK2 seems to alter PR function indirectly by increasing the recruitment of SRC-1 to liganded PRs. Clearly, there is an intimate relationship that integrates PR signaling and growth factor/cytoplasmic signaling that is to some extent controlled by the ability of cytoplasmic kinases to phosphorylate PRs.
2.1. Protein kinases and PR phosphorylation sites
PR phosphorylation has been studied for more than 30 years and is the best characterized of the post-translational modifications. As first reported in 1981 [57], liganded chicken PR-B and PR-A are phosphorylated in vitro by cAMP-dependent protein kinase A (PKA); a step that can be visualized by a decrease in PR protein mobility on SDS-PAGE gels. What has evolved since then is a picture of mind-blowing complexity involving numerous sites, multiple kinases, ligand-independent vs. -dependent stages, differences between in vitro vs. in vivo sites, species and isoform-specificity differences; all influenced by other post-translational states. We review this focusing on human PRs.
PR-B, the longest of the human PR isoforms, are 933 amino acids in length and contain at least 14 phosphorylation sites; mostly at serine (Ser, S) residues located in the N-terminus (Figure 1) [16]. Ser81, 162, 190 and 400 are considered to be basal sites phosphorylated in the absence of hormone. Ser102, 294 and 345 are ligand-dependent sites phosphorylated 1–2 hrs after binding of hormone at the LBD [34, 55, 58–63]. In turn, ligand-dependent phosphorylation is subdivided into DNA binding-independent and DNA binding-dependent stages [64]. Specific kinases responsible for phosphorylation of select sites have been identified but others remain unknown. Ligand-dependent kinases include CDK2, MAPK, PKA and tetradecanoyl 12-phorbol 12-acetate (PKC activator) [49, 56, 60, 65–68]. Kinases that phosphorylate PR in vitro include PKA, MAPK, casein kinase II and CDK2. For example, in vitro, Ser81 is phosphorylated by casein kinase II [69]; Ser162 and Ser294 are phosphorylated by MAPK [20]; 8 of the 14 sites (Ser25, 162, 190, 213, 400, 554, 676 and threonine (Thr) 430 are phosphorylated by cyclin A/CDK2 complexes [54, 55]. Five of the latter (Ser162, 190, 213, 400 and 676) have been confirmed as authentic in vivo sites [54, 55, 69–71].
The role of individual sites is under intensive study yet the functions of many remain unknown. For example, mutation of all 6 Ser clusters unique to PR-B that completely dephosphorylate BUS nevertheless produces receptors retaining the strong transactivating capacity of their wild type counterparts when measured by transient transfection of a synthetic promoter-reporter [58]. Thus either phosphorylation of those sites is immaterial, or the incorrect outcome is being measured, or transient transfection and synthetic promoters are inadequate tools to measure outcome. Mutation of Ser190 in the N-terminus inhibits PR transcriptional activity by 20–50% depending on cell or promoter context. Mutation of Ser676 in the hinge region also reduces transcriptional activity [58]. Functional roles for phospho-Ser345 and Ser400 have also been described. Ser345 for example, is phosphoryated by progestin-dependent “rapid” membrane signaling cascades that activate EGFR-, c-Src and MAPK pathways and allow PR to target growth promoting genes that lack canonical PREs [72].
Ser400 phosphorylation by CDK2 is linked to enhanced ligand-independent transcriptional activity (reviewed in [7]). All in all, interpretation of such results is extremely difficult. Most assays assume that transcription is the end point. This may or may not be the case. And, with regard to transcription assays, they are subject to spurious interpretations if experimental conditions yield anomalous receptor protein levels; faulty cellular compartmentalization; use irrelevant artificial response elements; or test promoters lacking chromatin structure, among other things. For instance transfected Ser190 and Ser676 mutants are expressed at lower levels than transfected wild-type PR, which alone could explain the observed reduction in their activity [58]. Since the phosphorylation state of individual sites may control transcription of only a subset of endogenous genes, under restricted physiological conditions and in tissue specific ways, discovering the true in vivo function of any post-translational modification on a site-by-site basis is a prodigious task. Of course there is always the possibility that nature is playing an enormous joke and that in some cases the phosphorylation-state is truly irrelevant. Given these constraints, below we review some information on these site-specific effects, with respect to the kinase responsible for phosphorylation of that site.
2.2. Mitogen Activated protein kinase (MAPK)
MAPK modulates PR activity by phosphorylating PR-B on Ser294 and Ser345 [38, 72]; N-terminal amino acids also found on PR-A. MAPK-dependent Ser294 phosphorylation is required for rapid nuclear translocation of unliganded PR. The conclusion is that MAPK signaling regulates PR action by altering nucleo-cytoplasmic shuttling and primes the receptors for robust transcriptional activation in response to ligand [49, 73]. Several other functions of Ser294 phosphorylation are proposed: PR activation by MAPK signaling is required for progestin-induced breast cancer cell entry into S-phase [72]. Ser294-phosphorylated PRs are transcriptionally hypersensitive to low concentrations of ligand on select promoters [49]. Ser294 may also play a role in the interplay between phosphorylation and other post-translational modifications as for example, PR ubiquitination. It is thought (but see below) that this augments receptor downregulation while antagonizing PR SUMOylation at Lys388 [37, 38].
2.3. Cyclin dependent kinase 2 (CDK2)
In vitro, CDK2 phosphorylates PRs at Ser25, 162, 190, 213, 400, Thr430 and Ser554, 676 [54, 55, 65]. Additionally, while Ser294 is phosphorylated by MAPK it can also be phosphorylated by CDK2 [74]. Site-directed mutagenesis has been used to study effects of these phosphorylations on PR function. Mutation of Ser190, Ser676 and a cluster of serines just upstream of the DBD (Ser549, 552, 554, 558 and 561) have modest inhibitory effects on transcription by PR-A or PR-B in transient transfection assays [58]. In these studies, transcription was the end-point. Rather than mutating the PR sites, other studies focus on expression of a constitutively active CDK2. This cellular modification induces nuclear translocation of wild-type PR-B and upregulates hormone independent transcription; i.e. it increases “basal” transcriptional activity. Under the same conditions a PR-B S400A mutant fails to translocate and has no effect on basal activity [61]. These studies would suggest that CDK2 and S400 regulate ligand-independent PR-B activity. A different study indicates that overexpression of wild type CDK2 enhanced the hormone-dependent activity of exogenous PR-B or PR-A. Although CDK2 can phosphrylate PR, elimination of these phosphorylation sites has little effect on the ability of CDK2 to stimulate PR activity [56].
2.4. CK2
Phosphorylation of PR-B at Ser81 is CK2 dependent and progestin-regulated in intact cells. It can also occur in the absence of progestins upon entrance of cells into G1/S. Unlike other PR-B phosphorylation sites, Ser81 phosphorylation is unresponsive to growth factor or serum treatment of cells [75]. Mutation of Ser81 inhibits ligand-independent cell survival, as measured by soft-agar colony formation, and impairs recruitment of PR-B to target genes important for proliferation [7]. Regulation of select genes by PR-B but not by PR-A may also require Ser79/81 phosphorylation [75]. In this case, PR-B interacts with dual-specificity phosphatase 6 via a “common docking domain” located in BUS [76]. Mutation of this domain attenuates cell cycle progression and expression of some PR-B target genes [76]. Note that this domain is lacking in PR-A. For more detailed discussions of PR phosphorylation the reader is referred to recent reviews [65, 77, 78].
3. Ubiquitination
Ubiquitin is a 76 amino acid peptide that binds covalently to lysine (Lys; K) residues on substrate proteins via a sequential enzymatic cascade involving a ubiquitin activating enzyme (E1), a conjugating enzyme (E2) and a ligase (E3) [79, 80]. The process can be reversed by multiple ubiquitin-specific proteases (USP/UBP) [81]. Ubiquitination targets proteins to varied fates. Mono-ubiquitination of a membrane protein for example, can lead to its internalization and activation [82]. Ligation of a single ubiquitin moiety to the activation domain of VP16 is required to activate transcription [83]. In this regard, it is of considerable interest that the general transcription factors p300/CBP, originally identified as histone acetylases, also possess ubiquitin-ligase activity [84, 85]. In contrast, poly-ubiquitination usually targets proteins for proteasomal degradation. With regard to transcription factors, there is a surprising inverse and closely linked relationship between protein degradation and transcriptional activity [83, 86, 87]. For example, increasing the number of activation domains on VP16 increases its transcriptional activity while at the same time signaling ubiquitination that decreases its half-life. Several other unstable transcription factors including E2F-1, fos, jun and p53 have overlapping activation and protein destruction motifs [87]; clear evidence of the close link between transcriptional activity and protein degradation.
Nuclear receptors are common targets of ubiquitination. The E2 enzymes UBCH5 and UBCH7 are critical for ER, Retinoic Acid (RAR) and Thyroid (TR) Receptor-dependent transcriptional activities [88]. PRs are well-known ubiquitin substrates [38]. PR activity is stimulated by the yeast E3 ubiquitin ligase RSP5, and its human homologs hRPF1 [89] and E6-AP [90]; coexpression of UBCH7 and E6-AP enhance transcription by PR synergistically; and SRC-1 coactivation of PR requires UBCH7 [91]. Also of interest is the fact that the breast cancer susceptibility gene BRCA1 expresses E3 ligase activity. Through this activity, BRCA1 promotes both ligand-dependent and –independent PR degradation. Additionally, at PR target genes, recruitment of a BRCA1/BARD1 complex to DNA-bound PR modifies local levels of monoubiquitinated histone H2A and contributes to epigenetic promoter silencing [92].
But the most common effect of PR polyubiquitination is “ligand-dependent downregulation” or “receptor processing” (a term we initially coined to describe ER and PR protein degradation [93]. In breast cancer cells for example, the half-life of unliganded PRs is ~21 hrs, which falls to ~6 hrs for liganded PRs [94] due to their accelerated degradation by proteasomes [95–98]. Paradoxically, receptor downregulation is a stimulatory switch that accelerates “on” and “off” cycling of receptors from pre-initiation complexes required for active transcription. Indeed, inhibition of proteasome activity prevents receptor degradation and suppresses PR-dependent transcription [36]. Thus, like other transcription factors, PR degradation is closely linked to high activity. Besides targeting the receptors, proteasomal degradation influences multiple other factors critical to transcriptional activity including recruitment of RNA polymerase II to receptor-bound promoters.
4. SUMOylation
Post-translational modification by Small Ubiquitin-related Modifiers (SUMO) is a major regulator of transcription [99], modulating target gene promoter selectivity and causing increased expression from some promoters while silencing others [100]. We and others [14, 37, 101] have shown that PRs have one SUMO binding consensus sequence at lysine (K) 388 in the N-terminal domain and that SUMOylation of this site is hormone dependent. PR SUMOylation has a suppressive effect on transcription [14, 101]. SUMOylated wild-type PRs have relatively low transcriptional activity compared to non-SUMOylated K388 mutants that have 6–10 fold higher activity. PR SUMOylation is especially important in regulating activity of promoters with multiple PREs rather than promoters with a single PRE [102]. However these studies use artificial promoters that may not reflect the structure of endogenous PREs [9].
4.1. Small ubiquitin-related modifier (SUMO)
SUMO-1,-2 and -3 are ~100 amino acid, 10–11 kDa peptides [103, 104] that reversibly modify hundreds of substrate proteins containing a specific K residue embedded in the consensus sequence ψKxD/E [103, 105]. SUMO-1 is ubiquitin-like with ~18% identity to ubiquitin and a remarkably similar secondary structure. It differs from ubiquitin in its surface-charge distribution in part explaining its specificity [104]. K48 in ubiquitin allows for the formation of polyubiquitin chains on proteins. Absence of a similar K in SUMO-1 explains why polySUMOylation does not occur [104]. Conjugation of SUMO to its client proteins involves an enzymatic cascade analogous to that required for ubiquitination including: SUMO activating enzymes E1 (Sua1/Uba2/SAE1/SAE2); a conjugation enzyme E2 (Ubc9); and E3 (PIAS) ligases [106–108]. SUMO-specific proteases, also called Sentrin-specific proteases (SENP 1–3 and SENP 5–7), deconjugate SUMO-modified proteins and are critical for maintaining physiological ratios of SUMOylated to deSUMOylated substrates.
Unlike ubiquitin, SUMOylation does not target proteins for degradation. Rather, SUMOylation play multiple roles in protein stabilization, subcellular localization, nuclear translocation, nuclear body formation and modulation (usually inhibition) of transcriptional activity [100, 103, 104]. Although the molecular mechanism(s) by which SUMO regulates transcription factor activity are not fully understood, one consequence of SUMOylation is to promote transcription factor, including nuclear receptor, interaction with corepressors. For example, ligand-dependent SUMOylation of the LBD of PPARγ, targets the receptors for binding of the corepressor (NCoR)-histone deacetylase-3 (HDAC3) complex. This in turn prevents recruitment of the ubiquitination/proteasome machinery that normally controls removal of corepressor complexes required for activation. As a result, NCoR complexes are not cleared from the promoter and target genes are maintained in a repressed state [109].
4.2. Ubc9 SUMO-conjugation enzyme
The enzymatic components of the SUMO-pathway apparently possess alternate properties separable from their SUMOylation functions. For example Ubc9, the only known SUMO E2 conjugating enzyme, interacts with all five steroid receptors and increases their transcriptional activity. However, a mutated Ubc9 that has lost its SUMO-1 conjugating activity nevertheless retains its ability to heighten transcription by AR and mineralocorticoid (MR) receptors [110–112]. These findings suggest that Ubc9 also acts as a coregulator, perhaps through recruitment of coactivators; properties that are independent of its enzymatic activity. Depending on receptor levels, Ubc9 exhibits unequal effects on glucocorticoid receptors (GR) and PRs [113].
4.3. E3 Ligase: PIAS
The human PIAS (Protein inhibitor of activated STAT) family of E3 ligases [114–117] consists of five homologous proteins – PIAS1, PIAS3, PIASxα, PIASxβ and PIASy – containing a RING-finger domain necessary for enzymatic activity. All PIAS family members including PIAS1, PIAS3, PIASxα, and PIASxβ modulate PR transcriptional activity with the degree of activation dependent on the receptors, the promoter and the cell type [118]. PIAS1 acts as a SUMO E3 ligase for PRs. This inhibits their transcriptional activity. Silencing of endogenous PIAS1 with an siRNA enhances the activity of wild-type PRs but has little effect on the activity of SUMOylation-deficient K388R PR mutants [119].
We find no evidence that PIAS3, PIASxα, PIASxβ or PIASy are involved in PR SUMOylation [119]. In contrast, Man et al. [120] report that PIAS3 induces PR-B SUMOylation at K7, K388 and K531 but that SUMOylation at K7 and K531 is dependent on SUMOylation at K388. They also report that PIAS3 significantly inhibits gene transcription by liganded PR-B, and that reduction of endogenous PIAS3 by siRNAs enhances transcription. However, this effect of PIAS3 appears to be independent of PR-B SUMOylation because PR-B mutants lacking SUMOylation ability are still repressed by PIAS3. We speculate that the inhibitory effect of PIAS3 may involve anomalous nuclear translocation or DNA binding of PR-B. There is no question that PIAS3 can interact with PRs, and that this is enhanced by ligand. This has been demonstrated both in vitro and in vivo [120]. However, depending on the promoter or cells being tested, PIAS3 can either activate (ie. PRE2-Luc) ([118], Abdel-Hafiz unpublished) or repress (ie. MMTV-Luc) transcription ([120], Abdel-Hafiz unpublished). Clearly, these results must be classified as preliminary and need to be analyzed in vivo on natural promoters. In the end, it is likely that no solid rules will apply to SUMOylation and its effects on PR. Rather effects will be dependent on cell and tissue types, and genes being regulated under physiological conditions.
4.4. SENP and deSUMOylation
Studies in knockout mice demonstrate that normal embryonic development requires a fine balance between SUMOylation and deSUMOylation [121]. This balance may be altered in malignancies. Persistent elevation of SENP1 facilitates transformation of the normal prostate to a dysplastic state in transgenic mice. Increased SENP expression is observed in other malignancies including thyroid adenomas, colon and prostate cancers [122–126].
Removal of SUMO from transcription factors by mutation of the SUMO-conjugation site or by overexpression of a deSUMOylating enzyme like SENP1 generally increases their activity. This has been shown for AR, as well as for C/EBP, Elk-1, Sp3 and Smad4 [127–133]. With regard to AR, SENP1 is stimulatory, but two different mechanisms have been proposed: Cheng et al. [134] suggest that the transactivating effects of SENP1 do not involve SUMO deconjugation of the receptors but rather cleavage of SUMO from HDAC1 thereby alleviated its repressive effects on AR activity. In contrast, Kaikkonen et al. [135] demonstrate that effects of SENP1 and SENP2 require intact SUMO acceptor sites in AR, indicating that the coactivating effects of the enzymes are directly on the receptors. Clearly, more studies are required to clarify this keeping in mind that both mechanisms are possible.
We have shown that deSUMOylation of PR-A and PR-B by mutating the K388 SUMOylation motif increases their ligand-dependent transcriptional activity ~10-fold. In a similar manner, deSUMOylation of wild-type PRs by SENP1 or SENP2 heightens the transcriptional activity [37, 136] of exogenous PR in human cervical carcinoma (HeLa) cells, and endogenous PR in human breast cancer (T47Dco) cells. The stimulatory effects of SENP1 are dependent on its enzymatic activity; requires an intact PR SUMO conjugation site; and functions only at promoters containing multiple PREs. Trichostatin A (TSA) is a potent and specific inhibitor of histone deacetylase (HDAC) and recruitment of HDAC appears to be involved in the inhibitory effects of SUMOylation [134]. However inhibition of HDAC by TSA does not prevent SENP1 stimulation of wild-type PR since SUMOylation-deficient PR are similarly affected by TSA. This indicates that other mechanisms are responsible for the suppressive effects of SUMOylation on PR activity and is in agreement with a recent report that wild type and SUMOylation deficient AR are similarly influenced by TSA [135].
5. Acetylation
Like other post-translational modifications, acetylation regulates many transcription factors including the tumor suppressor p53 [137–139], GATA-1 and -2 [140] [141], NFkB, p65, SRC3 and poly(ADP-ribose)polymerase 1 [138, 142, 143]. Acetylation can enhance or inhibit transcription depending on the target protein. It activates by enhancing DNA binding activity; by stimulating interactions with positive regulators such as chromatin remodeling factors or coactivators; by inhibiting interaction with negative regulators; by increasing the stability of regulatory factors; or by altering their subcellular localization [143]. It inhibits by similar mechanisms that in general reduce protein/DNA or protein/protein interactions [143].
Steroid receptors, including AR, GR and ER are also modified by acetylation on Lys residues [144]. PR acetylation was first inferred using the HDAC inhibitor TSA, which showed that in the presence of progesterone, chromatin remodeling and PR levels were enhanced [145]. Direct PR acetylation has been studied by Daniel et al. [146]. They showed that PRs are acetylated at a conserved KxKK motif located at amino acids 638-641 in the NLS/hinge region. In line with this, TAF-I beta and pp32, two proteins involved in deacetylation, bind PRs in pull-down assays [144, 147, 148]. Disruption of the acetylation motif results in receptors that are clearly cytoplasmic in the absence of ligand and require at least 4 hrs of progesterone treatment to accumulate in the nucleus. This is in contrast to 15 min required for liganded wild-type PR to translocate to nuclei [146]. Daniel et al. [146] showed that PR acetylation negatively regulates transcription of PR Acetylation mutant K-A PR-B displayed a marked increase in progestin-induced transcriptional activity relative to wt PR-B using PRE2-Luc reporter.
6. Interplay between PR post-translational modifications
6.1. Phosphorylation and ubiquitination
As briefly reviewed above, post-translational modifications regulate protein activity by multiple mechanisms. However additional complexity is generated by linkage between these modifications [149]. For instance, PR ubiquitination and phosphorylation may be linked but data on this remain unclear. As discussed above PR transcriptional activity is dependent on ligand-dependent receptor “downregulation” [38] involving degradation of ubiquitinated receptors by proteasomes. Lange et al. [49] believe that this is linked to PR phosphorylation. They find that compared to wild-type PR-B stably transfected into human breast cancer cells, PR-B S294A phosphorylation-deficient mutants, despite retaining DNA binding ability, fail to be ubiquitinated. This renders them highly stable but transcriptionally deficient. They conclude that phosphorylation of S294 is required for efficient PR ubiquitination, downregulation and transcription. However, other investigators using similar cells show that like wild-type PR-B, PR-B S294A mutants do undergo ligand-dependent downregulation, albeit the mutants do so with reduced efficiency [39, 150, 151]. Other studies based on transient transfection methods that allow testing of multiple protein concentrations of wild-type PR-B and PR-B S294A, and that probe a variety of cells, conclude that there are no significant differences between the wild-type and mutant PR. Such studies show that at similar protein concentrations their ligand-dependent downregulation and transcriptional activities are similar [49, 102]. This attests to the importance of experimental conditions and intimates that the extent of ligand-dependent downregulation is controlled by receptor concentrations; perhaps more so than by phosphorylation [102]. In our opinion, S294 phosphorylation does not follow an “all or none” rule with regard to PR degradation and activity. Rather, S294 phosphorylation is simply one of multiple factors that regulate PR turnover.
6.2. Phosphorylation and SUMOylation
SUMOylation is often regulated by phosphorylation. SUMOylation of p53, c-Jun, IκBα, KAP1 and PML is repressed by phosphorylation [128, 152–154]. Phosphorylation also positively regulates SUMOylation of HSF1, STAT1 HSF4b, MEF2A and GATA-1 [155]. This requires presence of a phosphorylation-dependent SUMOylation motif (PDSM) on the proteins characterized by a SUMO consensus site adjacent to a proline-directed phosphorylation site (ΨKxExxSP).
With regard to PR, Daniel et al. [37] report that the SUMOylated receptors are exceptionally stable and have low transcriptional activity, while Ser294-phosphorylated and deSUMOylated PR are rapidly downregulated and transcriptionally hyperactive. They conclude that there is an association between hormone-dependent PR phosphorylation and PR SUMOylation. However, in our opinion these two steps are unrelated. PR mutated at their Ser294/344/345 phosphorylation sites are as efficiently SUMOylated at K388 as wild-type phosphorylated PR [102]. That PR phosphorylation at S294 does not affect PR SUMOylation is also consistent with our data showing that there are no significant differences between the transcriptional activities of wild-type and S294A PR mutants [49, 102]. On the other hand, MAPK overexpression has complex, concentration-dependent effects on PR SUMOylation. At low concentrations, MAPK induces ligand-independent PR SUMOylation and increases basal PR-dependent transcription. At high concentrations, MAPK suppresses hormone-dependent PR SUMOylation. These contrasting dual activities suggest that some effects of MAPK on PR SUMOylation are indirect, by altering the activity of the general SUMOylation machinery. Molecular mechanisms by which MAPK may indirectly influence PR SUMOylation include changes in the amounts and/or the activities of the E3 ligases and cleavage enzymes [156, 157]. Qiu et al. [49] have also reported robust transcription with a PR S294A mutant. Analogously, Kaikkonen et al. [135] show that AR phosphorylation has no effects on AR SUMOylation. Indeed, there are no phosphorylation-dependent SUMOylation PDSM motifs in either AR or PR. The reasons for differences in conclusions among similar studies are unclear, but are likely to be related to experimental conditions including use of DNA concentrations for receptor expression at which squelching effects are observed [102].
There are other indications that PR phosphorylation and SUMOylation are unlinked: 1. SENP1 deSUMOylates PR and increases their ligand-dependent activity. High levels of MAPK reportedly also deSUMOylate PR. However, this does not increase their ligand-dependent activity; rather it increases their ligand-independent activity [60]. 2. Removal of the PR LBD yields an N-terminus/DBD fragment that is constitutively active. It cannot be SUMOylated (which requires ligand) but can be activated by MAPK (independent of ligand). 3. SUMOylation has no effect on PR-dependent transcription of the MMTV promoter [136], while MAPK enhances PR-dependent transcription of this promoter [136]. 4. According to Khan et al. [39] differential stabilization of PR-A vs. PR-B is MAPK regulated but independent of PR S294 phosphorylation. They conclude that S294 phosphorylation is not a major sensor of PR downregulation [38]. Taken together, most results suggest that despite their importance, effects of MAPK do not depend on modulating PR SUMOylation.
6.3. Ubiquitination and SUMOylation
CUE domain-containing 2 interacts with PR-B and promotes progesterone-dependent downregulation via the ubiquitin-proteasome pathway. According to Zhang et al. [158], mutation of the PR K388 SUMOylation site suppresses progesterone-dependent PR degradation leading the authors to suggest that K388 is both a SUMOylation and ubiquitination site and that the two modifications compete with one another. Nevertheless, we [102] and others [37] have shown that PR K388 mutants still undergo progesterone-dependent downregulation suggesting to us that PR must be ubiquitinated at residues other than K388. We have no explanations for the discrepant results.
7. Summary and conclusions
PRs are subject to post-translational modifications that control their actions in response to progesterone or synthetic progestins. These modifications modulate protein/protein interactions, subcellular receptor localization, hormone sensitivity, receptor stability, isoform ratios, etc. Together they fine-tune how PRs regulate gene transcription. Cross-talk among various signaling pathways and post-translational events play a key role. It is clear that the post-translational modifications maintain tight control over the functions of these important receptors. Understanding the detailed mechanism(s) therefore, could theoretically lead to the discovery of novel, selective drugs for physiological and medical conditions for which progestins acting through PR are critical. But the controls by PR and their post-translational modifications vary with the cell and tissue under study, the genes being regulated, subtle and not-so-subtle differences in experimental conditions, natural vs. synthetic ligands, ligand-independent vs. dependent effects, etc. The complexity is such that it behooves scientists in the field who truly wish to get to the bottom of these questions, to collaborate with one another, to exchange reagents and to agree on key experimental conditions that must be met to replicate results and validate conclusions. For example, if transient transfection assays cannot accurately reflect what happens with endogenous receptors on endogenous genes, then scientists in the field must concede that conclusions based on this assay can only be considered preliminary and come to some agreement about the best way to proceed.
Highlights.
Progesterone receptors (PR) are subject to posttranslational modifications (PTM).
PTMs include phosphorylation, ubiquitination, acetylation and SUMOylation.
These modifications control the classical transcriptional activity of PR.
PR PTM could be therapeutically targeted as part of breast cancer treatment.
Acknowledgments
We are grateful for the support of the NIH (RO1-CA026869-33), the Avon Foundation for Women, the Breast Cancer Research Foundation, and the National Foundation for Cancer Research.
8. Abbreviations
- AF-1
Activation domain 1
- AF-2
Activation domain 2
- AR
Androgen receptor
- Ap1
Activator protein 1
- BUS
B-upstream segment
- CBP
CREB-binding protein
- CDK2
Cyclin dependent kinase 2
- DBD
DNA-binding domain
- EGF
Epidermal growth factor
- ER
Estrogen receptor
- GR
Glucocorticoid receptor
- GRIP1
Glucocorticoid receptor-interacting protein 1
- HAT
Histone acetylase
- HDAC
Histone deacetylase
- HRE
Hormone response element
- hsp
Heat shock protein
- LBD
Ligand-binding domain
- MAPK
Mitogen-activated protein kinase
- MMTV
Mouse mammary tumor virus
- MR
Mineralocorticoid receptor
- NcoR
Nuclear receptor corepressor
- NLS
Nuclear localization signal
- NTD
N-terminal domain
- PIAS
Protein inhibitor of activated STAT
- PKA
cAMP-dependent protein kinase A
- PML
Promyelocytic leukemia protein
- PR
Progesterone receptor
- PRE
Progesterone response element
- RAR
Retinoid acid receptor
- Sp1
Specificity protein 1
- SR
Steroid receptor
- SRC
Steroid receptor coactivators
- Src
Tyrosine kinase-c
- STAT5
Signal transducers and activator of transcription
- SUMO-1
Small ubiquitin-related modifier 1
- Ubc9
ubiquitin-conjugating enzyme 9 (SUMO-conjugating E2 enzyme)
- USP
ubiquitin-specifc proteases
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Richer JK, Jacobsen BM, Manning NG, Abel MG, Wolf DM, Horwitz KB. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem. 2002;277(7):5209–5218. doi: 10.1074/jbc.M110090200. [DOI] [PubMed] [Google Scholar]
- 2.Mueck AO, Ruan X, Seeger H, Fehm T, Neubauer H. Genomic and non-genomic actions of progestogens in the breast. J Steroid Biochem Mol Biol. 2013 doi: 10.1016/j.jsbmb.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 3.Wei LL, Gonzalez-Aller C, Wood WM, Miller LA, Horwitz KB. 5′-Heterogeneity in human progesterone receptor transcripts predicts a new amino-terminal truncated “C”-receptor and unique A-receptor messages. Mol Endocrinol. 1990;4(12):1833–1840. doi: 10.1210/mend-4-12-1833. [DOI] [PubMed] [Google Scholar]
- 4.Conneely OM, Mulac-Jericevic B, DeMayo F, Lydon JP, O’Malley BW. Reproductive functions of progesterone receptors. Recent Prog Horm Res. 2002;57:339–355. doi: 10.1210/rp.57.1.339. [DOI] [PubMed] [Google Scholar]
- 5.Lydon JP, DeMayo FJ, Funk CR, Mani SK, Hughes AR, Montgomery CA, Jr, Shyamala G, Conneely OM, O’Malley BW. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 1995;9(18):2266–2278. doi: 10.1101/gad.9.18.2266. [DOI] [PubMed] [Google Scholar]
- 6.Mulac-Jericevic B, Lydon JP, DeMayo FJ, Conneely OM. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc Natl Acad Sci U S A. 2003;100(17):9744–9749. doi: 10.1073/pnas.1732707100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hagan CR, Daniel AR, Dressing GE, Lange CA. Role of phosphorylation in progesterone receptor signaling and specificity. Mol Cell Endocrinol. 2012 doi: 10.1016/j.mce.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hopp TA, Weiss HL, Hilsenbeck SG, Cui Y, Allred DC, Horwitz KB, Fuqua SA. Breast cancer patients with progesterone receptor PR-A-rich tumors have poorer disease-free survival rates. Clin Cancer Res. 2004;10(8):2751–2760. doi: 10.1158/1078-0432.ccr-03-0141. [DOI] [PubMed] [Google Scholar]
- 9.Jacobsen BM, Horwitz KB. Progesterone receptors, their isoforms and progesterone regulated transcription. Mol Cell Endocrinol. 2012;357(1–2):18–29. doi: 10.1016/j.mce.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thornton JW. Evolution of vertebrate steroid receptors from an ancestral estrogen receptor by ligand exploitation and serial genome expansions. Proc Natl Acad Sci U S A. 2001;98(10):5671–5676. doi: 10.1073/pnas.091553298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sartorius CA, Melville MY, Hovland AR, Tung L, Takimoto GS, Horwitz KB. A third transactivation function (AF3) of human progesterone receptors located in the unique N-terminal segment of the B-isoform. Mol Endocrinol. 1994;8(10):1347–1360. doi: 10.1210/mend.8.10.7854352. [DOI] [PubMed] [Google Scholar]
- 12.Tung L, Shen T, Abel MG, Powell RL, Takimoto GS, Sartorius CA, Horwitz KB. Mapping the unique activation function 3 in the progesterone B-receptor upstream segment. Two LXXLL motifs and a tryptophan residue are required for activity. J Biol Chem. 2001;276(43):39843–39851. doi: 10.1074/jbc.M106843200. [DOI] [PubMed] [Google Scholar]
- 13.Sheridan PL, Evans RM, Horwitz KB. Phosphotryptic peptide analysis of human progesterone receptor. New phosphorylated sites formed in nuclei after hormone treatment. J Biol Chem. 1989;264(11):6520–6528. [PubMed] [Google Scholar]
- 14.Abdel-Hafiz H, Takimoto GS, Tung L, Horwitz KB. The inhibitory function in human progesterone receptor N termini binds SUMO-1 protein to regulate autoinhibition and transrepression. J Biol Chem. 2002;277(37):33950–33956. doi: 10.1074/jbc.M204573200. [DOI] [PubMed] [Google Scholar]
- 15.Lavery DN, McEwan IJ. Structure and function of steroid receptor AF1 transactivation domains: induction of active conformations. Biochem J. 2005;391(Pt 3):449–464. doi: 10.1042/BJ20050872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Africander D, Verhoog N, Hapgood JP. Molecular mechanisms of steroid receptor-mediated actions by synthetic progestins used in HRT and contraception. Steroids. 2011;76(7):636–652. doi: 10.1016/j.steroids.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 17.Bourguet W, Germain P, Gronemeyer H. Nuclear receptor ligand-binding domains: three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol Sci. 2000;21(10):381–388. doi: 10.1016/s0165-6147(00)01548-0. [DOI] [PubMed] [Google Scholar]
- 18.Beato M, Klug J. Steroid hormone receptors: an update. Hum Reprod Update. 2000;6(3):225–236. doi: 10.1093/humupd/6.3.225. [DOI] [PubMed] [Google Scholar]
- 19.Condon JC, Hardy DB, Kovaric K, Mendelson CR. Up-regulation of the progesterone receptor (PR)-C isoform in laboring myometrium by activation of nuclear factor-kappaB may contribute to the onset of labor through inhibition of PR function. Mol Endocrinol. 2006;20(4):764–775. doi: 10.1210/me.2005-0242. [DOI] [PubMed] [Google Scholar]
- 20.Ward RD, Weigel NL. Steroid receptor phosphorylation: Assigning function to site-specific phosphorylation. Biofactors. 2009;35(6):528–536. doi: 10.1002/biof.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Owen GI, Richer JK, Tung L, Takimoto G, Horwitz KB. Progesterone regulates transcription of the p21(WAF1) cyclin- dependent kinase inhibitor gene through Sp1 and CBP/p300. J Biol Chem. 1998;273(17):10696–10701. doi: 10.1074/jbc.273.17.10696. [DOI] [PubMed] [Google Scholar]
- 22.Kalkhoven E, Beraldi E, Panno ML, De Winter JP, Thijssen JH, Van Der Burg B. Growth inhibition by anti-estrogens and progestins in TGF-beta-resistant and -sensitive breast-tumor cells. Int J Cancer. 1996;65(5):682–687. doi: 10.1002/(SICI)1097-0215(19960301)65:5<682::AID-IJC20>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 23.Bamberger AM, Bamberger CM, Gellersen B, Schulte HM. Modulation of AP-1 activity by the human progesterone receptor in endometrial adenocarcinoma cells. Proc Natl Acad Sci U S A. 1996;93(12):6169–6174. doi: 10.1073/pnas.93.12.6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Madauss KP, Grygielko ET, Deng SJ, Sulpizio AC, Stanley TB, Wu C, Short SA, Thompson SK, Stewart EL, Laping NJ, et al. A structural and in vitro characterization of asoprisnil: a selective progesterone receptor modulator. Mol Endocrinol. 2007;21(5):1066–1081. doi: 10.1210/me.2006-0524. [DOI] [PubMed] [Google Scholar]
- 25.Subtil-Rodriguez A, Millan-Arino L, Quiles I, Ballare C, Beato M, Jordan A. Progesterone induction of the 11beta-hydroxysteroid dehydrogenase type 2 promoter in breast cancer cells involves coordinated recruitment of STAT5A and progesterone receptor to a distal enhancer and polymerase tracking. Mol Cell Biol. 2008;28(11):3830–3849. doi: 10.1128/MCB.01217-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boonyaratanakornkit V, Bi Y, Rudd M, Edwards DP. The role and mechanism of progesterone receptor activation of extra-nuclear signaling pathways in regulating gene transcription and cell cycle progression. Steroids. 2008;73(9–10):922–928. doi: 10.1016/j.steroids.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 27.Heneghan AF, Berton N, Miura MT, Bain DL. Self-association energetics of an intact, full-length nuclear receptor: the B-isoform of human progesterone receptor dimerizes in the micromolar range. Biochemistry. 2005;44(27):9528–9537. doi: 10.1021/bi050609i. [DOI] [PubMed] [Google Scholar]
- 28.Heneghan AF, Connaghan-Jones KD, Miura MT, Bain DL. Coactivator assembly at the promoter: efficient recruitment of SRC2 is coupled to cooperative DNA binding by the progesterone receptor. Biochemistry. 2007;46(39):11023–11032. doi: 10.1021/bi700850v. [DOI] [PubMed] [Google Scholar]
- 29.Nettles KW, Greene GL. Ligand control of coregulator recruitment to nuclear receptors. Annu Rev Physiol. 2005;67:309–333. doi: 10.1146/annurev.physiol.66.032802.154710. [DOI] [PubMed] [Google Scholar]
- 30.McKenna NJ, Xu J, Nawaz Z, Tsai SY, Tsai MJ, O’Malley BW. Nuclear receptor coactivators: multiple enzymes, multiple complexes, multiple functions. J Steroid Biochem Mol Biol. 1999;69(1–6):3–12. doi: 10.1016/s0960-0760(98)00144-7. [DOI] [PubMed] [Google Scholar]
- 31.Heinlein CA, Chang C. Androgen receptor (AR) coregulators: an overview. Endocr Rev. 2002;23(2):175–200. doi: 10.1210/edrv.23.2.0460. [DOI] [PubMed] [Google Scholar]
- 32.Mrusek S, Classen-Linke I, Vloet A, Beier HM, Krusche CA. Estradiol and medroxyprogesterone acetate regulated genes in T47D breast cancer cells. Mol Cell Endocrinol. 2005;235(1–2):39–50. doi: 10.1016/j.mce.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 33.Kalkhoven E, Wissink S, van der Saag PT, van der Burg B. Negative interaction between the RelA(p65) subunit of NF-kappaB and the progesterone receptor. J Biol Chem. 1996;271(11):6217–6224. doi: 10.1074/jbc.271.11.6217. [DOI] [PubMed] [Google Scholar]
- 34.Beck CA, Zhang Y, Altmann M, Weigel NL, Edwards DP. Stoichiometry and site-specific phosphorylation of human progesterone receptor in native target cells and in the baculovirus expression system. J Biol Chem. 1996;271(32):19546–19555. doi: 10.1074/jbc.271.32.19546. [DOI] [PubMed] [Google Scholar]
- 35.Beck CA, Zhang Y, Weigel NL, Edwards DP. Two types of anti-progestins have distinct effects on site-specific phosphorylation of human progesterone receptor. J Biol Chem. 1996;271(2):1209–1217. doi: 10.1074/jbc.271.2.1209. [DOI] [PubMed] [Google Scholar]
- 36.Dennis AP, Lonard DM, Nawaz Z, O’Malley BW. Inhibition of the 26S proteasome blocks progesterone receptor-dependent transcription through failed recruitment of RNA polymerase II. J Steroid Biochem Mol Biol. 2005;94(4):337–346. doi: 10.1016/j.jsbmb.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 37.Daniel AR, Faivre EJ, Lange CA. Phosphorylation-dependent antagonism of sumoylation derepresses progesterone receptor action in breast cancer cells. Mol Endocrinol. 2007;21(12):2890–2906. doi: 10.1210/me.2007-0248. [DOI] [PubMed] [Google Scholar]
- 38.Lange CA, Shen T, Horwitz KB. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc Natl Acad Sci U S A. 2000;97(3):1032–1037. doi: 10.1073/pnas.97.3.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khan JA, Amazit L, Bellance C, Guiochon-Mantel A, Lombes M, Loosfelt H. p38 and p42/44 MAPKs differentially regulate progesterone receptor A and B isoform stabilization. Mol Endocrinol. 2011;25(10):1710–1724. doi: 10.1210/me.2011-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nilsen J, Brinton RD. Divergent impact of progesterone and medroxyprogesterone acetate (Provera) on nuclear mitogen-activated protein kinase signaling. Proc Natl Acad Sci U S A. 2003;100(18):10506–10511. doi: 10.1073/pnas.1334098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stellato C. Post-transcriptional and nongenomic effects of glucocorticoids. Proc Am Thorac Soc. 2004;1(3):255–263. doi: 10.1513/pats.200402-015MS. [DOI] [PubMed] [Google Scholar]
- 42.Wierman ME. Sex steroid effects at target tissues: mechanisms of action. Adv Physiol Educ. 2007;31(1):26–33. doi: 10.1152/advan.00086.2006. [DOI] [PubMed] [Google Scholar]
- 43.Grossmann C, Gekle M. New aspects of rapid aldosterone signaling. Mol Cell Endocrinol. 2009;308(1–2):53–62. doi: 10.1016/j.mce.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 44.Zhu Y, Hanna RN, Schaaf MJ, Spaink HP, Thomas P. Candidates for membrane progestin receptors--past approaches and future challenges. Comp Biochem Physiol C Toxicol Pharmacol. 2008;148(4):381–389. doi: 10.1016/j.cbpc.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 45.Vicent GP, Nacht AS, Zaurin R, Ballare C, Clausell J, Beato M. Minireview: role of kinases and chromatin remodeling in progesterone signaling to chromatin. Mol Endocrinol. 2010;24(11):2088–2098. doi: 10.1210/me.2010-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boonyaratanakornkit V, Edwards DP. Receptor mechanisms mediating non-genomic actions of sex steroids. Semin Reprod Med. 2007;25(3):139–153. doi: 10.1055/s-2007-973427. [DOI] [PubMed] [Google Scholar]
- 47.Orti E, Bodwell JE, Munck A. Phosphorylation of steroid hormone receptors. Endocr Rev. 1992;13(1):105–128. doi: 10.1210/edrv-13-1-105. [DOI] [PubMed] [Google Scholar]
- 48.Hill CS, Treisman R. Transcriptional regulation by extracellular signals: mechanisms and specificity. Cell. 1995;80(2):199–211. doi: 10.1016/0092-8674(95)90403-4. [DOI] [PubMed] [Google Scholar]
- 49.Qiu M, Lange CA. MAP kinases couple multiple functions of human progesterone receptors: degradation, transcriptional synergy, and nuclear association. J Steroid Biochem Mol Biol. 2003;85(2–5):147–157. doi: 10.1016/s0960-0760(03)00221-8. [DOI] [PubMed] [Google Scholar]
- 50.Haslam SZ, Counterman LJ, Nummy KA. Effects of epidermal growth factor, estrogen, and progestin on DNA synthesis in mammary cells in vivo are determined by the developmental state of the gland. J Cell Physiol. 1993;155(1):72–78. doi: 10.1002/jcp.1041550110. [DOI] [PubMed] [Google Scholar]
- 51.Richer JK, Lange CA, Manning NG, Owen G, Powell R, Horwitz KB. Convergence of progesterone with growth factor and cytokine signaling in breast cancer. Progesterone receptors regulate signal transducers and activators of transcription expression and activity. J Biol Chem. 1998;273(47):31317–31326. doi: 10.1074/jbc.273.47.31317. [DOI] [PubMed] [Google Scholar]
- 52.Lange CA, Richer JK, Shen T, Horwitz KB. Convergence of progesterone and epidermal growth factor signaling in breast cancer. Potentiation of mitogen-activated protein kinase pathways. J Biol Chem. 1998;273(47):31308–31316. doi: 10.1074/jbc.273.47.31308. [DOI] [PubMed] [Google Scholar]
- 53.Groshong SD, Owen GI, Grimison B, Schauer IE, Todd MC, Langan TA, Sclafani RA, Lange CA, Horwitz KB. Biphasic regulation of breast cancer cell growth by progesterone: role of the cyclin-dependent kinase inhibitors, p21 and p27(Kip1) Mol Endocrinol. 1997;11(11):1593–1607. doi: 10.1210/mend.11.11.0006. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Y, Beck CA, Poletti A, Clement JPt, Prendergast P, Yip TT, Hutchens TW, Edwards DP, Weigel NL. Phosphorylation of human progesterone receptor by cyclin-dependent kinase 2 on three sites that are authentic basal phosphorylation sites in vivo. Mol Endocrinol. 1997;11(6):823–832. doi: 10.1210/mend.11.6.0006. [DOI] [PubMed] [Google Scholar]
- 55.Knotts TA, Orkiszewski RS, Cook RG, Edwards DP, Weigel NL. Identification of a phosphorylation site in the hinge region of the human progesterone receptor and additional amino-terminal phosphorylation sites. J Biol Chem. 2001;276(11):8475–8483. doi: 10.1074/jbc.M009805200. [DOI] [PubMed] [Google Scholar]
- 56.Narayanan R, Adigun AA, Edwards DP, Weigel NL. Cyclin-dependent kinase activity is required for progesterone receptor function: novel role for cyclin A/Cdk2 as a progesterone receptor coactivator. Mol Cell Biol. 2005;25(1):264–277. doi: 10.1128/MCB.25.1.264-277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weigel NL, Tash JS, Means AR, Schrader WT, O’Malley BW. Phosphorylation of hen progesterone receptor by cAMP dependent protein kinase. Biochem Biophys Res Commun. 1981;102(1):513–519. doi: 10.1016/0006-291x(81)91549-7. [DOI] [PubMed] [Google Scholar]
- 58.Takimoto GS, Hovland AR, Tasset DM, Melville MY, Tung L, Horwitz KB. Role of phosphorylation on DNA binding and transcriptional functions of human progesterone receptors. J Biol Chem. 1996;271(23):13308–13316. doi: 10.1074/jbc.271.23.13308. [DOI] [PubMed] [Google Scholar]
- 59.Weigel NL, Bai W, Zhang Y, Beck CA, Edwards DP, Poletti A. Phosphorylation and progesterone receptor function. J Steroid Biochem Mol Biol. 1995;53(1–6):509–514. doi: 10.1016/0960-0760(95)00098-k. [DOI] [PubMed] [Google Scholar]
- 60.Shen T, Horwitz KB, Lange CA. Transcriptional hyperactivity of human progesterone receptors is coupled to their ligand-dependent down-regulation by mitogen-activated protein kinase-dependent phosphorylation of serine 294. Mol Cell Biol. 2001;21(18):6122–6131. doi: 10.1128/MCB.21.18.6122-6131.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pierson-Mullany LK, Lange CA. Phosphorylation of progesterone receptor serine 400 mediates ligand-independent transcriptional activity in response to activation of cyclin-dependent protein kinase 2. Mol Cell Biol. 2004;24(24):10542–10557. doi: 10.1128/MCB.24.24.10542-10557.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weigel NL, Moore NL. Kinases and protein phosphorylation as regulators of steroid hormone action. Nucl Recept Signal. 2007;5:e005. doi: 10.1621/nrs.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dressing GE, Hagan CR, Knutson TP, Daniel AR, Lange CA. Progesterone receptors act as sensors for mitogenic protein kinases in breast cancer models. Endocr Relat Cancer. 2009;16(2):351–361. doi: 10.1677/ERC-08-0281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takimoto GS, Tasset DM, Eppert AC, Horwitz KB. Hormone-induced progesterone receptor phosphorylation consists of sequential DNA-independent and DNA-dependent stages: analysis with zinc finger mutants and the progesterone antagonist ZK98299. Proc Natl Acad Sci U S A. 1992;89(7):3050–3054. doi: 10.1073/pnas.89.7.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moore NL, Narayanan R, Weigel NL. Cyclin dependent kinase 2 and the regulation of human progesterone receptor activity. Steroids. 2007;72(2):202–209. doi: 10.1016/j.steroids.2006.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weigel NL, Zhang Y. Ligand-independent activation of steroid hormone receptors. J Mol Med. 1998;76(7):469–479. doi: 10.1007/s001090050241. [DOI] [PubMed] [Google Scholar]
- 67.Edwards DP, Weigel NL, Nordeen SK, Beck CA. Modulators of cellular protein phosphorylation alter the trans-activation function of human progesterone receptor and the biological activity of progesterone antagonists. Breast Cancer Res Treat. 1993;27(1–2):41–56. doi: 10.1007/BF00683192. [DOI] [PubMed] [Google Scholar]
- 68.Beck CA, Weigel NL, Edwards DP. Effects of hormone and cellular modulators of protein phosphorylation on transcriptional activity, DNA binding, and phosphorylation of human progesterone receptors. Mol Endocrinol. 1992;6(4):607–620. doi: 10.1210/mend.6.4.1316549. [DOI] [PubMed] [Google Scholar]
- 69.Zhang Y, Beck CA, Poletti A, Edwards DP, Weigel NL. Identification of phosphorylation sites unique to the B form of human progesterone receptor. In vitro phosphorylation by casein kinase II. J Biol Chem. 1994;269(49):31034–31040. [PubMed] [Google Scholar]
- 70.Zhang C, Dowd DR, Staal A, Gu C, Lian JB, van Wijnen AJ, Stein GS, MacDonald PN. Nuclear coactivator-62 kDa/Ski-interacting protein is a nuclear matrix-associated coactivator that may couple vitamin D receptor-mediated transcription and RNA splicing. J Biol Chem. 2003;278(37):35325–35336. doi: 10.1074/jbc.M305191200. [DOI] [PubMed] [Google Scholar]
- 71.Zhang Y, Beck CA, Poletti A, Edwards DP, Weigel NL. Identification of a group of Ser-Pro motif hormone-inducible phosphorylation sites in the human progesterone receptor. Mol Endocrinol. 1995;9(8):1029–1040. doi: 10.1210/mend.9.8.7476977. [DOI] [PubMed] [Google Scholar]
- 72.Faivre EJ, Daniel AR, Hillard CJ, Lange CA. Progesterone receptor rapid signaling mediates serine 345 phosphorylation and tethering to specificity protein 1 transcription factors. Mol Endocrinol. 2008;22(4):823–837. doi: 10.1210/me.2007-0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Qiu M, Olsen A, Faivre E, Horwitz KB, Lange CA. Mitogen-activated protein kinase regulates nuclear association of human progesterone receptors. Mol Endocrinol. 2003;17(4):628–642. doi: 10.1210/me.2002-0378. [DOI] [PubMed] [Google Scholar]
- 74.Daniel AR, Lange CA. Protein kinases mediate ligand-independent derepression of sumoylated progesterone receptors in breast cancer cells. Proc Natl Acad Sci U S A. 2009;106(34):14287–14292. doi: 10.1073/pnas.0905118106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hagan CR, Regan TM, Dressing GE, Lange CA. ck2-dependent phosphorylation of progesterone receptors (PR) on Ser81 regulates PR-B isoform-specific target gene expression in breast cancer cells. Mol Cell Biol. 2011;31(12):2439–2452. doi: 10.1128/MCB.01246-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hagan CR, Knutson TP, Lange CA. A Common Docking Domain in Progesterone Receptor-B links DUSP6 and CK2 signaling to proliferative transcriptional programs in breast cancer cells. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hagan CR, Daniel AR, Dressing GE, Lange CA. Role of phosphorylation in progesterone receptor signaling and specificity. Mol Cell Endocrinol. 2012;357(1–2):43–49. doi: 10.1016/j.mce.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lange CA. Integration of progesterone receptor action with rapid signaling events in breast cancer models. J Steroid Biochem Mol Biol. 2008;108(3–5):203–212. doi: 10.1016/j.jsbmb.2007.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ciechanover A. The ubiquitin proteolytic system: from an idea to the patient bed. Proc Am Thorac Soc. 2006;3(1):21–31. doi: 10.1513/pats.200510-106JH. [DOI] [PubMed] [Google Scholar]
- 80.Ciechanover A. The ubiquitin proteolytic system: from a vague idea, through basic mechanisms, and onto human diseases and drug targeting. Neurology. 2006;66(2 Suppl 1):S7–19. doi: 10.1212/01.wnl.0000192261.02023.b8. [DOI] [PubMed] [Google Scholar]
- 81.Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, Bernards R. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123(5):773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 82.Haglund K, Dikic I. Ubiquitylation and cell signaling. EMBO J. 2005;24(19):3353–3359. doi: 10.1038/sj.emboj.7600808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Salghetti SE, Caudy AA, Chenoweth JG, Tansey WP. Regulation of transcriptional activation domain function by ubiquitin. Science. 2001;293(5535):1651–1653. doi: 10.1126/science.1062079. [DOI] [PubMed] [Google Scholar]
- 84.Goult BT, Bouaouina M, Elliott PR, Bate N, Patel B, Gingras AR, Grossmann JG, Roberts GC, Calderwood DA, Critchley DR, et al. Structure of a double ubiquitin-like domain in the talin head: a role in integrin activation. EMBO J. 2010;29(6):1069–1080. doi: 10.1038/emboj.2010.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grossman SR, Deato ME, Brignone C, Chan HM, Kung AL, Tagami H, Nakatani Y, Livingston DM. Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science. 2003;300(5617):342–344. doi: 10.1126/science.1080386. [DOI] [PubMed] [Google Scholar]
- 86.Salghetti SE, Kim SY, Tansey WP. Destruction of Myc by ubiquitin-mediated proteolysis: cancer-associated and transforming mutations stabilize Myc. EMBO J. 1999;18(3):717–726. doi: 10.1093/emboj/18.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Salghetti SE, Muratani M, Wijnen H, Futcher B, Tansey WP. Functional overlap of sequences that activate transcription and signal ubiquitin-mediated proteolysis. Proc Natl Acad Sci U S A. 2000;97(7):3118–3123. doi: 10.1073/pnas.050007597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Perissi V, Aggarwal A, Glass CK, Rose DW, Rosenfeld MG. A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell. 2004;116(4):511–526. doi: 10.1016/s0092-8674(04)00133-3. [DOI] [PubMed] [Google Scholar]
- 89.Imhof MO, McDonnell DP. Yeast RSP5 and its human homolog hRPF1 potentiate hormone-dependent activation of transcription by human progesterone and glucocorticoid receptors. Mol Cell Biol. 1996;16(6):2594–2605. doi: 10.1128/mcb.16.6.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nawaz Z, Lonard DM, Smith CL, Lev-Lehman E, Tsai SY, Tsai MJ, O’Malley BW. The Angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol Cell Biol. 1999;19(2):1182–1189. doi: 10.1128/mcb.19.2.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Verma S, Ismail A, Gao X, Fu G, Li X, O’Malley BW, Nawaz Z. The ubiquitin-conjugating enzyme UBCH7 acts as a coactivator for steroid hormone receptors. Mol Cell Biol. 2004;24(19):8716–8726. doi: 10.1128/MCB.24.19.8716-8726.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Calvo V, Beato M. BRCA1 counteracts progesterone action by ubiquitination leading to progesterone receptor degradation and epigenetic silencing of target promoters. Cancer Res. 2011;71(9):3422–3431. doi: 10.1158/0008-5472.CAN-10-3670. [DOI] [PubMed] [Google Scholar]
- 93.Wei LL, Krett NL, Francis MD, Gordon DF, Wood WM, O’Malley BW, Horwitz KB. Multiple human progesterone receptor messenger ribonucleic acids and their autoregulation by progestin agonists and antagonists in breast cancer cells. Mol Endocrinol. 1988;2(1):62–72. doi: 10.1210/mend-2-1-62. [DOI] [PubMed] [Google Scholar]
- 94.Nardulli AM, Greene GL, O’Malley BW, Katzenellenbogen BS. Regulation of progesterone receptor messenger ribonucleic acid and protein levels in MCF-7 cells by estradiol: analysis of estrogen’s effect on progesterone receptor synthesis and degradation. Endocrinology. 1988;122(3):935–944. doi: 10.1210/endo-122-3-935. [DOI] [PubMed] [Google Scholar]
- 95.Bochtler M, Ditzel L, Groll M, Hartmann C, Huber R. The proteasome. Annu Rev Biophys Biomol Struct. 1999;28:295–317. doi: 10.1146/annurev.biophys.28.1.295. [DOI] [PubMed] [Google Scholar]
- 96.Groll M, Heinemeyer W, Jager S, Ullrich T, Bochtler M, Wolf DH, Huber R. The catalytic sites of 20S proteasomes and their role in subunit maturation: a mutational and crystallographic study. Proc Natl Acad Sci U S A. 1999;96(20):10976–10983. doi: 10.1073/pnas.96.20.10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998;8(10):397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 98.Lee DH, Goldberg AL. Proteasome inhibitors cause induction of heat shock proteins and trehalose, which together confer thermotolerance in Saccharomyces cerevisiae. Mol Cell Biol. 1998;18(1):30–38. doi: 10.1128/mcb.18.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hannoun Z, Greenhough S, Jaffray E, Hay RT, Hay DC. Post-translational modification by SUMO. Toxicology. 2010;278(3):288–293. doi: 10.1016/j.tox.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 100.Gill G. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 2004;18(17):2046–2059. doi: 10.1101/gad.1214604. [DOI] [PubMed] [Google Scholar]
- 101.Chauchereau A, Amazit L, Quesne M, Guiochon-Mantel A, Milgrom E. Sumoylation of the progesterone receptor and of the steroid receptor coactivator SRC-1. J Biol Chem. 2003;278(14):12335–12343. doi: 10.1074/jbc.M207148200. [DOI] [PubMed] [Google Scholar]
- 102.Abdel-Hafiz H, Dudevoir ML, Horwitz KB. Mechanisms underlying the control of progesterone receptor transcriptional activity by SUMOylation. J Biol Chem. 2009;284(14):9099–9108. doi: 10.1074/jbc.M805226200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Melchior F. SUMO--nonclassical ubiquitin. Annu Rev Cell Dev Biol. 2000;16:591–626. doi: 10.1146/annurev.cellbio.16.1.591. [DOI] [PubMed] [Google Scholar]
- 104.Muller S, Hoege C, Pyrowolakis G, Jentsch S. SUMO, ubiquitin’s mysterious cousin. Nat Rev Mol Cell Biol. 2001;2(3):202–210. doi: 10.1038/35056591. [DOI] [PubMed] [Google Scholar]
- 105.Yeh ET, Gong L, Kamitani T. Ubiquitin-like proteins: new wines in new bottles. Gene. 2000;248(1–2):1–14. doi: 10.1016/s0378-1119(00)00139-6. [DOI] [PubMed] [Google Scholar]
- 106.Johnson ES, Schwienhorst I, Dohmen RJ, Blobel G. The ubiquitin-like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. Embo J. 1997;16(18):5509–5519. doi: 10.1093/emboj/16.18.5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Desterro JM, Rodriguez MS, Kemp GD, Hay RT. Identification of the enzyme required for activation of the small ubiquitin-like protein SUMO-1. J Biol Chem. 1999;274(15):10618–10624. doi: 10.1074/jbc.274.15.10618. [DOI] [PubMed] [Google Scholar]
- 108.Gong L, Kamitani T, Fujise K, Caskey LS, Yeh ET. Preferential interaction of sentrin with a ubiquitin-conjugating enzyme, Ubc9. J Biol Chem. 1997;272(45):28198–28201. doi: 10.1074/jbc.272.45.28198. [DOI] [PubMed] [Google Scholar]
- 109.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437(7059):759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chang YL, Huang CJ, Chan JY, Liu PY, Chang HP, Huang SM. Regulation of nuclear receptor and coactivator functions by the carboxyl terminus of ubiquitin-conjugating enzyme 9. Int J Biochem Cell Biol. 2007;39(5):1035–1046. doi: 10.1016/j.biocel.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 111.Poukka H, Aarnisalo P, Karvonen U, Palvimo JJ, Janne OA. Ubc9 interacts with the androgen receptor and activates receptor-dependent transcription. J Biol Chem. 1999;274(27):19441–19446. doi: 10.1074/jbc.274.27.19441. [DOI] [PubMed] [Google Scholar]
- 112.Yokota K, Shibata H, Kurihara I, Kobayashi S, Suda N, Murai-Takeda A, Saito I, Kitagawa H, Kato S, Saruta T, et al. Coactivation of the N-terminal transactivation of mineralocorticoid receptor by Ubc9. J Biol Chem. 2007;282(3):1998–2010. doi: 10.1074/jbc.M607741200. [DOI] [PubMed] [Google Scholar]
- 113.Szapary D, Song LN, He Y, Simons SS., Jr Differential modulation of glucocorticoid and progesterone receptor transactivation. Mol Cell Endocrinol. 2008;283(1–2):114–126. doi: 10.1016/j.mce.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kahyo T, Nishida T, Yasuda H. Involvement of PIAS1 in the sumoylation of tumor suppressor p53. Mol Cell. 2001;8(3):713–718. doi: 10.1016/s1097-2765(01)00349-5. [DOI] [PubMed] [Google Scholar]
- 115.Takahashi Y, Kahyo T, Toh EA, Yasuda H, Kikuchi Y. Yeast Ull1/Siz1 is a novel SUMO1/Smt3 ligase for septin components and functions as an adaptor between conjugating enzyme and substrates. J Biol Chem. 2001;276(52):48973–48977. doi: 10.1074/jbc.M109295200. [DOI] [PubMed] [Google Scholar]
- 116.Hochstrasser M. SP-RING for SUMO: new functions bloom for a ubiquitin-like protein. Cell. 2001;107(1):5–8. doi: 10.1016/s0092-8674(01)00519-0. [DOI] [PubMed] [Google Scholar]
- 117.Kotaja N, Karvonen U, Janne OA, Palvimo JJ. PIAS proteins modulate transcription factors by functioning as SUMO-1 ligases. Mol Cell Biol. 2002;22(14):5222–5234. doi: 10.1128/MCB.22.14.5222-5234.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kotaja N, Aittomaki S, Silvennoinen O, Palvimo JJ, Janne OA. ARIP3 (androgen receptor-interacting protein 3) and other PIAS (protein inhibitor of activated STAT) proteins differ in their ability to modulate steroid receptor-dependent transcriptional activation. Mol Endocrinol. 2000;14(12):1986–2000. doi: 10.1210/mend.14.12.0569. [DOI] [PubMed] [Google Scholar]
- 119.Jones MC, Fusi L, Higham JH, Abdel-Hafiz H, Horwitz KB, Lam EW, Brosens JJ. Regulation of the SUMO pathway sensitizes differentiating human endometrial stromal cells to progesterone. Proc Natl Acad Sci U S A. 2006;103(44):16272–16277. doi: 10.1073/pnas.0603002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Man JH, Li HY, Zhang PJ, Zhou T, He K, Pan X, Liang B, Li AL, Zhao J, Gong WL, et al. PIAS3 induction of PRB sumoylation represses PRB transactivation by destabilizing its retention in the nucleus. Nucleic Acids Res. 2006;34(19):5552–5566. doi: 10.1093/nar/gkl691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bawa-Khalfe T, Yeh ET. The in vivo functions of desumoylating enzymes. Subcell Biochem. 2011;54:170–183. doi: 10.1007/978-1-4419-6676-6_14. [DOI] [PubMed] [Google Scholar]
- 122.Bawa-Khalfe T, Yeh ET. SUMO Losing Balance: SUMO Proteases Disrupt SUMO Homeostasis to Facilitate Cancer Development and Progression. Genes Cancer. 2010;1(7):748–752. doi: 10.1177/1947601910382555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yeh ET. SUMOylation and De-SUMOylation: wrestling with life’s processes. J Biol Chem. 2009;284(13):8223–8227. doi: 10.1074/jbc.R800050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cheng J, Bawa T, Lee P, Gong L, Yeh ET. Role of desumoylation in the development of prostate cancer. Neoplasia. 2006;8(8):667–676. doi: 10.1593/neo.06445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jacques C, Baris O, Prunier-Mirebeau D, Savagner F, Rodien P, Rohmer V, Franc B, Guyetant S, Malthiery Y, Reynier P. Two-step differential expression analysis reveals a new set of genes involved in thyroid oncocytic tumors. J Clin Endocrinol Metab. 2005;90(4):2314–2320. doi: 10.1210/jc.2004-1337. [DOI] [PubMed] [Google Scholar]
- 126.Xu Y, Li J, Zuo Y, Deng J, Wang LS, Chen GQ. SUMO-specific protease 1 regulates the in vitro and in vivo growth of colon cancer cells with the upregulated expression of CDK inhibitors. Cancer Lett. 2011;309(1):78–84. doi: 10.1016/j.canlet.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 127.Poukka H, Karvonen U, Janne OA, Palvimo JJ. Covalent modification of the androgen receptor by small ubiquitin-like modifier 1 (SUMO-1) Proc Natl Acad Sci U S A. 2000;97(26):14145–14150. doi: 10.1073/pnas.97.26.14145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yang SH, Jaffray E, Hay RT, Sharrocks AD. Dynamic interplay of the SUMO and ERK pathways in regulating Elk-1 transcriptional activity. Mol Cell. 2003;12(1):63–74. doi: 10.1016/s1097-2765(03)00265-x. [DOI] [PubMed] [Google Scholar]
- 129.Kim J, Cantwell CA, Johnson PF, Pfarr CM, Williams SC. Transcriptional activity of CCAAT/enhancer-binding proteins is controlled by a conserved inhibitory domain that is a target for sumoylation. J Biol Chem. 2002;277(41):38037–38044. doi: 10.1074/jbc.M207235200. [DOI] [PubMed] [Google Scholar]
- 130.Subramanian L, Benson MD, Iniguez-Lluhi JA. A synergy control motif within the attenuator domain of CCAAT/enhancer-binding protein alpha inhibits transcriptional synergy through its PIASy-enhanced modification by SUMO-1 or SUMO-3. J Biol Chem. 2003;278(11):9134–9141. doi: 10.1074/jbc.M210440200. [DOI] [PubMed] [Google Scholar]
- 131.Ross S, Best JL, Zon LI, Gill G. SUMO-1 modification represses Sp3 transcriptional activation and modulates its subnuclear localization. Mol Cell. 2002;10(4):831–842. doi: 10.1016/s1097-2765(02)00682-2. [DOI] [PubMed] [Google Scholar]
- 132.Sapetschnig A, Rischitor G, Braun H, Doll A, Schergaut M, Melchior F, Suske G. Transcription factor Sp3 is silenced through SUMO modification by PIAS1. Embo J. 2002;21(19):5206–5215. doi: 10.1093/emboj/cdf510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Long J, Wang G, He D, Liu F. Repression of Smad4 transcriptional activity by SUMO modification. Biochem J. 2004;379(Pt 1):23–29. doi: 10.1042/BJ20031867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cheng J, Wang D, Wang Z, Yeh ET. SENP1 enhances androgen receptor-dependent transcription through desumoylation of histone deacetylase 1. Mol Cell Biol. 2004;24(13):6021–6028. doi: 10.1128/MCB.24.13.6021-6028.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kaikkonen S, Jaaskelainen T, Karvonen U, Rytinki MM, Makkonen H, Gioeli D, Paschal BM, Palvimo JJ. SUMO-specific protease 1 (SENP1) reverses the hormone-augmented SUMOylation of androgen receptor and modulates gene responses in prostate cancer cells. Mol Endocrinol. 2009;23(3):292–307. doi: 10.1210/me.2008-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Abdel-Hafiz HA, Horwitz KB. Control of progesterone receptor transcriptional synergy by SUMOylation and deSUMOylation. BMC Mol Biol. 2012;13:10. doi: 10.1186/1471-2199-13-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fu M, Rao M, Wu K, Wang C, Zhang X, Hessien M, Yeung YG, Gioeli D, Weber MJ, Pestell RG. The androgen receptor acetylation site regulates cAMP and AKT but not ERK-induced activity. J Biol Chem. 2004;279(28):29436–29449. doi: 10.1074/jbc.M313466200. [DOI] [PubMed] [Google Scholar]
- 138.Fu M, Wang C, Zhang X, Pestell RG. Acetylation of nuclear receptors in cellular growth and apoptosis. Biochem Pharmacol. 2004;68(6):1199–1208. doi: 10.1016/j.bcp.2004.05.037. [DOI] [PubMed] [Google Scholar]
- 139.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90(4):595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 140.Boyes J, Byfield P, Nakatani Y, Ogryzko V. Regulation of activity of the transcription factor GATA-1 by acetylation. Nature. 1998;396(6711):594–598. doi: 10.1038/25166. [DOI] [PubMed] [Google Scholar]
- 141.Hayakawa F, Towatari M, Ozawa Y, Tomita A, Privalsky ML, Saito H. Functional regulation of GATA-2 by acetylation. J Leukoc Biol. 2004;75(3):529–540. doi: 10.1189/jlb.0603389. [DOI] [PubMed] [Google Scholar]
- 142.Kim MY, Woo EM, Chong YT, Homenko DR, Kraus WL. Acetylation of estrogen receptor alpha by p300 at lysines 266 and 268 enhances the deoxyribonucleic acid binding and transactivation activities of the receptor. Mol Endocrinol. 2006;20(7):1479–1493. doi: 10.1210/me.2005-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kim Y, Sun Y, Chow C, Pommier YG, Simons SS., Jr Effects of acetylation, polymerase phosphorylation, and DNA unwinding in glucocorticoid receptor transactivation. J Steroid Biochem Mol Biol. 2006;100(1–3):3–17. doi: 10.1016/j.jsbmb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 144.Faus H, Haendler B. Post-translational modifications of steroid receptors. Biomed Pharmacother. 2006;60(9):520–528. doi: 10.1016/j.biopha.2006.07.082. [DOI] [PubMed] [Google Scholar]
- 145.Aoyagi S, Archer TK. Dynamic histone acetylation/deacetylation with progesterone receptor-mediated transcription. Mol Endocrinol. 2007;21(4):843–856. doi: 10.1210/me.2006-0244. [DOI] [PubMed] [Google Scholar]
- 146.Daniel AR, Gaviglio AL, Czaplicki LM, Hillard CJ, Housa D, Lange CA. The progesterone receptor hinge region regulates the kinetics of transcriptional responses through acetylation, phosphorylation, and nuclear retention. Mol Endocrinol. 2010;24(11):2126–2138. doi: 10.1210/me.2010-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Loven MA, Davis RE, Curtis CD, Muster N, Yates JR, Nardulli AM. A novel estrogen receptor alpha-associated protein alters receptor-deoxyribonucleic acid interactions and represses receptor-mediated transcription. Mol Endocrinol. 2004;18(11):2649–2659. doi: 10.1210/me.2003-0195. [DOI] [PubMed] [Google Scholar]
- 148.Loven MA, Muster N, Yates JR, Nardulli AM. A novel estrogen receptor alpha-associated protein, template-activating factor Ibeta, inhibits acetylation and transactivation. Mol Endocrinol. 2003;17(1):67–78. doi: 10.1210/me.2002-0280. [DOI] [PubMed] [Google Scholar]
- 149.Woodsmith J, Kamburov A, Stelzl U. Dual coordination of post translational modifications in human protein networks. PLoS Comput Biol. 2013;9(3):e1002933. doi: 10.1371/journal.pcbi.1002933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Daniel AR, Qiu M, Faivre EJ, Ostrander JH, Skildum A, Lange CA. Linkage of progestin and epidermal growth factor signaling: phosphorylation of progesterone receptors mediates transcriptional hypersensitivity and increased ligand-independent breast cancer cell growth. Steroids. 2007;72(2):188–201. doi: 10.1016/j.steroids.2006.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Skildum A, Faivre E, Lange CA. Progesterone receptors induce cell cycle progression via activation of mitogen-activated protein kinases. Mol Endocrinol. 2005;19(2):327–339. doi: 10.1210/me.2004-0306. [DOI] [PubMed] [Google Scholar]
- 152.Muller S, Matunis MJ, Dejean A. Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus. Embo J. 1998;17(1):61–70. doi: 10.1093/emboj/17.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Muller S, Berger M, Lehembre F, Seeler JS, Haupt Y, Dejean A. c-Jun and p53 activity is modulated by SUMO-1 modification. J Biol Chem. 2000;275(18):13321–13329. doi: 10.1074/jbc.275.18.13321. [DOI] [PubMed] [Google Scholar]
- 154.Yang SH, Jaffray E, Senthinathan B, Hay RT, Sharrocks AD. SUMO and transcriptional repression: dynamic interactions between the MAP kinase and SUMO pathways. Cell Cycle. 2003;2(6):528–530. doi: 10.4161/cc.2.6.597. [DOI] [PubMed] [Google Scholar]
- 155.Anckar J, Sistonen L. SUMO: getting it on. Biochem Soc Trans. 2007;35(Pt 6):1409–1413. doi: 10.1042/BST0351409. [DOI] [PubMed] [Google Scholar]
- 156.Yang SH, Sharrocks AD. PIASxalpha differentially regulates the amplitudes of transcriptional responses following activation of the ERK and p38 MAPK pathways. Mol Cell. 2006;22(4):477–487. doi: 10.1016/j.molcel.2006.03.037. [DOI] [PubMed] [Google Scholar]
- 157.Liu B, Yang Y, Chernishof V, Loo RR, Jang H, Tahk S, Yang R, Mink S, Shultz D, Bellone CJ, et al. Proinflammatory stimuli induce IKKalpha-mediated phosphorylation of PIAS1 to restrict inflammation and immunity. Cell. 2007;129(5):903–914. doi: 10.1016/j.cell.2007.03.056. [DOI] [PubMed] [Google Scholar]
- 158.Zhang PJ, Zhao J, Li HY, Man JH, He K, Zhou T, Pan X, Li AL, Gong WL, Jin BF, et al. CUE domain containing 2 regulates degradation of progesterone receptor by ubiquitin-proteasome. Embo J. 2007;26(7):1831–1842. doi: 10.1038/sj.emboj.7601602. [DOI] [PMC free article] [PubMed] [Google Scholar]