

Abstract
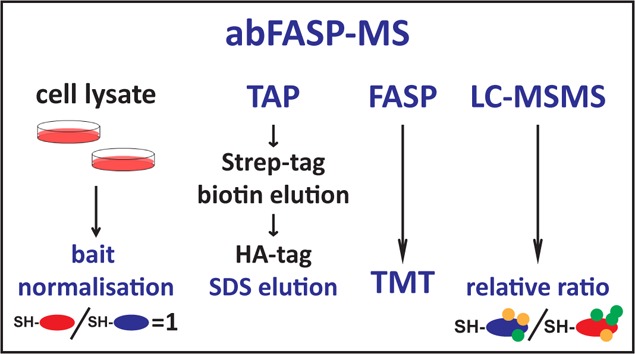
Affinity purification coupled to 1-D gel-free liquid chromatography mass spectrometry (LC–MS) is a well-established and widespread approach for the analyses of noncovalently interacting protein complexes. In this study, two proteins conjugated to a streptavidin-binding peptide and hemagglutinin double tag were expressed in the respective Flp-In HEK293 cell lines: green fluorescent protein (SH-GFP) and TANK binding kinase 1 (SH-TBK1_MOUSE). Fluorescent anti-HA immunoblots revealed that the expression level of SH-GFP was ∼50% lower than that of SH-TBK1_MOUSE. Subsequently, the input material was normalized to obtain a similar quantity of purified SH-tagged proteins. Optimization of the release of protein complexes from the anti-HA-agarose with different eluting agents was then assessed. With respect to the total number of protein groups identified in the purified complexes, elution with 2% SDS surpassed both 100 mM glycine and 100 mM formic acid. Relative quantitation of the purified protein complexes using TMT 6-plex reagents confirmed the higher efficiency of the 2% SDS elution followed by filter-aided sample preparation (FASP). The data presented in this study provide a new application of FASP to quantitative MS analysis of affinity-purified protein complexes. We have termed the approach abFASP-MS, or affinity-based filter-aided sample preparation mass spectrometry.
Keywords: affinity purification, orbitrap velos, SDS elution, normalization, bait expression, FASP, quantitation, TMT 6-plex
Introduction
Large-scale affinity purifications (APs) coupled to liquid chromatography mass spectrometry (LC–MS) have gained considerable momentum in the investigation of near-physiological protein complexes. Studies have ranged from the investigation of yeast,1,2 autophagy,3 immune modulators,4,5 kinases,6 to viral-human interactions.7,8 Thus, the broad applicability and power of such approaches has been firmly and undoubtedly established. Although outside the scope of these particular studies, interestingly, no quantitative approaches such as SILAC or chemical labeling were utilized in these large-scale investigations. Naturally, any study on protein complexes can only be further enhanced by understanding the quantitative nature of the protein complex formation. On a much smaller scale, it would be very important to be able to determine changes in protein quantities within a single complex after for example, treatment with either a stimulant or perturbant or between mutant versions of the same protein. It is imperative that there is a high degree of experimental normalization and standardization of the entire biochemical and analytical procedure to achieve this goal.
An inducible expression system in Flp-In HEK293 cells is routinely used in our laboratory to purify naturally folded, noncovalently interacting protein complexes.9 In our hands, the approach has proven to be robust and efficient for the production of recombinant proteins (baits) for tandem affinity purification (TAP) and qualitative LC–MS analysis of purified protein complexes.6,7,10,11 Two cell lines were used throughout the study. One expressing green fluorescent protein N-terminally tagged with streptavidin-binding peptide and hemagglutinin (SH-GFP) and the other expressing the similarly tagged mouse TANK binding kinase 1 (SH-TBK1_MOUSE). Because GFP is absent in human cells, any potential interaction established with human proteins is due to nonspecific physicochemical properties rather than true physiological interactions. Recombinant SH-GFP is thus used as a control for TAP-MS experiments to determine the proteins that nonspecifically bind to the 3-D structure of GFP, the SH-tag, and/or the sepharose beads used throughout the protein purification procedure. TBK1 plays an essential role in controlling the innate immune response, cell proliferation, and oncogenesis and is one of the central proteins that leads to induction of type-I interferon (IFN) in response to pathogens.12,13 TAP-MS experiments4,13−15 have identified TBK1 and three primary core complex adaptor components: TRAF family member associated NF-κB activator (TANK); TBK binding protein 1 (TBKBP1 or SINTBAD); and AZI2 (NAP1 or TBKBP2).
Experience has shown that HEK293 Flp-In cells express SH-tagged proteins at variable levels. Subsequently, it is quite challenging to directly qualitatively or quantitatively compare the TAP-MS data from different proteins or even the same protein under several conditions. Thus, to compensate for these difficulties, it is important to normalize to the expression level of the SH-tagged protein to obtain comparable results subsequent to the biochemical purification steps and the analytical analysis by LC–MS. Additionally, elution of the proteins in the second step of the TAP is sometimes less efficient compared with the first step of the purification. Often this reduced efficiency is due to the physicochemical properties of the bait; however, other contributing factors are lack of experience of the researcher performing the TAP, slight pH variations in the buffers, desiccated column beds, contamination from residual detergent in the buffers, and so on. As a consequence, such small variations throughout the procedure become compounded, thus affecting the number and nature of the proteins that are eluted. Ultimately, inefficient elution of the proteins can lead to a distorted view of the true composition of the protein complex in question. Such anomalies are further exacerbated following a quantitative experiment when incorrect assumptions are made concerning the variation in protein levels and the relative stoichiometric configuration of a complex. Thus, there is a need to find a means to elute the proteins more efficiently and consistently, thereby maximizing the information on protein complex composition and stoichiometry. This is necessary to ensure that the quantitative data generated from a chemical labeling experiment is reliable and trustworthy.
Therefore, the aims of this study were to: (i) normalize the quantity of protein input for different SH-tagged proteins with dissimilar expression or purification yield levels prior to the tandem affinity purification mass spectrometry experiment; (ii) compare alternative elution methods for the second step of the purification; and (iii) couple the most efficient elution condition to the quantitative analysis of protein complexes via chemical labeling.
Material and Methods
Materials
The following materials were used: iodoacetamide SigmaUltra, dl-dithiothreitol for molecular biology minimum 99% titration, triethylammonium bicarbonate 1 M pH 8.5 (TEAB), protease inhibitor cocktail, anti-HA agarose, sodium chloride ≥99.8% (NaCl), sodium fluoride ≥99% (NaF), sodium orthovanadate (Na3VO4), urea SigmaUltra, tris(hydroxymethyl)aminomethane (tris HCl) ultrapure grade ≥99.9%, phenylmethylsulfonyl fluoride (PMSF) (Sigma-Aldrich, St. Louis, MO); trypsin (Promega, Madison, WI); formic acid 98–100% (pro analysi) (HCOOH), 2-[4-(2-hydoxyethyl)-1-piperazinyl]ethanesulfonic acid (HEPES), ethylenediaminetetraacetic acid (EDTA), NP-40 alternative, glycine (NH2CH2COOH) (Merck, Darmstadt, Germany); StrepTactin sepharose (IBA TAGnologies, Göttingen, Germany); d-biotin (Alfa Aesar, Karlsruhe, Germany); bovine serum albumin (BSA) standard prediluted set, 6-plex TMT reagent (ThermoFisher Scientific, Waltham, MA); micro Bio-Spin chromatography columns (Bio-Rad, Hercules, CA); 30 kDa molecular weight cutoff filter (VIVACON 500) (Sartorius Stedim Biotech, Goettingen, Germany); goat antimouse IgG IRDye 800CW, Odyssey blocking buffer (LiCor, Lincoln, NE); HA.11 monoclonal antibody (Covance Research Products, Princeton, NJ); and sodium dodecyl sulfate (SDS) (SERVA Electrophoresis, Heidelberg, Germany).
Cell Cultivation and Collection
SH-TBK1_MOUSE- and SH-GFP-expressing HEK293 Flp-In cells were grown as adherent, sterile cell cultures. Cells were harvested at 70–80% confluence after 24 h of induction of recombinant protein expression with 1 μg/mL doxycycline. For each sample, cells from 5 × 15 cm diameter plates were collected in PBS, and centrifuged at 300g for 10 min, and the supernatant was removed. Pellets were snap frozen in liquid nitrogen and stored at −80 °C until required.
Affinity Purification
SH-TBK1_MOUSE- and SH-GFP-expressing HEK293 Flp-In cell pellets were lysed in freshly prepared buffer (50 mM HEPES pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, 50 mM NaF, 1.5 mM Na3VO4, 1.0 mM PMSF, and protease inhibitor cocktail) on ice for 20 min. The obtained suspension was centrifuged at 14 000g for 30 min, and the supernatant containing the protein extract was collected. Protein concentration was determined by the Bradford assay using BSA as a standard. Two-step affinity purifications were performed as previously described.15 In brief, 200 μL StrepTactin sepharose was added to a Bio-Spin column and washed with buffer. The cell lysate was applied to the column and gravity drained, and the column was washed. Bound proteins were eluted with 900 μL of freshly prepared 2.5 mM d-biotin in buffer. The biotin eluates were added to the prewashed 100 μL anti-HA agarose beads and rotated for 1 h at 4 °C. Unbound material was removed. The agarose beads were loaded into a fresh Bio-Spin column and washed with 3 × 1 mL buffer. The beads were then washed with 2 × 1 mL buffer containing only 50 mM HEPES pH 8.0, 150 mM NaCl, and 5 mM EDTA to remove NP-40. For the elution under acidic condition, bound proteins were eluted from the column directly into a glass HPLC vial with 500 μL of 100 mM formic acid or 500 μL of 100 mM glycine and immediately neutralized with 125 μL of 1 M TEAB.15 Two hundred microliters were removed for SDS-PAGE followed by silver stain visualization of the proteins and/or immunoblot analysis. For the elution with SDS, the anti-HA agarose was washed as described above. To elute bound proteins, the beads were incubated for 20 min at room temperature in 150 μL of buffer containing 50 mM HEPES, pH 8.0, 150 mM NaCl, 5 mM EDTA, and 2% SDS (referred to throughout as 2% SDS buffer or 2% SDS elution). Fifty microliters were removed for SDS-PAGE followed by silver stain visualization of the proteins and/or immunoblot analysis. The remaining samples were frozen at −20 °C until further processing.
Gel Electrophoresis and Immunoblotting
Aliquots of the TAP protein eluates were denatured in Laemmli sample buffer (1×) by boiling at 95 °C for 5 min, and the proteins were separated by SDS-PAGE. Proteins were transferred to a nitrocellulose membrane by electrophoresis, and nonspecific binding sites were blocked with Odyssey blocking buffer. To detect the SH-tagged proteins, we incubated the membranes with a primary mouse anti-HA-tag antibody (HA.11) (1:3000) and then with a secondary goat antimouse antibody (1:15 000) that emits green fluorescence at an excitation wavelength of 800 nm. The intensity of band fluorescence on the immunoblot was measured and quantitated using the LiCor Odyssey VIS spectrophotometer software. During calculation, the nonfluorescent blot background was set to zero.
Tryptic Digestion and Sample Preparation for LC–MS/MS Analysis
TEAB-neutralized formic acid and glycine protein eluates were reduced with dithiothreitol, alkylated with iodoacetamide, and digested with trypsin.15 Proteins eluted with buffer containing 2% SDS were digested according to the FASP protocol16,17 using a 30 kDa molecular weight cutoff filter. In brief, 100 μL of each protein eluate was reduced with 20 μL of 500 mM DTT and incubation for 5 min at 99 °C. After cooling to RT, the samples were mixed in the filter unit with 8 M urea in 100 mM Tris HCl (pH 8.5) (UA) and centrifuged at 14 000g for 15 min at 20 °C to remove SDS. Any remaining SDS was exchanged by urea with 200 μL of UA. The proteins were alkylated by addition of 100 μL of 50 mM iodoacetamide in UA and incubation for 30 min at RT. Afterward, three washing steps with 100 μL of UA solution were performed, followed by three washing steps with 100 μL of 50 mM TEAB buffer. Proteins were digested with trypsin overnight at 37 °C. Peptides were recovered from the filter using 40 μL of 50 mM TEAB buffer followed by 50 μL of 0.5 M NaCl. Fifteen percent of the digest volume was desalted and concentrated with customized reversed-phase stage tips.18 The volume of the eluted sample was reduced to ∼2 μL in a vacuum centrifuge and reconstituted to 24 μL with 5% formic acid.
Tandem Mass Tag Derivatization and Reversed-Phase Peptide Fractionation
For the TMT labeling experiment, the tryptically digested samples were derivatized with 6-plex TMT reagent according to the instructions provided by the manufacturer. The tryptic peptides from the tandem affinity purified protein complexes of SH-GFP and SH-TBK1_MOUSE eluted with 100 mM formic acid, 100 mM glycine, and 2% SDS were labeled with TMT 126, 127, and 128 for GFP and 129, 130, and 131 for TBK1, respectively. All six samples were pooled, the peptides were separated by reversed-phase liquid chromatography, pH 10,19 and 20 fractions were collected. Acidified fractions were analyzed by LC–MS as technical duplicates. Details of the procedure are essentially as previously described.20
Reversed-Phase Liquid Chromatography Mass Spectrometry
Mass spectrometry was performed on a hybrid linear trap quadrupole (LTQ) Orbitrap Velos mass spectrometer (ThermoFisher Scientific, Waltham, MA) using the Xcalibur version 2.1.0 coupled to an Agilent 1200 HPLC nanoflow system (dual pump system with one precolumn and one analytical column) (Agilent Biotechnologies, Palo Alto, CA) via a nanoelectrospray ion source using liquid junction (Proxeon, Odense, Denmark). Solvents for LC–MS separation of the digested samples were as follows: solvent A consisted of 0.4% formic acid in water and solvent B consisted of 0.4% formic acid in 70% methanol and 20% isopropanol. From a thermostatic microautosampler, 8 μL of the tryptic peptide mixture were automatically loaded onto a trap column (Zorbax 300SB-C18 5 μm, 5 × 0.3 mm, Agilent Biotechnologies) with a binary pump at a flow rate of 45 μL/min. 0.1% TFA was used for loading and washing the precolumn. After washing, the peptides were eluted by back-flushing onto a 16 cm fused silica analytical column with an inner diameter of 50 μm packed with C18 reversed phase material (ReproSil-Pur 120 C18-AQ, 3 μm, Dr. Maisch, Ammerbuch-Entringen, Germany). The peptides were eluted from the analytical column with a 27 min gradient ranging from 3 to 30% solvent B, followed by a 25 min gradient from 30 to 70% solvent B and, finally, a 7 min gradient from 70 to 100% solvent B at a constant flow rate of 100 nL/min.
The analyses were performed in a data-dependent acquisition mode using a top 15 collision-induced dissociation (CID) method for peptide identification alone or a top-10 high-energy collision-induced dissociation (HCD) method for peptide identification plus relative quantitation of TMT reporter ions. Dynamic exclusion for selected ions was 60 s. A single lock mass at m/z 445.120024 was employed.21 The maximal ion accumulation time allowed for MS mode in the orbitrap was 500 ms. For CID and HCD, the accumulation times were 50 and 200 ms, respectively. Automatic gain control (AGC) was used to prevent overfilling of the ion traps. In MS mode, AGC was set to 106 ions, and in MS2 mode, AGC was set to 5000 and 105 ions for CID and HCD, respectively. For CID analyses, peptides were detected in MS mode at 60 000 (at m/z 400); and for HCD, peptides were detected in MS and MS2 mode at 30 000 (at m/z 400) and 7500 resolution, respectively. The threshold for switching from MS to MS2 was 2000 counts. All samples were analyzed as technical, back-to-back replicates.
Data Analysis
The acquired raw MS data files were processed with msconvert (ProteoWizard Library v2.1.2708) and converted into Mascot generic format (mgf) files. The resultant peak lists were searched against the human SwissProt database version v2013.01_20130110 (37 398 sequences including isoforms obtained from varsplic.pl and appended with SH-tagged-TBK1_MOUSE and other known contaminants) with the search engines Mascot (v2.3.02, MatrixScience, London, U.K.) and Phenyx (v2.5.14, GeneBio, Geneva, Switzerland).22 Submission to the search engines was via a Perl script that performs an initial search with relatively broad mass tolerances (Mascot only) on both the precursor and fragment ions (±10 ppm and ±0.6 Da, respectively). High-confidence peptide identifications were used to recalibrate all precursor and fragment ion masses prior to a second search with narrower mass tolerances (CID, ± 4 ppm and ±0.3 Da; HCD, ± 4 ppm and ±0.025 Da). One missed tryptic cleavage site was allowed. Carbamidomethyl cysteine and TMT 6-plex (N-terminii and lysine) were set as fixed modifications, and oxidized methionine was set as a variable modification. To validate the proteins, we processed Mascot and Phenyx output files by internally developed parsers. Proteins with ≥2 unique peptides above a score T1 or with a single peptide above a score T2 were selected as unambiguous identifications. Additional peptides for these validated proteins with score >T3 were also accepted. For Mascot and Phenyx, T1, T2, and T3 peptide scores were equal to 16, 40, 10 and 5.5, 9.5, 3.5, respectively (P value <10–3). The validated proteins retrieved by the two algorithms were merged, and any spectral conflicts were discarded and grouped according to shared peptides. A false positive detection rate (FDR) of <1 and <0.1% (including the peptides exported with lower scores) was determined for proteins and peptides, respectively, by applying the same procedure against a reversed database. Comparisons between analytical methods involved comparisons between the corresponding sets of identified proteins. This was achieved by an internally developed program that simultaneously computes the protein groups in all samples and extracts statistical data such as the number of distinct peptides, number of spectra, and sequence coverage.
Tandem Mass Tag Quantitation
The quantitation module of Proteome Discoverer 1.4, version 1.4.0.288 (ThermoFisher Scientific, Waltham, MA) was used to assess the ratios for the individually tagged TAP samples. The intensity of the TMT 6-plex reporter ions was integrated using the default settings for centroid peak detection at the highest confidence and a mass tolerance of 20 ppm. Correction for isotopic impurities was not performed. In addition, spectra with reporter ion intensities below 100 counts and spectra with coisolation of contaminating peptides exceeding 40% of the selected precursor ion were excluded from the protein ratio calculations. The median ratios for all peptides were calculated relative to the 126 channel (127–128/126) or to the 129 channel (130–131/129). Ratios 129/126, 130/127, and 131/128 were also calculated depending on the sample comparison required. Shared peptides were excluded from quantitation. Protein ratios for the two combined technical replicates were calculated using the arithmetic mean of the protein ratios (median ratio of all used peptide ratios) for each replicate.
Results and Discussion
Normalization of Protein Input for Affinity Purification to the Level of SH-Tagged Protein Expression
To compare the expression levels of SH-GFP and SH-TBK1_MOUSE in HEK293 Flp-In cells, we lysed pellets from 5 × 15 cm diameter plates according to the procedure outlined in the Materials and Methods. Following a protein assay, 20 μg of the protein extracts were separated by SDS-PAGE and immunoblotted using a primary mouse HA.11 antibody and a secondary fluorescent antimouse antibody (Figure 1). Blots were analyzed at an excitation wavelength of 800 nm, and the fluorescence intensity of the SH-GFP bands was estimated relative to the SH-TBK1_MOUSE at 100%. Despite the same protein input, the difference in the fluorescence intensity of SH-GFP and SH-TBK1 revealed that the expression level of SH-GFP was approximately 45–50% lower compared with SH-TBK1_MOUSE. Taking this into consideration, the next step was to normalize the starting input material such that comparable quantities of the SH-tagged proteins were used and subsequently comparable quantities of the tagged proteins were retrieved following the TAP procedure.
Figure 1.
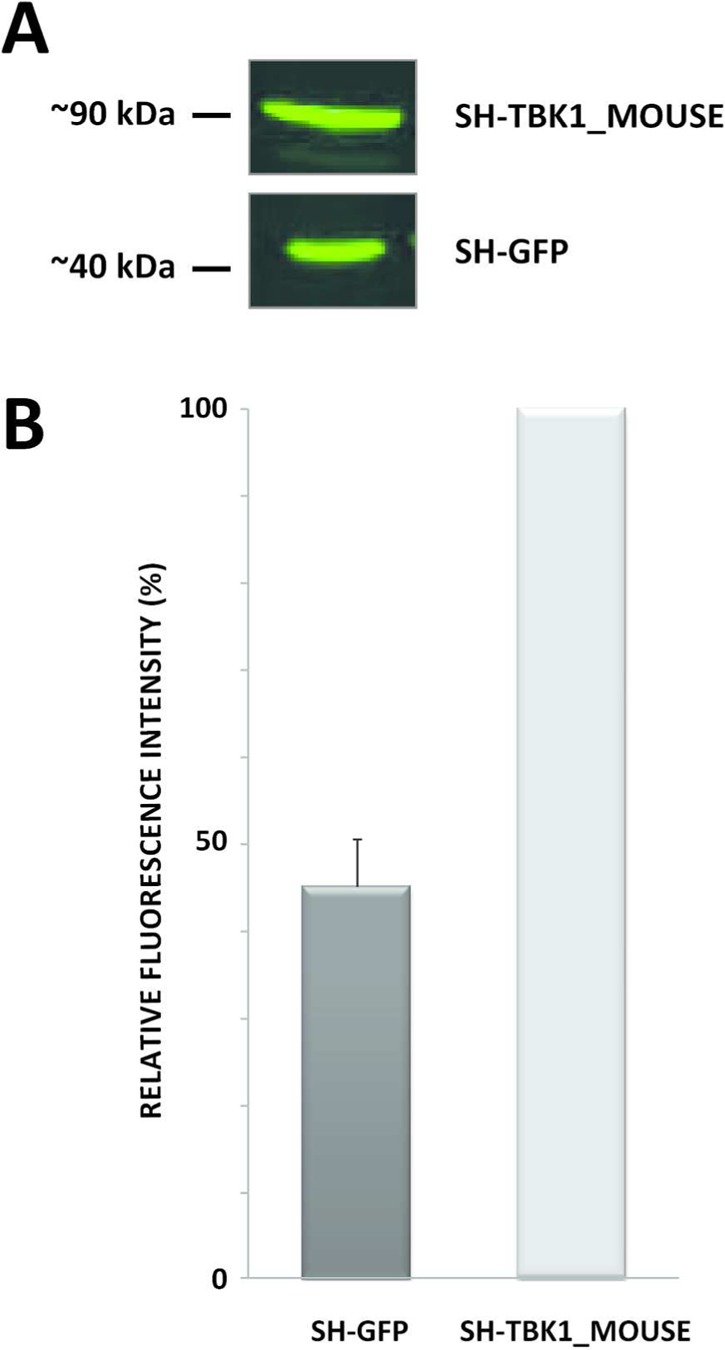
Comparison of the amount of SH-GFP and SH-TBK1_MOUSE in the cell lysates of the respective HEK293 Flp-In cell lines. Twenty micrograms of total protein from each cell lysate were separated by SDS-PAGE. SH-proteins were visualized and quantitated by immunoblot performed with mouse HA.11 antibody (1:3000) followed by goat antimouse antibody (800 nm) (1:15 000). Experiments were performed as biological triplicates. (A) Representative immunoblot of SH-GFP and SH-TBK1_MOUSE in the cell lysates; (B) Relative quantitation of the SH-GFP and SH-TBK1_MOUSE bands from the triplicate immunoblot analyses. The fluorescence intensity of the SH-TBK1_MOUSE band was set to 100%. SH, streptavidin hemeagglutinin; GFP, green fluorescent protein; TBK1, TANK binding kinase 1; HA, hemeagglutinin; SDS-PAGE, sodium dodecylsulphate polyacrylamide gel electrophoresis.
An unequal expression level of different SH-tagged proteins in the HEK293 Flp-In cells resulted in a dissimilar relative abundance of the recombinant protein in the total protein extract from the lysed cells. To assess the purification yield obtained after the two-step affinity purification using different levels of the SH-tagged expressed protein, we investigated different total protein inputs of the SH-GFP and SH-TBK1_MOUSE protein extracts. Two-step affinity purifications were performed with 10, 20, and 40 mg of protein extract. Because of the lower expression of GFP compared with TBK1, a 60 mg total protein for SH-GFP was also included. The anti-HA immunoblot of the eluates from the second step of the affinity purification is shown in Figure 2A. Fluorescent bands of SH-GFP (lanes 4–7) and SH-TBK1_MOUSE (lanes 1–3) in the corresponding eluates are given. The fluorescence intensities of the SH-proteins in the purified products are compared on the same immunoblot and calculated as a percentage of the intensity of the SH-TBK1_MOUSE band (lane 3, set to 100%) detected in the purified product from 40 μg total cell lysate (Figure 2B). Evaluation of the immunoblot data revealed that the fluorescence intensity of the SH-TBK1_MOUSE band (i.e., the purified product from 40 mg protein input) was comparable to that of the SH-GFP band (i.e., the purified product from 60 mg protein input) (Figure 2B). Thus, all further experiments were conducted using 60 and 40 mg total protein input of lysates from SH-GFP- and SH-TBK1_MOUSE-expressing cells, respectively.
Figure 2.

Comparison of the eluates from the anti-HA agarose beads for SH-GFP and SH-TBK1_MOUSE proteins performed from different quantities of total protein input. Proteins were eluted from the anti-HA-agarose with 100 mM formic acid. One percent (v/v) from each eluate was separated by SDS-PAGE. SH-proteins were visualized and quantitated by immunoblot performed with mouse HA.11 antibody (1:3000) followed by goat antimouse antibody (800 nm) (1:15 000). (A) Representative immunoblot of SH-GFP and SH-TBK1 in the eluates after AP. Product of the purification from 10, 20, and 40 mg SH-TBK1_MOUSE cell lysate (lanes 1–3, 6 μL eluate loaded). Product of the purification from 10, 20, 40, and 60 mg SH-GFP cell lysate (lanes 4–7, 6 μL eluate loaded). (B) Fluorescence intensities for SH-GFP and SH-TBK1_MOUSE relative to the intensity of the band from the SH-TBK1_MOUSE purified product from 40 mg input set to 100%. Protein inputs are: (blue square) 10 mg; (red diamond) 20 mg; (green triangle) 40 mg; and (purple circle) 60 mg. Fluorescence intensity comparable to the intensity of SH-TBK1 from 40 mg protein input was obtained for SH-GFP from 60 mg protein input. AP, affinity purification; SH, streptavidin hemeagluttinin; GFP, green fluorescent protein; TBK1, TANK binding kinase 1; HA, hemeagluttinin; TAP, tandem affinity purification.
Comparative Elution Methods for Two-Step Affinity Purifications
Once the level of bait expression had been normalized and the minimum input of protein material for SH-GFP and SH-TBK1_MOUSE had been ascertained, the next step was to compare three alternative approaches to release the protein complex(es) from the anti-HA agarose beads after the second step of the purification. The aim was to determine the most efficient and specific elution method that could then be coupled to relative protein quantitation using chemical labels such as iTRAQ or TMT. In addition to the standard 100 mM formic acid elution routinely used in our laboratory;15 elution with 100 mM glycine6,9 and with 2% SDS was also assessed. For SH-TBK1_MOUSE, an anti-HA immunoblot was performed on the second step eluates (Figure 3A). Comparison between the three elution methods showed that SDS (lane 4) had a higher elution efficiency than formic acid (lane 2) and glycine (lane 3). The whole cell extract (100 μg) is also given in lane 1.
Figure 3.

(A) Fluorescent anti-HA immunoblot of the second step eluates from the anti-HA agarose beads for SH-TBK1_MOUSE. Elutions were performed with 100 mM formic acid (lane 2), 100 mM glycine (lane 3), and 2% SDS (lane 4). 100 μg whole cell extract (lane 1); lanes 2–4, 1% v/v loaded. Immunoblotting was performed with mouse HA.11 antibody (1:3000) followed by goat antimouse antibody (800 nm) (1:15 000). (B) Total number of protein groups identified for the SH-GFP and SH-TBK1_MOUSE purified protein complexes for the three elution conditions: formic acid, glycine, and 2% SDS (n = 4). (C) Average protein sequence coverage (%) for the purified protein complexes from the three elution conditions: formic acid, glycine, and 2% SDS (n = 4). Data presented are for SH-TBK1_MOUSE; the core protein complex interactors, TBK1_HUMAN, TANK_HUMAN, TBKB1_HUMAN, AZI2_HUMAN; plus the additional interactors OPTN_HUMAN, TRAF2_HUMAN, CACO2_HUMAN, and LETM1_HUMAN. GFP, green fluorescent protein; TBK1, TANK binding kinase 1; HA, hemagglutinin; SDS, sodium dodecylsulphate.
According to our previous findings,10 3% of the total affinity-purified eluate (equivalent to 5% of the digested sample) from the second step of the TAP maximizes the protein identification from a 1D gel-free LC–MS analysis on our laboratory and thus was used to evaluate the three different elution conditions. The proteins identified in all analyses are given in Supplementary Table S1 in the Supporting Information. The total number of proteins ± standard deviation (SD) that were identified for the three elution strategies are given in Figure 3B (n = 4). The data is an average of two biochemical and two technical replicates and shows that the efficiency of eluting the highest number of proteins for both SH-GFP and SH-TBK1_MOUSE cell lysates was 2% SDS > 100 mM glycine >100 mM formic acid. Note that the standard deviation for the 2% SDS-eluted SH-GFP is rather high. This was attributed to storage of the SH-GFP cell lysate for a period of time between performing the first and second purification and emphasizes the necessity of conducting simultaneous back-to-back replicates to ensure optimal reproducibility. For the three elution conditions, all proteins that were identified in the TBK1 experiments that were ≥2-fold more abundant in median spectral counts compared with the GFP control are given in Supplementary Table S2 in the Supporting Information. Summarized in Figure 3C is the average sequence coverage ± SD for the main core interactors of SH-TBK1_MOUSE plus a selection of additional interactors: OPTN, TRAF2, CACO2, and LETM1. Several points are apparent from this Figure. First, from the 2% SDS eluted sample, the average sequence coverage for SH-TBK1_MOUSE, TBK1_HUMAN, and TANK_HUMAN all decrease; although the spectral counts are approximately two to three times higher compared with the elution with either formic acid or glycine (Supplementary Table S2C in the Supporting Information). Second, the sequence coverage for TBKB1_HUMAN remains relatively similar regardless of the elution strategy. Third, and most importantly, the sequence coverage of AZI2_HUMAN increases dramatically from 23 (formic acid) to 29 (glycine) to 38% (SDS). OPTN, TRAF2, and CACO2 showed a similar trend. Finally, LETM1_HUMAN was only observed in the SDS elution. These data indicate that the efficiency of protein elution from the anti-HA agarose in the second step of the tandem affinity purification is markedly enhanced by using 2% SDS coupled to FASP and that less abundant interactors are identified more confidently. We have termed our new approach affinity-based filter-aided sample preparation mass spectrometry, or abFASP-MS.
Relative Quantitative Comparison of Elution Conditions
The final step was to quantitate the identified proteins between the three elution conditions. To achieve this goal, we conducted a TMT 6-plex experiment. As indicated in the Materials and Methods section, the tandem affinity purifications of SH-GFP and SH-TBK1_MOUSE eluted with 100 mM formic acid, 100 mM glycine, and 2% SDS were labeled with TMT 126, 127, and 128 and 129, 130, and 131, respectively. The quantitative data summarized in Table 1 (n = 2) revealed that the average ratio for SH-GFP (peptide-to-spectrum-match, PSM = 1970) eluted with glycine compared with formic acid was 1.88 (TMT 127/126). The average ratio, however, for SH-GFP eluted with 2% SDS compared with formic acid was 6.94 (TMT 128/126) (Table 1A). This finding clearly indicated that elution of SH-GFP with 2% SDS was substantially more efficient than the elution with either formic acid or glycine. The average ratio for SH-TBK1_MOUSE (PSM = 4780) eluted with glycine and 2% SDS compared with formic acid was 1.88 (TMT 130/129) and 2.28 (TMT 131/129), respectively. Co-elution of peptides with similar m/z values during an LC–MS/MS analysis of isobaric, chemically labeled peptides leads to an inherent fold change compression.23 Such compression issues are evident in our data when peptides from SH-TBK1_MOUSE and the core interacting proteins are compared with the SH-GFP data (i.e., TMT labels 129/126, 130/127, and 131/128) (Table 1B). Despite this, however, the data do show the improved elution efficiency for glycine and 2% SDS compared with formic acid. The average ratio for PRKDC (a nonspecific protein identified in both SH-GFP and SH-TBK1_MOUSE purifications; PSM = 126) was compared for the three elution conditions (Table 1C). The ratio of PRKDC between the SH-TBK1_MOUSE and SH-GFP purified products for the formic acid, glycine, and 2% SDS elutions was 1.09 (TMT 129/126), 0.94 (TMT 130/127), and 0.92 (TMT 131/128), respectively. Namely, for each elution condition for two unrelated SH-tagged proteins, similar quantities of PRKDC were eluted.
Table 1. Average Quantitative Ratios Obtained from TMT 6-Plex Labelling of SH-GFP and SH-TBK1_MOUSE Protein Complexes Eluted with 100 mM Formic Acid, 100 mM Glycine, and 2% SDS, Respectively (n = 2)a.
| A | |||
|---|---|---|---|
| 127/126 | 128/126 | PSM | |
| SH-GFP | 1.88 | 6.94 | 1970 |
| B | ||||
|---|---|---|---|---|
| 129/126 | 130/127 | 131/128 | PSM | |
| SH-TBK1_MOUSE | 3.43 | 6.00 | 10.63 | 4780 |
| TBK1 | 3.90 | 4.42 | 5.75 | 3355 |
| TANK | 3.17 | 10.35 | 10.11 | 338 |
| TBKB1 | 4.37 | 8.58 | 6.44 | 280 |
| AZI2 | 2.58 | 6.25 | 8.36 | 136 |
| OPTN | 2.44 | 4.44 | 0.60 | 135 |
| TRAF2 | 2.09 | 6.25 | 16.48 | 114 |
| CACO2 | 2.48 | 4.26 | 11.74 | 64 |
| LETM1 | 2.21 | 8.74 | 8.28 | 34 |
| C | ||||
|---|---|---|---|---|
| 129/126 | 130/127 | 131/128 | PSM | |
| PRKDC | 1.09 | 0.94 | 0.92 | 126 |
(A) SH-GFP; (B) SH-TBK1_MOUSE, core interactors plus additional interacting proteins; and (C) PRKDC, one of the contaminant proteins present in both the SH-GFP and SH-TBK1_MOUSE affinity purifications. All SH-TBK1_MOUSE AP ratios are relative to the corresponding TMT label for SH-GFP AP and the equivalent elution agent. AP, affinity purification; GFP, green fluorescent protein; TBK1, TANK binding kinase 1; DNAPK, DNA-dependent protein kinase catalytic subunit; PSM, peptide-to-spectrum matches.
An ideal consequence of the comparative nature of the quantitative experiment would be that all nonspecific proteins should show a similar trend to PRKDC. This is the case for the majority of the contaminants identified in the TMT experiment (Supplementary Table S3 in the Supporting Information). The structure and physicochemical properties offered by control proteins (in this case, GFP), however, are usually insufficient to encompass all possible nonspecific interactions that occur during the purification. Thus, some ‘background’ proteins may preferentially interact with certain baits but not with others and be released from the agarose beads in quantities comparable to some specific interactors (primarily low- to medium-abundance binding proteins). The question that arose was whether the high protein elution efficiency shown by the abFASP method would translate into a clearer separation between specific and nonspecific interactors. As shown by the distribution densities reported in Figure 4, the abFASP-MS provides more accurate protein quantitation (average log2 TMT 131/129 of ∼2) than the acid elution-based MS data but also a broader TMT 131/129 distribution. This suggested that specific interactions should be retained even after aggressive filtering of the data. In the analysis of the TMT data shown in Figure 5, for each elution condition, emphasis was placed on proteins identified with at least two unique peptides and that were >3-fold higher in the SH-TBK1 TAP compared with the corresponding levels in the SH-GFP TAP (TMT 129/126, 130/127, and 131/128). Note that among the protein isoforms identified with only one unique peptide, many showed a total peptide number of >2 (for the corresponding protein groups). For example, both isoform 1 and 2 of OPTN were identified with a total of 38 shared peptides and only a single unique peptide each. Naturally, these proteins would not be excluded from the final detailed list of specific interactors. For our purposes, however, proteins such as OPTN were not evaluated in the quantitative analysis of the comparative elution conditions. As is common practice in quantitative proteomic experiments to obtain higher confidence data, only the unique peptides were used to calculate the relative ratios. As shown in Figure 5A, 37 proteins were common to all three elution conditions; 18 proteins were common to glycine and SDS only; 4 proteins were common to formic acid and glycine only; and 1 protein was common to formic acid and SDS only. 7, 9, and 12 proteins were uniquely identified in the formic acid, glycine, and SDS-eluted samples, respectively. The proteins in each group are shown in Supplementary Table S4 in the Supporting Information. As expected, the bait, the main interactors, and the pathway-related proteins were the most abundant in all three elution methods and were identified with a relatively higher number of unique peptides than other interactors (also shown in Figure 5B, left side of the plot, >10 peptides). The only exceptions were AZI2, TRAF2, and CACO2, which did not pass the threshold in the formic acid elution condition (although still present at a 129/126 ratio of >2-fold). In general and with few exceptions, proteins that passed the filtering with one elution method only showed ratios near the chosen >3-fold threshold. Interestingly, STX5 was one of the most abundant proteins in the SDS elution only (131/128 = 11.24). Other members of the syntaxin family were observed under all three elution conditions with high TMT ratios, thus indicating a possible functional role in the TBK1 network. Other Golgi-resident proteins involved in vesicular trafficking were also above the threshold under the SDS condition only (YKT6), in the overlap between glycine and SDS conditions (SC22B, BET1), and under the formic acid condition only (MPRD). In Figure 5B, the relative TMT ratios of the 37 SH-TBK1 shared interactors for each elution method are compared (all ratios are relative to the TMT 129 label). The direct comparison confirms that the elution of SH-TBK1_MOUSE protein complex with 2% SDS was considerably more efficient than elution with formic acid and more efficient than elution with glycine with respect to the majority of the identified interactors. The data presented in Figure 5 and Table S2 in the Supporting Information further emphasize the improvement that is achieved when eluting proteins with 2% SDS coupled to FASP. In general, this approach provides the most efficient conditions for removing proteins from the anti-HA agarose and subsequently the observation of higher TMT ratio values. Our data also complement the recent publication on the coupling FASP with chemical-labeling-based protein quantitation.24
Figure 4.
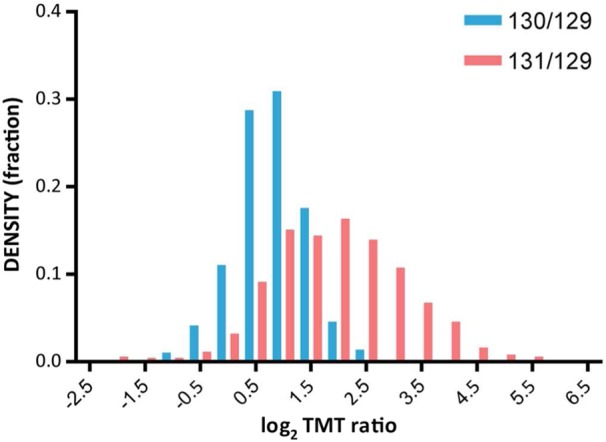
Density distribution (log2) of the TMT ratios for the SH-TBK1_MOUSE glycine and SDS elution conditions (TMT 6-plex channels 130 and 131, respectively) compared with the formic acid elution of the SH-TBK1 TAP (TMT 6-plex channel 129). TMT, tandem mass tag; TAP, tandem affinity purification; SDS, sodium dodecylsulphate.
Figure 5.

Analysis of the TMT data. (A) Venn diagram of the number of proteins identified in the SH-TBK1_MOUSE protein complex from the three elution conditions: 100 mM formic acid, 100 mM glycine, and 2% SDS. Only proteins identified with at least two unique peptides and that were more than >3-fold higher compared with the corresponding SH-GFP elution (channels 126, 127, and 128 TMT 6-plex) are included. (B) TMT ratios of the SH-TBK1 interactors resulting from the intersection of the three elution conditions assessed. All ratios were obtained by comparison with the formic acid elution of the SH-TBK1 TAP (channel 129 of the TMT 6-plex). SDS, sodium dodecylsulphate.
Concluding Remarks
The broad applicability and power of affinity purifications coupled to liquid chromatography mass spectrometry is indisputable. Such data sets can only be further enhanced by applying quantitative approaches such as SILAC or chemical labeling, for example, iTRAQ or TMT-plexing. To achieve this, however, it is imperative that there is a high degree of experimental normalization and standardization of the entire biochemical and analytical procedure. Namely, it is essential to: (i) assess the expression level of the SH-tagged proteins of interest in the cell lysate by anti-HA immunoblotting, (ii) assess the level of the SH-tagged proteins following affinity purification and subsequent normalization of the total protein input, and (iii) maximize elution of the SH-tagged proteins and interactors with a strong denaturing agent such as 2% SDS coupled to digestion by FASP. The new application of FASP to protein complexes is termed abFASP-MS or affinity-based filter-aided sample preparation mass spectrometry. abFASP-MS is directly compatible with chemical labeling of peptides and can provide reliable relative quantitation on protein interactor abundance between unrelated SH-tagged proteins. It is envisaged that abFASP-MS will also provide accurate information on proteins with a low expression profile and low-abundance interactors, SH-tagged proteins under altered conditions, for example, drug treatment, or comparison of protein complexes associated with a wild-type and mutant protein.
Acknowledgments
We thank Tilmann Bürckstümmer from Haplogen GmbH, Vienna for providing the mouse SH-TBK1_MOUSE entry vector; and Alexandra Zisser, Melanie Six and Melanie Planyavsky from the Bennett laboratory at CeMM for technical assistance with cell culture and immunoblots. Research in our laboratory is supported by the Austrian Academy of Sciences, the Austrian Federal Ministry for Science and Research (Gen-Au projects, APP-III and BIN-III), and the Austrian Science Fund FWF. E.L.R. and M.L.H are supported by the GenAu APP-III program (no. 820965), R.S. is supported by the FWF (no. P24217–B13), K.P. is supported by the European Research Council (ERC-2009-AdG-250179-i-FIVE), and A.S. is supported by the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme FP7/2007-2013/under REA grant agreement (no. 289611).
Glossary
Abbreviations
- AP
affinity purification
- AP-MS
affinity purification mass spectrometry
- CID
collision-induced dissociation
- PRKDC
DNA-dependent protein kinase catalytic subunit
- FASP
filter-aided sample preparation
- HCD
higher-energy collision-induced dissociation
- LC–MS
liquid chromatography mass spectrometry
- PBS
phosphate-buffered saline
- PBST
phosphate-buffered saline Tween-20
- PSM
peptide-to-spectrum matches
- SD
standard deviation
- SDS
sodium dodecylsulphate
- SDS-PAGE
sodium dodecylsulphate polyacrylamide gel electrophoresis
- SH-GFP
streptavidin-binding peptide and hemeagglutinin N-terminally tagged green fluorescent protein
- SH-TBK1_MOUSE
streptavidin-binding peptide and hemeagglutinin N-terminally tagged TANK binding kinase 1 (mouse)
- SILAC
stable isotope labeling by amino acids in cell culture
- TMT
tandem mass tag
- TANK
TRAF family member associated NF-κB activator
- TAP-MS
tandem affinity purification mass spectrometry
- TBKBP1
TBK binding protein 1
Supporting Information Available
Supplementary Table S1: All proteins identified by LC–MS from the tandem affinity purifications of SH-GFP and SH-TBK1_MOUSE for the formic acid, glycine and 2% SDS elutions. Supplementary Table S2: All proteins identified by LC–MS in the SH-TBK1_MOUSE experiments that were ≥2-fold more abundant in median spectral counts compared to the GFP control. Supplementary Table S3: Proteome Discoverer raw output file for the TMT experiment conducted on SH-GFP and SH-TBK1_MOUSE protein complexes eluted with 100 mM formic acid, 100 mM glycine and 2% SDS. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† E.L.R.: Center for Physiology and Pathophysiology, Institute of Medical Chemistry, Medical University of Vienna, Vienna, Austria.
Author Contributions
‡ M.L.H. and R.S. contributed equally to the work.
The authors declare no competing financial interest.
Supplementary Material
References
- Gavin A. C.; Bosche M.; Krause R.; Grandi P.; Marzioch M.; Bauer A.; Schultz J.; Rick J. M.; Michon A. M.; Cruciat C. M.; Remor M.; Hofert C.; Schelder M.; Brajenovic M.; Ruffner H.; Merino A.; Klein K.; Hudak M.; Dickson D.; Rudi T.; Gnau V.; Bauch A.; Bastuck S.; Huhse B.; Leutwein C.; Heurtier M. A.; Copley R. R.; Edelmann A.; Querfurth E.; Rybin V.; Drewes G.; Raida M.; Bouwmeester T.; Bork P.; Seraphin B.; Kuster B.; Neubauer G.; Superti-Furga G. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 2002, 4156868141–7. [DOI] [PubMed] [Google Scholar]
- Ho Y.; Gruhler A.; Heilbut A.; Bader G. D.; Moore L.; Adams S. L.; Millar A.; Taylor P.; Bennett K.; Boutilier K.; Yang L.; Wolting C.; Donaldson I.; Schandorff S.; Shewnarane J.; Vo M.; Taggart J.; Goudreault M.; Muskat B.; Alfarano C.; Dewar D.; Lin Z.; Michalickova K.; Willems A. R.; Sassi H.; Nielsen P. A.; Rasmussen K. J.; Andersen J. R.; Johansen L. E.; Hansen L. H.; Jespersen H.; Podtelejnikov A.; Nielsen E.; Crawford J.; Poulsen V.; Sorensen B. D.; Matthiesen J.; Hendrickson R. C.; Gleeson F.; Pawson T.; Moran M. F.; Durocher D.; Mann M.; Hogue C. W.; Figeys D.; Tyers M. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 2002, 4156868180–3. [DOI] [PubMed] [Google Scholar]
- Behrends C.; Sowa M. E.; Gygi S. P.; Harper J. W. Network organization of the human autophagy system. Nature 2010, 466730268–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwmeester T.; Bauch A.; Ruffner H.; Angrand P. O.; Bergamini G.; Croughton K.; Cruciat C.; Eberhard D.; Gagneur J.; Ghidelli S.; Hopf C.; Huhse B.; Mangano R.; Michon A. M.; Schirle M.; Schlegl J.; Schwab M.; Stein M. A.; Bauer A.; Casari G.; Drewes G.; Gavin A. C.; Jackson D. B.; Joberty G.; Neubauer G.; Rick J.; Kuster B.; Superti-Furga G. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat. Cell Biol. 2004, 6297–105. [DOI] [PubMed] [Google Scholar]
- Li S.; Wang L.; Berman M.; Kong Y. Y.; Dorf M. E. Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 2011, 353426–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varjosalo M.; Sacco R.; Stukalov A.; van Drogen A.; Planyavsky M.; Hauri S.; Aebersold R.; Bennett K. L.; Colinge J.; Gstaiger M.; Superti-Furga G. Interlaboratory reproducibility of large-scale human protein-complex analysis by standardized AP-MS. Nat. Methods 2013, 10, 307–314. [DOI] [PubMed] [Google Scholar]
- Pichlmair A.; Kandasamy K.; Alvisi G.; Mulhern O.; Sacco R.; Habjan M.; Binder M.; Stefanovic A.; Eberle C. A.; Goncalves A.; Burckstummer T.; Muller A. C.; Fauster A.; Holze C.; Lindsten K.; Goodbourn S.; Kochs G.; Weber F.; Bartenschlager R.; Bowie A. G.; Bennett K. L.; Colinge J.; Superti-Furga G. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature 2012, 4877408486–90. [DOI] [PubMed] [Google Scholar]
- Jager S.; Cimermancic P.; Gulbahce N.; Johnson J. R.; McGovern K. E.; Clarke S. C.; Shales M.; Mercenne G.; Pache L.; Li K.; Hernandez H.; Jang G. M.; Roth S. L.; Akiva E.; Marlett J.; Stephens M.; D’Orso I.; Fernandes J.; Fahey M.; Mahon C.; O’Donoghue A. J.; Todorovic A.; Morris J. H.; Maltby D. A.; Alber T.; Cagney G.; Bushman F. D.; Young J. A.; Chanda S. K.; Sundquist W. I.; Kortemme T.; Hernandez R. D.; Craik C. S.; Burlingame A.; Sali A.; Frankel A. D.; Krogan N. J. Global landscape of HIV-human protein complexes. Nature 2012, 4817381365–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatter T.; Wepf A.; Aebersold R.; Gstaiger M. An integrated workflow for charting the human interaction proteome: insights into the PP2A system. Mol. Syst. Biol. 2009, 5, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haura E. B.; Sacco R.; Li J.; Müller A. C.; Grebien F.; Superti-Furga G.; Bennett K. L. Optimisation of downscaled tandem affinity purifications to identify core protein complexes. J. Integr. OMICS 2012, 2155–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haura E. B.; Muller A.; Breitwieser F. P.; Li J.; Grebien F.; Colinge J.; Bennett K. L. Using iTRAQ combined with tandem affinity purification to enhance low-abundance proteins associated with somatically mutated EGFR core complexes in lung cancer. J. Proteome Res. 2011, 101182–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau T. L.; Gioia R.; Gatot J. S.; Patrascu F.; Carpentier I.; Chapelle J. P.; O’Neill L.; Beyaert R.; Piette J.; Chariot A. Are the IKKs and IKK-related kinases TBK1 and IKK-epsilon similarly activated?. Trends Biochem. Sci. 2008, 334171–80. [DOI] [PubMed] [Google Scholar]
- Soulat D.; Burckstummer T.; Westermayer S.; Goncalves A.; Bauch A.; Stefanovic A.; Hantschel O.; Bennett K. L.; Decker T.; Superti-Furga G. The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. EMBO J. 2008, 27152135–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves A.; Burckstummer T.; Dixit E.; Scheicher R.; Gorna M. W.; Karayel E.; Sugar C.; Stukalov A.; Berg T.; Kralovics R.; Planyavsky M.; Bennett K. L.; Colinge J.; Superti-Furga G. Functional Dissection of the TBK1 Molecular Network. PLoS One 2011, 69e23971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudashevskaya E. L.; Sacco R.; Kratochwill K.; Huber M. L.; Gstaiger M.; Superti-Furga G.; Bennett K. L. A method to resolve the composition of heterogeneous affinity-purified protein complexes assembled around a common protein by chemical cross-linking, gel electrophoresis and mass spectrometry. Nat. Protoc. 2013, 8175–97. [DOI] [PubMed] [Google Scholar]
- Manza L. L.; Stamer S. L.; Ham A. J.; Codreanu S. G.; Liebler D. C. Sample preparation and digestion for proteomic analyses using spin filters. Proteomics 2005, 571742–5. [DOI] [PubMed] [Google Scholar]
- Wisniewski J. R.; Zougman A.; Nagaraj N.; Mann M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 65359–62. [DOI] [PubMed] [Google Scholar]
- Rappsilber J.; Ishihama Y.; Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003, 753663–70. [DOI] [PubMed] [Google Scholar]
- Gilar M.; Olivova P.; Daly A. E.; Gebler J. C. Two-dimensional separation of peptides using RP-RP-HPLC system with different pH in first and second separation dimensions. J. Sep. Sci. 2005, 28141694–703. [DOI] [PubMed] [Google Scholar]
- Bennett K. L.; Funk M.; Tschernutter M.; Breitwieser F. P.; Planyavsky M.; Ubaida Mohien C.; Muller A.; Trajanoski Z.; Colinge J.; Superti-Furga G.; Schmidt-Erfurth U. Proteomic analysis of human cataract aqueous humour: Comparison of one-dimensional gel LCMS with two-dimensional LCMS of unlabelled and iTRAQ(R)-labelled specimens. J. Proteomics 2011, 742151–66. [DOI] [PubMed] [Google Scholar]
- Olsen J. V.; de Godoy L. M.; Li G.; Macek B.; Mortensen P.; Pesch R.; Makarov A.; Lange O.; Horning S.; Mann M. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics 2005, 4122010–21. [DOI] [PubMed] [Google Scholar]
- Colinge J.; Masselot A.; Giron M.; Dessingy T.; Magnin J. OLAV: towards high-throughput tandem mass spectrometry data identification. Proteomics 2003, 381454–63. [DOI] [PubMed] [Google Scholar]
- Ow S. Y.; Salim M.; Noirel J.; Evans C.; Rehman I.; Wright P. C. iTRAQ underestimation in simple and complex mixtures: ″the good, the bad and the ugly. J. Proteome Res. 2009, 8115347–55. [DOI] [PubMed] [Google Scholar]
- McDowell G. S.; Gaun A.; Steen H. iFASP: Combining Isobaric Mass Tagging with Filter-Aided Sample Preparation. J. Proteome Res. 2013, 12, 3809–3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.