

Abstract
The modulation of drug metabolism enzyme (DME) expression by therapeutic agents is a central mechanism of drug–drug interaction and should be assessed as early as possible in preclinical drug development. Direct measurement of DME levels is typically achieved by Western blotting, qPCR, or microarray, but these techniques have their limitations; antibody cross-reactivity among highly homologous subfamilies creates ambiguity, while discordance between mRNA and protein expression undermines observations. The aim of this study was to design a simple targeted workflow by combining in vivo SILAC and label-free proteomics approaches for quantification of DMEs in mouse liver, facilitating a rapid and comprehensive evaluation of metabolic potential at the protein level. A total of 197 peptides, representing 51 Phase I and Phase II DMEs, were quantified by LC-MS/MS using targeted high resolution single ion monitoring (tHR/SIM) with a defined mass-to-charge and retention time window for each peptide. In a constitutive androstane receptor (Car) activated mouse model, comparison of tHR/SIM-in vivo SILAC with Western blotting for analysis of the expression of cytochromes P450 was favorable, with agreement in fold-change values between methods. The tHR/SIM-in vivo SILAC approach therefore permits the robust analysis of multiple DME in a single protein sample, with clear utility for the assessment of the drug–drug interaction potential of candidate therapeutic compounds.
Keywords: drug metabolism, drug−drug interaction, constitutive androstane receptor, protein quantification, targeted in vivo SILAC
Introduction
The United States Food and Drug Administration and European Medicines Agency advise that, to improve safety in clinical development and postapproval, the potential for a new therapeutic agent to interact with established medications (drug–drug interaction, DDI) should be assessed as early as possible during preclinical development.1,2 One major mechanism of DDI is the ability of chemical agents to regulate the expression of drug metabolism enzymes and transporters, often through the modulation of nuclear hormone receptor (NHR) activity, thereby altering the efficacy of both themselves and other compounds. It is valuable, therefore, to have a comprehensive understanding of the levels of expression of these protein factors and how they are modulated during therapy.
In the liver, the primary site of drug metabolism, the majority of phase I (modification) reactions are carried out by cytochrome P450 (CYP), with additional contributions made by alcohol dehydrogenase (ADH), aldehyde dehydrogenase (ALDH), aldo-keto reductase (AKR), epoxide hydrolase (EPHX), and flavin-containing monooxygenase (FMO) superfamilies.3 UDP glucuronosyltransferases (UGT) and glutathione S-transferases (GST) enact most phase II (conjugation) events.3 During laboratory study, if a broad expression profile of these DMEs is required, DNA microarray or high-density RT-PCR arrays are typically employed. While providing a practicable platform for this type of analysis, there is a significant discordance between mRNA and protein expression.4−6 A recent study by Ohtsuki and colleagues demonstrated a poor correlation between protein and mRNA levels for multiple CYP, UGT, and drug transporters, with only a handful of exceptions.7 In this study, the direct measurement of protein expression, as opposed to the measurement of mRNA, correlated far better with enzymatic activity and is therefore a more appropriate readout for the evaluation of drug–drug interaction potential.7
Western blotting and other antibody-based approaches are the mainstay of protein expression analysis for DME, but our lab and others routinely struggle to interpret data due to the high degree of sequence homology of superfamily members, and hence cross-reactivity of antibody preparations. In order to circumvent this issue, recent developments have been made in stable isotope dilution mass spectrometry-based proteomics to simultaneously detect and quantify CYP and other drug metabolism related proteins.7−13 These studies utilize an absolute quantification technique (AQUA) where known quantities of multiple synthetic stable isotope peptides for a protein/proteins of interest are spiked into proteolytic protein digests derived from liver samples, prior to LC-MS/MS analysis in a multiple reaction monitoring (SRM) mode.14 The “heavy” stable isotope peptides and “light” unlabeled peptides co-elute, co-ionize, and are only differentiated by the difference in mass through LC-MS/MS analysis. Using peak intensity ratios of the pairs of light and heavy isotopes, concentrations of analyte peptides can be calculated based on known concentrations of the stable isotope peptides. A modified version of this procedure using stable isotope-labeled proteins expressed in and purified from Escherichia coli as internal standards has also been developed which, post-translational modification and extraction variability excepted, accounts for efficiency of enzymatic digestion.15,16 But for comprehensive proteomic analysis, stable isotope labeling by amino acids in cell culture (SILAC) or in whole organisms (in vivo SILAC, SILAM) permits the simultaneous quantification of thousands of proteins with a much improved confidence due to the fact that both light and heavy analytes share near-identical chemical properties and environment.17−22
Label-free shotgun proteomics is an alternative commonly used mass spectrometry based proteomics strategy.23 There are two mainstream label-free based LC-MS/MS approaches based on either spectral counting or ion intensity. The former compares the number of MS2 spectra assigned to a protein between samples, while the latter compares intensities of each precursor ion between samples. Ion intensity is generally considered to provide more detailed quantitative information. This approach aligns precursor ions between all LC-MS/MS runs using defined retention time and m/z windows. The signal intensities in each window for each sample are then integrated, normalized and compared between samples or groups. Precursor ions showing significant differences or all precursors with MS2 spectra can then be identified. The success of this approach relies on tight control in sample processing and consistency in retention time in LC separation.
In the current study, we harness the advantages of in vivo SILAC materials in conjunction with the label-free shotgun proteomics concept to quantify DMEs in mouse liver, although the approach could be used for targeted quantification of any detectable protein or protein group of interest. Peptides from DMEs were identified in a metabolically labeled spike-in standard lysate and a complementary list of unlabeled peptides was constructed. This list of peptide pairs was stress-tested with a range of heavy to light input sample ratios and only the most reliable were retained. The final peptide list allowed us to quantify changes in expression of 51 DMEs and was used to generate a constitutive androstane receptor (Car) activation signature.
Materials and Methods
Reagents
1,4-Bis-[2-(3,5-dichloropyridyloxy)]benzene, 3,3′,5,5′-tetrachloro-1,4-bis(pyridyloxy)benzene (TCPOBOP), corn oil, dl-dithiothreitol, and iodoacetamide were purchased from Sigma (Dorset, UK). Trypsin Gold was purchased from Promega (Madison, WI). Lys(6)-SILAC-mouse tissue was purchased from Silantes (Munich, Germany).
Animal Husbandry and Dosing
All mice were maintained under standard animal house conditions, with free access to food and water, and a 12 h light/12 h dark cycle. All animal work was carried out on male 8-week-old C57BL/6J mice in accordance with the Animal Scientific Procedures Act (1986) and after local ethical review. Mice were administered either TCPOBOP (a single intraperitoneal injection at 3 mg/kg) in corn oil vehicle at 10 μL/g body weight, or the same volume of corn oil vehicle alone. Animals were sacrificed 7 days after dosing, and liver tissue excised and snap-frozen in liquid nitrogen for storage at −80 °C.
Sample Preparation
For LC-MS/MS, frozen liver tissue was thawed by addition of 9 volumes of SDT lysis buffer (4% SDS, 0.1 M DTT, 100 mM Tris-HCl pH7.6) then homogenized by rotor-stator (2 × 5 s at 20k revolutions). Homogenate was heated to 95 °C for 5 min, sonicated (2 × 5 s), and then centrifuged at 16 000 g for 10 min. Supernatant (protein sample for analysis) was removed, aliquoted and stored at −80 °C until use. Protein samples (total of 30 μg/well) were electrophoresed through a 12% bis-tris gel in MOPS running buffer supplemented with antioxidant (all Life Technologies, Paisley, UK) alongside a Spectra multicolour broad range protein ladder (Thermo Fisher Scientific, Waltham, MA). Gels were stained with Coomassie blue, destained, and then rehydrated with Milli-Q water. Gel regions containing proteins of interest, as described in the Results section, were removed with a clean scalpel, sliced finely (ca. 1 × 1 mm cubes), and added to 1.5 mL PCR-clean eppendorf tubes (Eppendorf, Hamburg, Germany). In-gel trypsin digest and peptide extraction was carried out according to the method of Schevchenko and colleagues.24 Peptide sample concentration was determined by Nanodrop (Thermo Fisher Scientific) and adjusted to 0.2 mg/mL in water containing 0.1% (v/v) trifluoroacetic acid. For Western blotting, microsomal lysates were prepared and analyzed as described previously.25,26 Briefly, tissue was homogenized in 3 volumes of KCl buffer (1.15% w/v potassium chloride, 10 mM potassium phosphate, pH7.4) by rotor-stator (2 × 5 s at 20k revolutions) followed by centrifugation at 11 000g for 15 min. Supernatant was ultracentrifuged at 100 000g for 1 h and the resulting pellet resuspended in KCl buffer containing 0.25 M sucrose. Protein concentration was adjusted to 1 mg/mL in LDS sample buffer (Life Technologies) before electrophoresis through 10% acrylamide gels for 1 h at 200 V, followed by transfer onto nitrocellulose membranes (1 h at 100 V). Ponseau S and/or coomassie staining were used to ensure even loading. Our in-house panel of rabbit polyclonal antibodies for Cyp detection has been summarized previously.27 Fold changes were calculated from chemiluminescent signal intensity on a Fujifilm LAS-3000 imager (Fujifilm, Tokyo, Japan).
Liquid Chromatography and Mass Spectrometry
A nanoflow liquid chromatograph (Agilent 1200, Agilent, Santa Clara, CA) with a LTQ-Orbitrap XL (Thermo Fisher Scientific) was used to analyze the protein digests. Approximately 0.4 μg total peptide was loaded onto a trap column at a flow rate of 10 μL/min for 3 min and the flow was then reversed to an Agilent Zorbex nano C18 column (0.0075 mm ID; 15 cm; 3 μm particle size). The peptides were resolved with a 3 h binary gradient at a flow rate of 300 nL/min as follows: 0% buffer B for 5 min followed by 2–30% buffer B for 140 min, 30–90% buffer B for 15 min, 90–0% buffer B for 10 min, and 0% buffer B for 10 min. Buffer A contained 2% acetonitrile and 0.1% formic acid in water, and buffer B contained 0.1% formic acid in acetonitrile. The column was periodically cleaned with a 2 μL injection of buffer containing 50% acetonitrile and 0.1% formic acid in water. A Proxeon nanospray source with a stainless steel emitter (Thermo Fisher Scientific) was used to interface the Agilent nanoLC and LTQ-Orbitrap. Spray voltage was set at 1.8 kV. The Orbitrap was tuned using Glu-Fibrinogen B peptide. For the protein/peptide identification, a method that consisted of full scans between 330 and 1500 amu (in Orbitrap) and data dependent MS/MS with top six precursor ions (2+ to 4+ charged) in LTQ was employed. Orbitrap was operated in a profile mode at the resolution of 30 000 or 60 000 with a lock mass set at 445.1200 (polycyclodimethylsiloxane28), and LTQ was operated in a centroid mode with isolation width = 1 (m/z), normalized collision energy = 0.25, and activation time = 30 ms. The max fill times for Orbitrap and LTQ were set at 500 and 50 ms, respectively. A dynamic exclusion of 30 s was used to maximize the acquisition of MS2 on peptides with lower intensity. For tHR/SIM analysis, a method that consisted of full scans between 330 and 1500 a.m.u (in Orbitrap) and data dependent MS/MS scans with or without defined precursors was employed. A dynamic exclusion of 30 s and a threshold of 500 counts to trigger MS2 were also applied for MS/MS scans. Nontargeted data dependent MS/MS was performed when there was no targeted precursor found in the MS scan. Further details can be found in the method and tune files (Supporting Information Files S-1 and S-2).
Data Analysis
Protein and peptide database search was carried out using PEAKS version 6 (Bioinformatics Solutions, Waterloo, Canada) with an IPI-mouse database (version 3.87, European Bioinformatics Institute, Hinxton, UK). The precursor mass tolerance was set at 7 ppm, and fragment ion mass tolerance set at 0.5 amu. The only permitted post-translational modifications were N-terminal acetylation and cysteine carbamidomethylation, while a maximum of two miscleavages were allowed. Quantification of the predefined targeted peptides was carried out using SIEVE version 2.0 (Thermo Fisher Scientific) using two seed files, one for each DME region of interest, containing retention time and m/z information. The precursor mass tolerance was set to 5 ppm, and the minimal intensity for alignment was set at 100 000, with intensities derived from the first monoisotopic peak. Data from SIEVE were exported to Excel 2010 (Microsoft, Redmond, WA) for calculation of light to heavy peptide ratios. For protein quantification, light to heavy protein ratios were calculated within samples by summing average intensity values for all light peptides for each protein, then dividing by the corresponding heavy value. Light to heavy protein ratios for technical replicates were averaged then biological replicates normalized to the average of control, before calculation of fold changes. For calculation of statistical significance, normalized values were log2-transformed and then analyzed by unpaired Student’s t test (FDR = 0.5%) using Prism 6 (GraphPad, La Jolla, CA); *p < 0.05, **p < 0.01, ***p < 0.001.
Results
Concept of tHR/SI-in Vivo SILAC Workflow
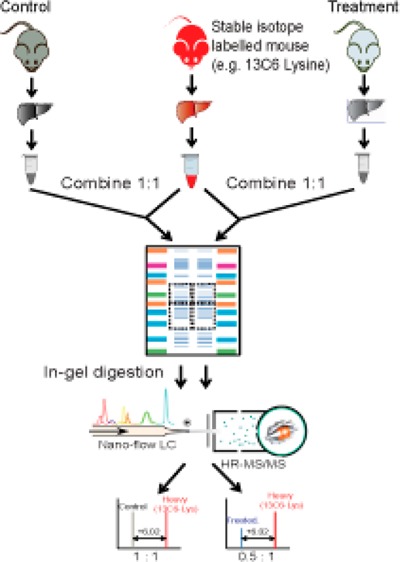
A workflow schematic of the tHR/SIM in vivo SILAC approach is shown in Figure 1A. Liver lysates were combined 1:1 with a liver lysate from metabolically labeled in vivo SILAC animals, which served as an internal standard for each experimental sample. Gel electrophoresis was selected for sample preparation, as it allowed for fractionation and enrichment of DMEs at the protein level. This selection was based on a pilot comparison of the SDS-PAGE in-gel digestion method to FASP. In the former, DMEs were resolved by SDS-PAGE into two defined molecular weight regions which were excised for analysis; DME region 1 (66–40 kDa) was expected to contain Adh, Aldh, Cyp, Fmo, Ugt, and Ephx enzymes, while DME region 2 (40–22 kDa) was expected to contain Akr, Gst, N-acetyltransferase (Nat), and sulfotransferase (Sult) enzymes, based on calculated MW values (Figure 1B). Protein in excised bands was reduced, alkylated, and digested with trypsin according to published protocols.24 Peptides were extracted and analyzed by LC–MS/MS, wherein MS2 data were obtained. A total of 64 DMEs were identified by PEAKS analysis after the two-sample in-gel procedure, compared to 49 following the single-sample FASP procedure (Supporting Information Tables S-1–S-3).
Figure 1.

tHR/SIM-in vivo SILAC workflow. (A) Metabolically labeled mouse liver tissue lysates were combined 1:1 with experimental sample lysates for SDS-PAGE. Excised bands were processed by in-gel trypsin digestion for LS–MS/MS analysis. Heavy/light ratios from the first monoisotopic peaks were calculated for each peptide of interest in individual samples, with fold changes across samples calculated using the ratio/ratio value. (B) The gel bands excised, region 1 (66–40 kDa) and region 2 (40–22 kDa), are predicted to contain the DMEs of interest.
Characterization of in Vivo SILAC Liver
Similar to the principles of stable isotope dilution LC–MS methods, the accuracy of measurements in tHR/SIM are highly dependent on the reliable detection of heavy stable isotope signals. It was therefore important to characterize the liver proteome of the spike-in reference lysate to determine which DMEs could be monitored. As detailed above, SDS-PAGE and in-gel digestion of an in vivo SILAC liver sample permitted identification of a total of 64 DMEs (2 Adh, 7 Akr, 11 Aldh, 23 Cyp, 1 Ephx, 2 Fmo, 9 Gst, 0 Nat, 2 Sult, and 7 Ugt) by unique peptides (FDR = 0.1%). In addition, due to the high degree of identity shared by certain enzymes, some were unidentifiable by unique peptides but were nonetheless retained as protein groups, for example, Cyp2a4/2a5. Peptides without lysine were removed from the list, as were those with −10Log P values <17.3 (FDR = 0.1%). This “heavy-only” peptide list was converted to a list of SILAC peptide pairs by addition of the predicted counterpart light members. The utility of these peptide pairs for quantification was then stress-tested as follows. An unlabeled liver lysate was mixed with the in vivo SILAC liver lysate at ratios of 1:1, 1:4, and 1:16 in duplicate for processing and analysis by LC–MS/MS. The MS data were analyzed in SIEVE using the SILAC peptide pair list as a seed file (retention time window of ±5 min). Light to heavy peptide ratios were calculated using average intensity values. The average ratio value from technical replicates was then log4 transformed to evenly distribute the three data points, allowing calculation of an equally weighted R2 value. Only those peptides showing strong linearity (R2 > 0.9) were retained. The data output from SIEVE, along with calculations of R2 values, can be found in Supporting Information Tables S-4 and S-5. Approximately 57% of identified peptides survived this filtration step (Supporting Information Figure S-1). Seed files containing m/z and retention time windows for each peptide can be found in Supporting Information Tables S-6 (DME region 1) and S-7 (DME region 2).
An example of tHR/SIM in vivo SILAC is shown in Figure 2. Three Cyp1a2 peptides were used to define Cyp1a2 levels, one of which eluted at 37.9 min and another at 94.7 min (Figure 2A). Although some interfering signals were observed, these could be separated from the signals of interest by retention time gating and the matching of internal standard signals. For one of these peptides, NSIQDITSALFK, an R2 value for linearity of 0.998 was observed (Figure 2B) and the identities of both the light and heavy forms could be confirmed by their MS2 (Figure 2C and D).
Figure 2.

Analytical performance of Cyp1a2 tryptic peptides. (A) Using a ±5 min retention time window (gray boxes), nontarget signals could be effectively gated from NSIQDITSALFK and FLTNNNSAIDK peptide pair quantification. (B) Dilution linearity of the NSIQDITSALFK peptide pair achieved R2 of 0.998, while MS2 spectra were acquired for both (C) light and (D) heavy forms, confirming their identity.
Validation of tHR/SIM by Comparison to Western Blotting
To compare the output of tHR/SIM in vivo SILAC to Western blotting, we assessed the Cyp induction profile in mice treated with a potent and specific inducer of the constitutive androstane receptor, TCPOBOP. Animals (n = 3) were administered the compound in corn oil, and, after 7 days, livers were harvested for analysis. Western blotting for Cyp indicated strong induction of multiple family members, compared to vehicle control (Figure 3A). Depending on the origin of antisera, as well as results of previous studies (not shown), the exact identity of individual enzymes may or may not be known. With our in-house panel of antibodies, up-regulation could be demonstrated for Cyp1a1 (46.5-fold), Cyp1a2 (5.1-fold), Cyp2b10 (362.9-fold), and 3a11 (5.7-fold), while down-regulation could be demonstrated for Cyp2e1 (0.8-fold), as these superfamily members are predictably and reproducibly identified. For other antisera, such as Cyp2c and Cyp2d, the precise identities of the proteins detected are unknown, so, although inductions are observed, these are not attributable to any family member in particular. For t-HR/SIM-in vivo SILAC, data for individual animals were acquired and processed separately, and then average fold changes ± SD calculated at the last stage, to account for biological variability. Cyp1a2 (3.5-fold), Cyp2a4/5 (16.2-fold), Cyp2c29 (15.7-fold), Cyp2c37 (9.5-fold), Cyp2d22 (2.5-fold), Cyp3a13 (5.3-fold), and Cyp8b1 (3.1-fold) were significantly up-regulated (Figure 3B). Although not statistically significant (p = 0.08), Cyp2e1 levels were decreased (0.87-fold). Due to the low abundance of Cyp1a1, Cyp2b10, and Cyp3a11 in the spike-in standard, these enzymes could not be quantified by LC–MS/MS. Nevertheless, there was close agreement between methods of changes in Cyp1a2 and Cyp2e1 expression. Moreover, Western blotting suggested induction of unconfirmed Cyp2c, Cyp2d, and Cyp3a family members, while LC–MS/MS demonstrated induction of Cyp2c29, Cyp2c37, Cyp2d22, and Cyp3a13.
Figure 3.
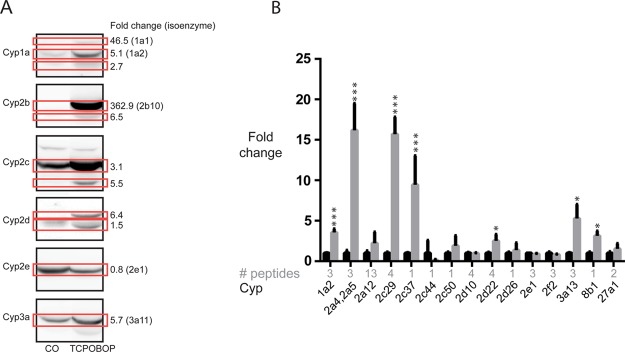
tHR/SIM-in vivo SILAC compared to Western blotting. Cyp isoform in TCPOBOP-treated mice (n = 3), as compared to corn oil vehicle-treated control animals (n = 3), were measured by (A) Western blot and (B) tHR/SIM-in vivo SILAC (black bars, corn oil; gray bars, TCPOBOP). Following Western blot of pooled samples, chemilluminescent signal was used to quantify fold changes in particular bands (red boxes), with values presented alongside. If known, the isoform detected is given in brackets. Note that, in the control, some protein levels were below the limit of detection by Western blotting (e.g., Cyp2b10) and therefore the fold changes calculated may not be accurate. For tHR/SIM-in vivo SILAC, the number of unique peptides used to calculate fold change is given (gray text), and as samples for individual animals were analyzed separately, error bars are a reflection of biological variability, as well as technical error.
To provide additional confidence that the assay was reliable, we extracted data for four proteins/protein families which are not expected to change significantly following TCPOBOP dosing: pro/albumin, calreticulin, α-tubulin, and β-tubulin. None of these changed significantly (Supporting Information Figure S2).
Additional DME Changes in Response to TCPOBOP
Of the other quantifiable phase I enzymes, Aldh1a1 (3.1-fold) and Fmo5 (3.1-fold) were significantly up-regulated, while Aldh2 (0.4-fold) and Aldh6a1 (0.5-fold) were significantly down-regulated (Figure 4A). For phase II, Gsta3 (1.6-fold), Gstm1 (7.5-fold), Gstt3 (7.0-fold), Sult3a1 (4.7-fold), Ugt1a1/2 (2.9-fold), Ugt2b34 (3.8-fold) and Ugt2b36 (1.8-fold) were significantly up-regulated (Figure 4B). Ugt2a3 (0.8-fold) was the only enzyme for which significant down-regulation occurred.
Figure 4.

tHR/SIM-in vivo SILAC analysis of modulation of additional DMEs by TCPOBOP. (A) Other phase I and (B) phase II enzymes were quantified. Black bars, corn oil; gray bars: TCPOBOP.
Discussion
Through the combination of in vivo SILAC and label-free approaches we have created a streamlined and simple targeted proteomics workflow permitting the quantitative analysis of 51 DMEs in the mouse. This technique has utility in the investigation of pharmacodynamics, particularly when the potential for, or occurrence of, drug–drug interaction is in question. Upstream, the use of metabolically labeled isotopic tissue as an internal standard reduces experimental variation derived from sample processing and ionization. This is especially important when error-prone methods such as in-gel digestion are used, and when the difference between experimental groups is relatively small. Downstream, the strategy resembles that of stable isotope dilution LC-MS for quantification of small molecules. The narrow retention time and m/z windows reduce the influence of potential contaminant signals during data processing, while data analysis is simplified in comparison to more conventional SILAC procedures as only a predetermined list of proteins/peptides of interest is measured, permitting immediate and intuitive interpretation.
One limitation of the tHR/SIM in vivo SILAC approach is that changes in expression cannot be monitored in the DME for which heavy isotope labeled MS2 are not acquired. A potential solution to this problem would be to rerun a database search on the experimental MS files and, where MS2 for light peptides from nontargeted DME can be detected, manually interrogate the primary raw file, referencing the light signal to other heavy peptides eluted within the same time frame, or to total ion current as typically performed with label-free proteomics.29 Caution must be reserved that such quantification may be subject to sources of peptide bias, such as differential recovery during sample processing and differential efficiency during ionization. Although in the current study we believe high resolution SIM mode is sufficient to monitor the levels of 51 DME proteins, mainly due to their higher relative abundance, the method can be easily adapted to an MRM mode where the precursor and fragment ion transitions are monitored.
We have applied the tHR/SIM-in vivo SILAC workflow to the analysis of DME changes in response to the Car activator, TCPOBOP. With an EC50 of approximately 100 nM, maximally effective dose of 3 mg/kg, and response duration of greater than 20 weeks in the mouse, this compound is the most potent car-specific inducer known.29−31 It acts as a mitogen, nongenotoxic tumor-promoter and complete carcinogen.32−34 In the liver, in studies variously employing TCPOBOP and/or Car knockout mice as tools to characterize the Car-dependent gene battery, up-regulation of transcription of mRNA has been demonstrated for Cyp1a1, 1a2, 2a4, 2a5, 2b9, 2b10, 2b13, 2c29, 2c37, 2c55, 2c65, 2f2, and 3a11, with decreases seen for 4a10 and 4a31.35−39 At the protein level, widespread Cyp induction has been demonstrated by Western blot40 and in a postdigest 18O labeling study, where Cyp1a2, 2a4/5, 2b10, 2b20, 2c29, 2c37, 2c38, 3a11, and 39a1 were shown to be up-regulated, with Cyp2c40, 2e1, 3a41, and 27a1 down-regulated.41 In the current study, we detected significant up-regulation of Cyp1a2, 2a4/2a5, 2c29, 2c37, 2d22, 3a13, and 8b1 and nonsignificant down-regulation of Cyp2e1, broadly in agreement with the literature, although we did not see the previously reported changes in Cyp2f2. This could be due to post-transcriptional or post-translational regulation, or variation in genetic background. Our data conflict with the report of Cyp27a1 down-regulation,41 as we detected a nonsignificant increase. We did not observe any significant changes in Adh or Akr but, to our knowledge, the only previously reported Car/TCPOBOP target within these superfamilies is Akr1b7,42 which we did not detect and were therefore unable to quantify. The only other reported Car/TCPOBOP-regulated members of the phase I metabolism superfamilies under study are Aldh1a1, Aldh1a7, and Fmo5, which are up-regulated at the mRNA level.36,43−45 Our data agree with those for Aldh1a1 and Fmo5, although we could not detect Aldh1a7 protein. In previous reports, Aldh1b1, 2, 6a1, and 7a1 have shown a Car/TCPOBOP-dependent down-regulation.43,44 Our data agree completely with these observations, although only the more pronounced decreases in Aldh2 and 6a1 achieve significance.
For phase II, at the mRNA level, Car/TCPOBOP can upregulate Gsta1, a2, a4, m1, m2, m3, m4, t1 and t3, Sult1a1, 1e1, 2a1, 2a2, 3a1 and 5a1, Ugt1a1, 1a9, 2b34, 2b35 and 2b36, and down-regulate Ugt2a3 and 3a1/2.36,44,46,47 In agreement with the literature, we observed significant induction of Gsta3, m1, t3 and Sult3a1, and nonsignificant induction of Gstm2 and t1, and Sult1a1. For Ugt, we detected induction of Ugt1a1/2, 2b34, and 2b36, with repression of 2a3, all in agreement with previous (mRNA) reports.36,44,47 Our findings agree with the only report we are aware of regarding phase II protein induction by Car/TCPOBOP, that of Ugt1a1.36 Therefore, as the vast majority of Car targets have been established as such from mRNA studies, it is noteworthy that, for many of these targets, we have provided the first evidence for their modulation at the protein level. Moreover, despite the influence of post-transcriptional and post-translational regulation, we have demonstrated that the Car activation signature is essentially conserved from mRNA to protein.
While we found that the tHR/SIM approach is satisfactory for the analysis of 51 DME proteins using a LTQ-Orbitrap, other recently developed MS2 based targeted quantitative proteomics approaches such as SWATH,48 MSE,49 and parallel reaction monitoring (PRM)50 may provide an alternative if a quadrupole-Orbitrap or quadrupole-time-of-flight mass spectrometer is available. In particular, the PRM approach has recently been shown to provide better sensitivity and specificity than SIM.50 PRM quantifies high resolution fragment ions signals derived from “light” and “heavy” precursor ion pairs that are preisolated by a quadrupole, accumulated and fragmented in a C-trap. It is therefore anticipated that the number of DME proteins measured could be potentially improved using the PRM approach.
In conclusion, the application of tHR/SIM-in vivo SILAC for quantification of 51 DME presented here constitutes a comprehensive means of profiling altered capacity for drug metabolism in the mouse. An appreciation of this capacity is valuable when carrying out pharmacokinetic studies in this model organism, when up- or down-regulation of DME expression due to drug treatment or genetic manipulation has a bearing on results. Moreover, this approach could provide an additional measure of the DDI potential of a candidate therapeutic compound during preclinical development. As modulation of DMEs typically requires upstream activation of one or more NHR by direct binding to chemical agents, species differences in drug/receptor affinity mean that caution must be reserved in drawing conclusions from studies in model organisms. Conceivably, tHR/SIM-in vivo SILAC could be used to measure DDI potential in NHR-humanized mice, such as the Car/Pxr double humanized line,51 constituting a more-accurately modeled in vivo approach and thereby reducing the gap between preclinical and clinical evaluation of DDI.
Acknowledgments
The authors would like to thank Dr. Sheila Sharp at Biomarker and Drug Analysis Core Facility for assistance with LC-MS/MS analysis and Mrs. Julia Carr for animal work. This study was supported by Cancer Research UK, Programme Grant C4639/A12330.
Glossary
Abbreviations
- ADH/Adh
alcohol dehydrogenase
- ALDH/Aldh
aldehyde dehydrogenase
- AKR/Akr
aldo-keto reductase
- AQUA
absolute quantification
- Car
constitutive androstane receptor
- CYP
Cyp cytochrome P450
- DDI
drug–drug interaction
- DME
drug metabolizing enzyme
- DTT
dithiothreitol
- EC50
effective concentration 50
- EPHX/Ephx
epoxide hydrolase
- FASP
filter-aided sample preparation
- FDR
false discovery rate
- FMO/Fmo
flavin-containing monooxygenase
- GST/Gst
glutathione S-transferase
- Nat
N-acetyl transferase
- NHR
nuclear hormone receptor
- SD
standard deviation
- SILAC
stable isotope labeling by amino acids in cell culture
- SILAM
stable isotope labeling in mammals
- Sult
sulfotransferase
- TCPOBOP
1,4-bis-[2-(3,5-dichloropyridyloxy)]benzene, 3,3′,5,5′-tetrachloro-1,4-bis(pyridyloxy)benzene
- tHR/SIM
targeted high resolution single ion monitoring
- UDP
uridine diphosphate
- UGT/Ugt
uridine diphosphate-glucuronosyltransferase
Supporting Information Available
Distribution of the number of peptides in each DME protein; image of Coomassie Brilliant Blue R-250 staining to confirm microsomal protein sample loading; effect of TCPOBOP treatment on the expression of albumin, calrecutin, α-tubulin, and β-tubulin in liver. Tables containing additional data: Peptides/proteins identified in 13C6 liver lysate following in-gel digestion protocol; peptides/proteins identified in 13C6 liver lysate following FASP protocol; summary of DME identified by in-gel digestion versus FASP derived from Tables S-1 and S-2; analysis of linearity of drug metabolism enzyme DME region 1 peptides; Analysis of linearity of drug metabolism enzyme DME region 2 peptides; DME region 1 seed file; DME region 2 seed file. This material is available free of charge via the Internet at http://pubs.acs.org. Raw data are available in ProteomeExchange.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf.
- http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf.
- Evans W. E.; Relling M. V. Pharmacogenomics: translating functional genomics into rational therapeutics. Science 1999, 2865439487–91. [DOI] [PubMed] [Google Scholar]
- Cox J.; Mann M. Is proteomics the new genomics?. Cell 2007, 1303395–8. [DOI] [PubMed] [Google Scholar]
- Huber M.; Bahr I.; Kratzschmar J. R.; Becker A.; Muller E. C.; Donner P.; Pohlenz H. D.; Schneider M. R.; Sommer A. Comparison of proteomic and genomic analyses of the human breast cancer cell line T47D and the antiestrogen-resistant derivative T47D-r. Mol. Cell. Proteomics 2004, 3143–55. [DOI] [PubMed] [Google Scholar]
- Pascal L. E.; True L. D.; Campbell D. S.; Deutsch E. W.; Risk M.; Coleman I. M.; Eichner L. J.; Nelson P. S.; Liu A. Y. Correlation of mRNA and protein levels: cell type-specific gene expression of cluster designation antigens in the prostate. BMC Genomics 2008, 9, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuki S.; Schaefer O.; Kawakami H.; Inoue T.; Liehner S.; Saito A.; Ishiguro N.; Kishimoto W.; Ludwig-Schwellinger E.; Ebner T.; Terasaki T. Simultaneous absolute protein quantification of transporters, cytochromes P450, and UDP-glucuronosyltransferases as a novel approach for the characterization of individual human liver: comparison with mRNA levels and activities. Drug Metab. Dispos. 2011, 40183–92. [DOI] [PubMed] [Google Scholar]
- Kawakami H.; Ohtsuki S.; Kamiie J.; Suzuki T.; Abe T.; Terasaki T. Simultaneous absolute quantification of 11 cytochrome P450 isoforms in human liver microsomes by liquid chromatography tandem mass spectrometry with in silico target peptide selection. J. Pharm. Sci. 2010, 1001341–52. [DOI] [PubMed] [Google Scholar]
- Langenfeld E.; Zanger U. M.; Jung K.; Meyer H. E.; Marcus K. Mass spectrometry-based absolute quantification of microsomal cytochrome P450 2D6 in human liver. Proteomics 2009, 992313–23. [DOI] [PubMed] [Google Scholar]
- Li N.; Nemirovskiy O. V.; Zhang Y.; Yuan H.; Mo J.; Ji C.; Zhang B.; Brayman T. G.; Lepsy C.; Heath T. G.; Lai Y. Absolute quantification of multidrug resistance-associated protein 2 (MRP2/ABCC2) using liquid chromatography tandem mass spectrometry. Anal. Biochem. 2008, 3802211–22. [DOI] [PubMed] [Google Scholar]
- Li N.; Palandra J.; Nemirovskiy O. V.; Lai Y. LC-MS/MS mediated absolute quantification and comparison of bile salt export pump and breast cancer resistance protein in livers and hepatocytes across species. Anal. Chem. 2009, 8162251–9. [DOI] [PubMed] [Google Scholar]
- Seibert C.; Davidson B. R.; Fuller B. J.; Patterson L. H.; Griffiths W. J.; Wang Y. Multiple-approaches to the identification and quantification of cytochromes P450 in human liver tissue by mass spectrometry. J. Proteome Res. 2009, 841672–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson B. L.; Purkayastha S.; Hunter C. L.; Nuwaysir L.; Hill J.; Easterwood L. Quantitative protein determination for CYP induction via LC-MS/MS. Proteomics 2010, 11133–41. [DOI] [PubMed] [Google Scholar]
- Gerber S. A.; Rush J.; Stemman O.; Kirschner M. W.; Gygi S. P. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. U.S.A. 2003, 100126940–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun V.; Dupuis A.; Adrait A.; Marcellin M.; Thomas D.; Court M.; Vandenesch F.; Garin J. Isotope-labeled protein standards: toward absolute quantitative proteomics. Mol. Cell. Proteomics 2007, 6122139–49. [DOI] [PubMed] [Google Scholar]
- Hanke S.; Besir H.; Oesterhelt D.; Mann M. Absolute SILAC for accurate quantitation of proteins in complex mixtures down to the attomole level. J. Proteome Res. 2008, 731118–30. [DOI] [PubMed] [Google Scholar]
- Ong S. E.; Blagoev B.; Kratchmarova I.; Kristensen D. B.; Steen H.; Pandey A.; Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 2002, 15376–86. [DOI] [PubMed] [Google Scholar]
- Kruger M.; Moser M.; Ussar S.; Thievessen I.; Luber C. A.; Forner F.; Schmidt S.; Zanivan S.; Fassler R.; Mann M. SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell 2008, 1342353–64. [DOI] [PubMed] [Google Scholar]
- McClatchy D. B.; Liao L.; Park S. K.; Xu T.; Lu B.; Yates Iii J. R. Differential proteomic analysis of mammalian tissues using SILAM. PLoS One 2011, 61e16039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther D. M.; Mann M. Accurate quantification of more than 4000 mouse tissue proteins reveals minimal proteome changes during aging. Mol. Cell. Proteomics 2011, 102M110 004523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanivan S.; Meves A.; Behrendt K.; Schoof E. M.; Neilson L. J.; Cox J.; Tang H. R.; Kalna G.; van Ree J. H.; van Deursen J. M.; Trempus C. S.; Machesky L. M.; Linding R.; Wickstrom S. A.; Fassler R.; Mann M. In vivo SILAC-based proteomics reveals phosphoproteome changes during mouse skin carcinogenesis. Cell Rep. 2013, 32552–66. [DOI] [PubMed] [Google Scholar]
- Mann M.; Kelleher N. L. Precision proteomics: the case for high resolution and high mass accuracy. Proc. Natl. Acad. Sci. U.S.A. 2008, 1054718132–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J. T.; McKenna T.; Hughes C.; Leweke F. M.; Schwarz E.; Bahn S. CSF biomarker discovery using label-free nano-LC-MS based proteomic profiling: technical aspects. J. Sep. Sci. 2007, 302214–25. [DOI] [PubMed] [Google Scholar]
- Shevchenko A.; Tomas H.; Havlis J.; Olsen J. V.; Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 2006, 162856–60. [DOI] [PubMed] [Google Scholar]
- Meehan R. R.; Forrester L. M.; Stevenson K.; Hastie N. D.; Buchmann A.; Kunz H. W.; Wolf C. R. Regulation of phenobarbital-inducible cytochrome P-450s in rat and mouse liver following dexamethasone administration and hypophysectomy. Biochem. J. 1988, 2543789–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrester L. M.; Henderson C. J.; Glancey M. J.; Back D. J.; Park B. K.; Ball S. E.; Kitteringham N. R.; McLaren A. W.; Miles J. S.; Skett P.; et al. Relative expression of cytochrome P450 isoenzymes in human liver and association with the metabolism of drugs and xenobiotics. Biochem. J. 1992, 281Pt 2359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R. D.; McLaughlin L. A.; Ronseaux S.; Rosewell I.; Houston J. B.; Henderson C. J.; Wolf C. R. Defining the in Vivo Role for cytochrome b5 in cytochrome P450 function through the conditional hepatic deletion of microsomal cytochrome b5. J. Biol. Chem. 2008, 2834631385–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J. V.; de Godoy L. M.; Li G.; Macek B.; Mortensen P.; Pesch R.; Makarov A.; Lange O.; Horning S.; Mann M. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics 2005, 4122010–21. [DOI] [PubMed] [Google Scholar]
- Poland A.; Mak I.; Glover E.; Boatman R. J.; Ebetino F. H.; Kende A. S. 1,4-Bis[2-(3,5-dichloropyridyloxy)]benzene, a potent phenobarbital-like inducer of microsomal monooxygenase activity. Mol. Pharmacol. 1980, 183571–80. [PubMed] [Google Scholar]
- Tzameli I.; Pissios P.; Schuetz E. G.; Moore D. D. The xenobiotic compound 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene is an agonist ligand for the nuclear receptor CAR. Mol. Cell. Biol. 2000, 2092951–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei P.; Zhang J.; Egan-Hafley M.; Liang S.; Moore D. D. The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature 2000, 4076806920–3. [DOI] [PubMed] [Google Scholar]
- Ledda-Columbano G. M.; Pibiri M.; Loi R.; Perra A.; Shinozuka H.; Columbano A. Early increase in cyclin-D1 expression and accelerated entry of mouse hepatocytes into S phase after administration of the mitogen 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene. Am. J. Pathol. 2000, 156191–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragani T. A.; Manenti G.; Galliani G.; Della Porta G. Promoting effects of 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene in mouse hepatocarcinogenesis. Carcinogenesis 1985, 62225–8. [DOI] [PubMed] [Google Scholar]
- Diwan B. A.; Lubet R. A.; Ward J. M.; Hrabie J. A.; Rice J. M. Tumor-promoting and hepatocarcinogenic effects of 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP) in DBA/2NCr and C57BL/6NCr mice and an apparent promoting effect on nasal cavity tumors but not on hepatocellular tumors in F344/NCr rats initiated with N-nitrosodiethylamine. Carcinogenesis 1992, 13101893–901. [DOI] [PubMed] [Google Scholar]
- Honkakoski P.; Moore R.; Gynther J.; Negishi M. Characterization of phenobarbital-inducible mouse Cyp2b10 gene transcription in primary hepatocytes. J. Biol. Chem. 1996, 271169746–53. [DOI] [PubMed] [Google Scholar]
- Maglich J. M.; Stoltz C. M.; Goodwin B.; Hawkins-Brown D.; Moore J. T.; Kliewer S. A. Nuclear pregnane x receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Mol. Pharmacol. 2002, 623638–46. [DOI] [PubMed] [Google Scholar]
- Locker J.; Tian J.; Carver R.; Concas D.; Cossu C.; Ledda-Columbano G. M.; Columbano A. A common set of immediate-early response genes in liver regeneration and hyperplasia. Hepatology 2003, 382314–25. [DOI] [PubMed] [Google Scholar]
- Hernandez J. P.; Mota L. C.; Huang W.; Moore D. D.; Baldwin W. S. Sexually dimorphic regulation and induction of P450s by the constitutive androstane receptor (CAR). Toxicology 2009, 2561–253–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tojima H.; Kakizaki S.; Yamazaki Y.; Takizawa D.; Horiguchi N.; Sato K.; Mori M. Ligand dependent hepatic gene expression profiles of nuclear receptors CAR and PXR. Toxicol. Lett. 2012, 2123288–97. [DOI] [PubMed] [Google Scholar]
- Smith G.; Henderson C. J.; Parker M. G.; White R.; Bars R. G.; Wolf C. R. 1,4-Bis[2-(3,5-dichloropyridyloxy)]benzene, an extremely potent modulator of mouse hepatic cytochrome P-450 gene expression. Biochem. J. 1993, 289Pt 3807–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane C. S.; Wang Y.; Betts R.; Griffiths W. J.; Patterson L. H. Comparative cytochrome P450 proteomics in the livers of immunodeficient mice using 18O stable isotope labeling. Mol. Cell. Proteomics 2007, 66953–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M. J.; Takahashi Y.; Wada T.; He J.; Gao J.; Tian Y.; Li S.; Xie W. The aldo-keto reductase Akr1b7 gene is a common transcriptional target of xenobiotic receptors pregnane X receptor and constitutive androstane receptor. Mol. Pharmacol. 2009, 763604–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alnouti Y.; Klaassen C. D. Tissue distribution, ontogeny, and regulation of aldehyde dehydrogenase (Aldh) enzymes mRNA by prototypical microsomal enzyme inducers in mice. Toxicol. Sci. 2008, 101151–64. [DOI] [PubMed] [Google Scholar]
- Aleksunes L. M.; Klaassen C. D. Coordinated regulation of hepatic phase I and II drug-metabolizing genes and transporters using AhR-, CAR-, PXR-, PPARalpha-, and Nrf2-null mice. Drug Metab. Dispos. 2012, 4071366–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld J. M.; Vargas R. Jr.; Xie W.; Evans R. M. Genetic profiling defines the xenobiotic gene network controlled by the nuclear receptor pregnane X receptor. Mol. Endocrinol. 2003, 1771268–82. [DOI] [PubMed] [Google Scholar]
- Knight T. R.; Choudhuri S.; Klaassen C. D. Induction of hepatic glutathione S-transferases in male mice by prototypes of various classes of microsomal enzyme inducers. Toxicol. Sci. 2008, 1062329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley D. B.; Klaassen C. D. Induction of mouse UDP-glucuronosyltransferase mRNA expression in liver and intestine by activators of aryl-hydrocarbon receptor, constitutive androstane receptor, pregnane X receptor, peroxisome proliferator-activated receptor alpha, and nuclear factor erythroid 2-related factor 2. Drug Metab. Dispos. 2009, 374847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet L. C.; Navarro P.; Tate S.; Rost H.; Selevsek N.; Reiter L.; Bonner R.; Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 2012, 116O111 016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin Y.; Hradetzky E.; Bahn S. Quantification of proteins using data-independent analysis (MSE) in simple andcomplex samples: a systematic evaluation. Proteomics 2011, 11163273–87. [DOI] [PubMed] [Google Scholar]
- Gallien S.; Duriez E.; Crone C.; Kellmann M.; Moehring T.; Domon B. Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol. Cell. Proteomics 2012, 11121709–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheer N.; Ross J.; Rode A.; Zevnik B.; Niehaves S.; Faust N.; Wolf C. R. A novel panel of mouse models to evaluate the role of human pregnane X receptor and constitutive androstane receptor in drug response. J. Clin. Invest. 2008, 11893228–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.