

Abstract
Background
Hepatocellular carcinoma (HCC) is often diagnosed at an advanced stage, when it is not amenable for aggressive therapies such as surgical resection or liver transplantation. Current therapeutic options achieve clinical responses in only a small percentage of cases. As a consequence, effective approaches for prevention and treatment are greatly needed. Altered lipid metabolism has been recently linked to HCC pathogenesis. The aims of this study were to define the cellular and molecular mechanisms linking stearoyl-CoA desaturase (SCD), the rate-limiting enzyme and an essential regulator of lipid homeostasis in liver cells, to carcinogenesis in HCC.
Material and methods
HCC and normal liver specimens were collected. Human HCC cell lines: HepG2, Hep3B, and PLC/PLF/5 were used for immunoblot, cell viability, proliferation, and apoptosis assays. Small interfering RNAs were used for genetic inhibition, and 10, 12 conjugated linoleic acid was used for pharmacologic SCD inhibition.
Results
SCD was strongly expressed in surgically resected HCC (n = 64) and various human HCC cell lines (HepG2, Hep3B, and PLC/PLF/5). The levels of SCD negatively correlated with degree of tumor differentiation (P < 0.01). Treatment of these HCC cell lines with a panel of chemotherapeutic drugs resulted in a time-dependent, phosphatidylinositol 3 kinase- and c-Jun N-terminal kinases1/2–mediated upregulation of SCD expression, which paralleled the degree of resistance to drug-induced apoptosis. Specific genetic or pharmacologic SCD suppression resulted in inhibition of cell proliferation (P < 0.001) and significantly increased sensitivity to chemotherapy-induced apoptosis.
Conclusions
Our data suggest that increased SCD expression plays an important role in HCC development and resistance to chemotherapy-induced apoptosis, and this is in part mediated by phosphatidylinositol 3 kinase/c-Jun N-terminal kinases activation. Specific targeted interruption of this pathway in HCC could be a desirable approach in designing novel therapeutic strategies.
Keywords: Lipid metabolism, Apoptosis, Stearoyl-CoA desaturase, Carcinogenesis, Chemotherapy
1. Introduction
Hepatocellular carcinoma (HCC), the third leading cause of cancer death worldwide, is often diagnosed at an advanced stage, when it is not amenable for aggressive therapies such as surgical resection or liver transplantation [1–3]. There is currently no reliably effective therapy for patients with advanced or metastatic disease, and standard chemotherapeutic agents such as doxorubicin or 5-fluorouracil (5-FU) have relatively low response rates of only 10%–20% with no effect on survival [1–6].
Long-chain free fatty acids (FFA) are biologically active molecules that not only serve as a source of metabolic energy and substrates for cell and organelle membranes but also play a key role in fundamental cell processes such as cell growth and proliferation as well as apoptotic cell death [7,8]. The apoptosis effect appears to be specific for saturated fatty acid (SFA) [9,10]. Altered lipid metabolism, characterized by increased endogenous fatty acid (FA) synthesis, has been linked to HCC pathogenesis and several other human malignancies including breast, prostate, colon, and lung cancers [11–13].
Stearoyl-CoA desaturase (SCD), predominantly located in endoplasmic reticulum, is a key regulator of intracellular FA composition and catalyzes the conversion of SFA into monounsaturated FAs [14,15]. This enzyme facilitates the channeling of both exogenous (uptake from circulation) and endogenous (from de novo lipogenesis) FFA into triglyceride storage, phospholipids, and cholesterol ester synthesis [16]. By doing this, SCD may have a dual role in cell survival by protecting against SFA-induced lipotoxicity and at the same time favor cell growth and proliferation [17,18]. However, very little is known regarding the relationship between SCD, HCC survival, and resistance to apoptotic cell death.
We hypothesized that SCD plays an important role in cell growth and proliferation in HCC and confers resistance to chemotherapy when HCC is exposed to conventional chemotherapeutic agents. In this study, we sought to investigate the expression and role of SCD in human HCC and its role in resistance to chemotherapy-induced apoptosis.
2. Materials and methods
Institutional review board approval was obtained as per the institution protocols.
2.1. Cell lines, tissues, and reagents
Human hepatoma cell lines HepG2, Hep3B, and PLC/PRF/5 were purchased from ATCC (Manassas, VA). The cells were grown in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (Sigma, St. Louis, MO), 100 U/mL of penicillin, and 100 mg/L of streptomycin at 37°C in 5% CO2 in a humidified incubator. The cells were plated at 600,000 cells per 100-mm tissue culture dish and allowed to adhere overnight (18 h) before any treatment began. The cells were incubated with three chemotherapeutic agents: Staurosporine (STS; 1 μmol/L) (Calbiochem, San Diego, CA), 5-FU (10 μg/mL; Sigma), and doxorubicin (1 μg/mL; Sigma) for up to 24 h. Pooled human-liver microsomal protein from normal human hepatocytes (BD Biosciences, Woburn, MA) served as control. Sixty-four paraffin-fixed HCC tissue samples and 10 normal human liver tissue samples, obtained from adjacent to hepatic metastatic tumors, were obtained from the Department of Pathology, Cleveland Clinic. Selective SCD inhibitor 10, 12 conjugated linoleic acid (10, 12 CLA) and 9, 11 CLA were from Matreya (Pleasant Gap, PA). The c-Jun N-terminal kinases (JNK)1/2 inhibitor SP600125 was from Biomol International (Plymouth, PA), the p38 mitogen-activated protein kinases (MAPK) inhibitor SB203580 was from Calbiochem (San Diego, CA), and the phosphatidylinositol 3 kinase (PI3K) inhibitor LY294002 was purchased from Cell signaling Technology (Beverly, MA).
2.2. Immunoblot analysis
For whole cell lysates, cells were homogenized in a lysis buffer containing 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 10 μg/mL of aprotinin, 100 μg/mL of phenylmethylsulfonyl fluoride, 1 mmol/L of sodium ortho-vanadate, 50 mmol/L of sodium fluoride, 5 μg/mL of pepstein, 5 μg/mL of leupeptin, 2 mmol/L of Pefabloc (4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride; Sigma Aldrich), and phosphate-buffered saline (PBS), pH 7.4. Nuclear protein extracts were prepared as previously described [19]. Total or nuclear protein of 20 μg per lane was electrophoretically separated by SDS–8% polyacrylamide gels, transferred to nitrocellulose membrane, and probed with anti–SCD monoclonal antibody (Ab) or anti–sterol regulatory element–binding protein 1 (SREBP-1) monoclonal Ab (Abcam, Cambridge, MA). The blots were developed by an enhanced chemiluminescence system (Immune-Star HRP substrate kit, BioRad Laboratories, Hercules, CA). As a control for sample loading, the blot was stripped and reprobed with β-actin or anti–eglyceraldehyde-3-phosphate dehydrogenase (GAPDH) polyclonal Ab (Ambion, Austin, TX). Optical densities of bands in each blot were analyzed using the Image-Pro software.
2.3. Immunohistochemistry
Immunohistochemistry was performed using an anti–SCD monoclonal Ab (Abcam). The samples were incubated with primary Ab 1:100 in the blocking solution for 3 h at room temperature. After washing with PBS, the sections were incubated with the ready-to-use secondary Ab (biotin-labeled affinity-isolated goat anti-rabbit immunoglobulin; DAKO Corporation, Carpinteria, CA) for 30 min at room temperature. After washing in PBS, the samples were incubated with a ready-to-use streptavidin peroxidase conjugate in PBS-containing carrier protein and anti-microbial agents (DAKO corporation) for 30 min at room temperature. After washing with PBS, the samples were stained with 3,3′-diaminobenzidine (vector) for 2–5 min, washed in PBS, counterstained with hematoxylin for 2–3 min, and dehydrated by transferring them through increasing ethanol solutions (30%, 50%, 70%, 80%, 95%, and 100% ethanol). After dehydration, the slices were soaked twice in a xylene bath at room temperature for 5 min, respectively, mounted, and examined. Immunohistochemical results were semiquantitatively evaluated in a blinded fashion using a four-point scoring system (0, no staining; 1, positive staining in <30% of cells per high-power field; 2, positive staining in >30% but <70% of cells per high-power field; and 3, positive staining in >70% of cells per high-power field) and compared with the tumor differentiation, obtained from pathology report.
2.4. Small interfering RNA and transfection
Small interfering RNA (siRNA) was used to silence SCD expression. Inhibition of SCD protein expression was assessed by immunoblot analysis after transfection of HepG2 cells using siPORT NeoFX kit (Ambion, Austin, TX). Briefly, 25 μL of siPORT NeoFX was mixed with 25 μL of 10 nM siRNA, incubated, and distributed to culture wells in 24-well plate and overlaid with 4.5 × 104 cells per well in a total transfection volume of 0.5 mL for 24 h. The cells were then treated and analyzed as described in the following.
2.5. Cell viability and apoptosis assessment
The number of viable cells was determined using a fluorometric assay (CellTiter-Blue, Promega; Madison, WI) following the manufacture's instructions. Caspase activation and apoptosis were quantified using biochemical and fluorescent microscopic techniques: Caspase-Glo 3/7 Assay (Promega), the cell death ELISA kit (Roche Diagnostics, Mannheim, Germany), and assessing the characteristic nuclear changes of apoptosis (i.e., chromatin condensation and nuclear fragmentation) using the nuclear binding dye 4′,6-diamidino-2-phenylindole dihydrochloride (Molecular Probes Inc, Eugene, OR) and fluorescent microscopy.
2.6. Cell proliferation
To evaluate the role of SCD in HCC proliferation, we used two different approaches. First, cell proliferation was assessed by measuring the conversion of tetrazolium salt to water-soluble formazan dye using WST-1 cell proliferation assay (Roche Diagnostics, Mannheim, Germany). HepG2 cells were treated with graded doses of selective SCD inhibitor 10,12 CLA and 9,11 CLA, for the control group, for up to 48 h. After incubation, cells were washed with PBS, and the WST-1 reagent was added with cell culture medium and incubated for 4 h. Sample absorbance was analyzed using a bichromatic enzyme-linked immunosorbent assay reader at 450 nm. Second, proliferation effect of SCD was further assessed using bromodeoxyuridine (BrdU), a synthetic thymidine analog, immunofluorescence staining. Hepatoma cells grown in four-chamber slides were treated with 0, 50, and 100 μM of 10,12 CLA and 9,11 CLA as negative control for 48 h. BrdU staining was performed using 5-Bromo-2′-deoxy-uridine Labeling and Detection Kit I (Roche Diagnostics, Penzberg, Germany) according to manufacturer's protocol. Fluorescence microscopic images were obtained with microscope system and quantified using Image-Pro software. All experiments were performed in triplicate.
2.7. Statistical analysis
All data represent at least three independent experiments and were expressed as the mean ± standard deviation unless otherwise indicated. Differences between groups were compared by an analysis of variance followed by a post hoc Bonferroni test to correct for multiple comparisons. Differences were considered to be statistically significant at P < 0.05.
3. Results
3.1. SCD expression is enhanced in human HCC
SCD immunostaining was performed in the HCC specimens and compared with normal liver specimens. The immunoreactive product was readily identified in most grades 2 and 3 (moderately and poorly differentiated) HCC tissue samples and showed a diffuse cytosolic cellular staining pattern. It was present to a lesser extend in grade 1 (well differentiated) HCC tissue samples and was only weakly seen in control specimens (Fig. 1A). The immunoreactive product was selective for the SCD epitope as reactive product was not observed in the absence of the primary Ab (Fig. 1A). Semiquantitative analysis showed that SCD staining score was significantly higher in patients with grade 3 HCC compared with patients with grade 1 HCC and normal controls (2.6 ± 0.5 versus 1.7 ± 0.3 versus 0.2 ± 0.4, P < 0.01; Fig. 1B). These data suggest that upregulation of SCD may contribute to the HCC development.
Fig. 1.

SCD expression is enhanced in human HCC. Immunohistochemistry for SCD was performed in liver tissue from patients with HCC (n = 64) and normal liver (n = 10). (A) Representative histologic microphotographs of SCD immunostaining from grades 1–3 HCC and normal liver. (B) Quantitation of SCD staining in the different groups of patients. *P < 0.001 compared with normal liver. #P < 0.01 compared with well-differentiated (grade 1) HCC. IHC = Immunohistochemical.
3.2. SCD is expressed in different HCC cell lines and its levels increase in a time-dependent manner after exposure to a panel of chemotherapeutic drugs
The findings of increased SCD expression in human HCC tissue led us to further examine the role of SCD in HCC development using three different human HCC cell lines including HepG2, Hep3B, and PLC5. We initially examined the baseline expression of SCD in these cell lines and compared it with that of normal human hepatocytes. The cells were grown in appropriate growth media and harvested after 72 h, and SCD expression was analyzed using Western blot analysis (Fig. 2A). SCD expression was found to be significantly increased in all three HCC cell lines compared with that in normal hepatocytes (P < 0.001). Highest level of expression was seen in HepG2 cell line although the differences in expression among the three cell lines did not reach statistical significance (Fig. 2B). We next investigated the possible role of SCD in the resistance of HCC cell lines to chemotherapy. To address the effect of chemotherapy on cell viability and caspase 3 activation in HCC, HepG2 cells were incubated overnight in 96-well plates and treated with three different chemotherapeutic agents that are routinely used clinically as part of standard HCC chemotherapy at various time points. Cell viability and caspase 3 activity were assessed using CellTiter-Blue assay and Apo-ONE Homogenous Caspase-3/7 assay, respectively. Consistent with previous reports [20–22], HepG2 cells demonstrated low sensitivity to chemotherapy-induced apoptosis with cell viability of about 80%–85% at 24 h with all three chemotherapeutic agents despite significant caspase 3 activation (Fig. 2C). Similar results for chemoresistance were obtained using both Hep3B and PLC5 cell lines (data not shown). These changes were associated with a time-dependent upregulation of SCD expression, which parallels the degree of resistance to drug-induced apoptosis (Fig. 2D).
Fig. 2.

SCD is expressed in hepatoma cell lines and upregulated on exposure to chemotherapy. (A) Western blot analysis of SCD in normal pooled liver microsomal fraction and human hepatoma cell lines (HepG2, Hep3B, and PLC/PRF/5). β-actin served as a control for sample loading. Whole cell lysates containing 30 mg of protein were subjected to SDS–polyacrylamide gel electrophoresis, and immunoblot analysis was done using specific monoclonal Ab against SCD. (B) SCD expression was significantly increased in all three hepatoma cell lines compared with normal hepatocytes. Results are expressed as mean ± standard deviation from three independent experiments. *P < 0.001 compared with normal pooled liver microsomal fraction. (C) HepG2 cells were incubated with STS (1 μM), 5-FU (10 μg/mL) or doxorubicin (1 μg/mL) at 2, 6, 16, and 24 h after the cells reached 50% confluency. Cell viability and caspase activity were determined using CellTiter-Blue cell viability assay and Apo-ONE Homogenous Caspase-3/7 assay, respectively, at different time points after drug treatments. The cell viability was calculated as a percentage with respect to cells without drug treatment. The data are presented as the means ± standard deviation of three independent experiments. HepG2 cells demonstrated low sensitivity to chemotherapy-induced apoptosis with levels <15% at 24 h for all three drugs despite significant caspase 3 activation. (D) Expression of SCD in treated and untreated cells was measured by Western blot analysis. β-actin served as a control for sample loading. Treatment with all three chemotherapeutic agents resulted in a time-dependent upregulation of SCD expression, which parallels the degree of resistance to drug-induced apoptosis. *P < 0.001 compared with controls. NFL = normal pooled liver microsomal fraction.
3.3. SCD suppression results in increased sensitivity of HepG2 cells to chemotherapy
The findings of increased SCD expression and the strong association with HCC resistance to chemotherapy led us to further investigate whether manipulation of SCD expression possibly influences the apoptosis sensitivity of HCC cell lines to chemotherapy. HepG2 cells were transfected with three different isoforms of SCD-specific siRNA targeting the same gene sequence. Transfection resulted in >96% suppression of SCD expression (Fig. 3A). siRNA and siRNA against GAPDH were used as negative and positive controls, respectively. GAPDH was also used as an internal loading control. After 48 h of transfection, cells were then incubated in the absence or presence of STS or 5-FU for up to 24 h. Suppression of SCD expression resulted in significantly increased sensitivity of HepG2 cells to chemotherapy-induced apoptosis (Fig. 3B). Cell viability was reduced to 46% versus 88% after STS treatment and 65% versus 95% after 5-FU treatment after genetic suppression of SCD. To determine whether pharmacologic inhibition would have a similar effect compared with specific suppression of SCD expression, HepG2 cells were incubated with a selective SCD inhibitor, 10,12 CLA, or the inactive control compound, 9,11 CLA, for 48 h. The cells were then treated with STS or 5-FU for up to 24 h. Pharmacologic inhibition of SCD resulted in a similar increased sensitivity to chemotherapeutic agents as seen by siRNA suppression. Reduction in cell viability after SCD inhibition was 61.8% versus 98% with STS treatment and 66.8% versus 92.5% with 5-FU treatment (Fig. 3C).
Fig. 3.

SCD suppression by genetic or pharmacologic approach results in increased sensitivity to chemotherapy-induced apoptosis. HepG2 cells were transfected with three different isoforms of SCD-specific siRNA targeting the same gene sequence. (A) Transfection resulted in >96% suppression of SCD expression. siRNA and siRNA against GAPDH were used as negative and positive controls, respectively. GAPDH was also used as an internal loading control. (B) Cells were then incubated in the absence or presence of STS for up to 24 h. Cell viability was determined using CellTiter-Blue cell viability assay and calculated as a percentage with respect to cells without siRNA and drug treatment. Suppression of SCD expression resulted in a significant increase sensitivity to chemotherapy-induced apoptosis. *P < 0.05 compared with controls. (C) HepG2 cells were incubated with 45 mmol/L 10,12 CLA or the inactive control compound 9,11 CLA in 10% fetal bovine serum containing media for 48 h. Cells were then treated with STS or 5-FU for up to 24 h. Cell viability was determined using CellTiter-Blue cell viability assay and calculated as a percentage with respect to cells without inhibitor and drug treatment. HepG2 cells demonstrated significant increased susceptibility to chemotherapy-induced apoptosis after suppression of SCD activity. *P < 0.01 compared with controls.
3.4. Downregulation of SCD activity results in decreased proliferation of HepG2 cells
We next investigated the role of SCD in proliferation of HCC cells. After pharmacologic inhibition of SCD using 10,12 CLA or the inactive control compound 9,11 CLA, proliferation was measured using WST-1 proliferation assay and absorbance was detected at 450 nm. Growth rate of cells was found to be adversely affected after SCD inhibition compared with that of untreated cells in a dose-dependent manner (Fig. 4A and B). Inhibition of SCD resulted in >20% reduction in cell proliferation after 48 h. Proliferation was further assessed using BrdU immunofluorescence staining. BrdU incorporation was significantly reduced in the cells with suppression of SCD activity, a similar observation made by WST-1 assay. Cell growth was suppressed to >50% of original growth rates at 48 h compared with the negative control and the untreated cells (Fig. 4C).
Fig. 4.

Decreased proliferation of HepG2 cells after downregulation of SCD activity. (A) Time- and dose-dependent growth inhibitory effect after suppression of SCD activity using 10,12 CLA. HepG2 cells were treated with 25, 50, or 100 nmol/L of 10,12 CLA and cultured for up to 48 h. Cells were then subjected to WST-1 proliferation assay, and absorbance was detected at 450 nm with an ELISA plate reader. Control cells were treated with 100 nmol/L of 9,11 CLA. The growth rate of cells with suppression of SCD activity was significantly reduced compared with untreated and the control group. (B) Dose-dependent growth inhibitory effect of SCD suppression. Cells treated with various concentrations of 10,12 CLA were cultured for 48 h, and quantified by WST-1 assay. Data from three independent experiments are shown as mean ± standard deviation. *P < 0.01 compared with controls. (C) Proliferation of HepG2 cells was further assessed using BrdU immunofluorescence staining. HepG2 cells grown in four-chamber slides were treated with 50 or 100 nmol/L of 10,12 CLA and cultured for up to 48 h. Control group was treated with 100 nmol/L of 9,11 CLA (negative control). Immunofluorescence staining was done using anti-BrdU Ab, and nuclei were counterstained with 4′,6-diamidino-2-phenylindole dihydrochloride stain. Cells were examined under fluorescence microscope and analyzed using the Image-Pro software. BrdU incorporation was reduced in the cells with suppression of SCD activity. *P < 0.001 compared with untreated cells.
3.5. Increased expression of SCD is SREBP-1 dependent and mediated by JNK1/2 and PI3K activation but not by p38 MAPK
SREBP-1 is a key transcription factor that regulates FA synthesis by upregulating the expression of various lipogenic enzymes including SCD [23]. To identify whether alterations in SCD expression in HCC is dependent on SREBP-1, HepG2 cells were incubated in the absence or presence of 10 μg/mL of 5-FU for up to 24 h. TO901317, a selective liver X receptor agonist known to upregulate SREBP-1 expression was used as positive control. Nuclear protein was then extracted, and Western blot analysis was done using an anti–SREBP-1 Ab. SREBP-1 expression demonstrated a parallel increase as of the SCD expression after 5-FU treatment of HepG2 cells, as seen in our earlier experiments (Fig. 5A and B). To further explore the molecular mechanisms involved, the MAPK pathway, especially JNK, PI3K, and p38 kinases, was studied based on their described role in protection against 5-FU-induced apoptosis in hepatoma cell lines [23]. The potential contribution of these MAPKs in mediating alterations of SCD expression after 5-FU treatment was ascertained by using selective pharmacologic inhibitors for p38 MAPK, the JNK1/2, and the PI3K. Pre-incubation with the JNK1/2 inhibitor SP600125 (20 μM) and the PI3K inhibitor LY294002 (50 μM) resulted in significant reduction of 5-FU-induced SCD expression (Fig. 5C and D). Conversely, the p38 MAPK inhibitor SB203580 (20 μM) did not have any effect on the SCD expression (Fig. 5C and D). These results indicate that 5-FU induced increase in SCD expression via SREBP-1 and in a process dependent on JNK1/2 and PI3K activation but independent of p38 MAPK.
Fig. 5.
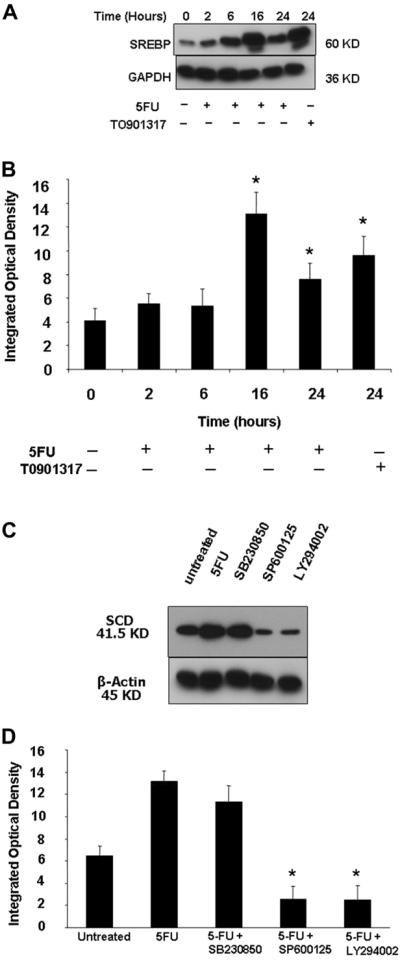
Increased expression of SCD, induced by 5-FU, is SREBP-1 dependent and mediated by activation of JNK1/2 and PI3K but not by p38 MAPK. (A) HepG2 cells were incubated in the absence or presence of 10 μg/mL of 5-FU or TO901317 (positive control) for up to 24 h. Nuclear protein was then extracted, and Western blot analysis was done using anti–SREBP-1 Ab. GAPDH was used as a loading control. Increase in SREBP-1 expression parallels the increase in SCD expression after 5-FU treatment as shown in earlier experiments. (B) Quantitated SREBP-1 bands normalized to GAPDH signals. *P < 0.05 compared with untreated cells. HepG2 cells were preincubated for 30 min with the p38 MAPK inhibitor SB203580 (20 μM), the JNK1/2 inhibitor SP600125 (20 μM), or the PI3K inhibitor LY294002 (50 μM) and then incubated in the absence or presence of 10 μg/mL of 5-FU for 16 h. (C) Immunoblot analysis was performed using specific monoclonal Ab against SCD and (D) the optical density of the bands quantified using Image-Pro software. *P < 0.01 compared with 5-FU–treated cells.
4. Discussion
The principle findings of this study relate to the mechanisms linking SCD expression to HCC development and resistance to chemotherapy-induced apoptosis. Our results demonstrate that (1) SCD, the rate-limiting enzyme in the biosynthesis of monounsaturated fats and an essential regulator of lipid homeostasis in liver cells, is expressed at high levels in both human HCC tissues and various human HCC cell lines; (2) treatment of these HCC cell lines with a panel of chemotherapeutic drugs results in a time-dependent upregulation of SCD expression which parallels the degree of resistance to drug-induced apoptosis; and (3) genetic or pharmacologic inhibition of SCD sensitizes HCC cells toward chemotherapy-induced apoptosis.
The incidence of HCC continues to rise worldwide, particularly in younger age cohorts in the United States and Europe [24]. In western countries, <40% of patients are eligible for potential curative treatment at the time of presentation [24]. Altered lipid metabolism, characterized by an increase in endogenous FA synthesis, has been linked to HCC pathogenesis and several other human malignancies including breast, prostate, colon, and lung cancers [11,13]. Our current data extend these previous observations by demonstrating a high level of SCD expression in HCC tissue. Moreover, we identified an inverse correlation between the increase in the expression of this enzyme and the degree of tumor differentiation. To further understand the mechanisms linking SCD expression to HCC pathogenesis, we used a variety of different human hepatoma cell lines. SCD is an enzyme that plays a key role as a regulator of intracellular FA composition by catalyzing the conversion of SFA into monounsaturated FAs [14,15]. The enzyme facilitates the channeling of both exogenous (uptake from circulation) and endogenous (from de novo lipogenesis) FFA into triglyceride storage, phospholipids, and cholesterol ester synthesis [16]. By doing this, SCD may have a dual role in cell survival by protecting against SFA-induced lipotoxicity and at the same time favor cell growth and proliferation [17,18]. Our in vitro studies demonstrate that dysregulated expression of SCD promotes tumor cell survival and resistance to chemotherapy in human HCC. We also found that SCD is present in various hepatoma cells, and its expression is significantly increased after incubation with a panel of chemotherapeutic agents routinely used in patients with HCC who are not amenable to surgery or local therapies. These changes strongly correlated with resistance to chemotherapy-induced apoptosis. The resistance to apoptotic cell death was seen despite significant caspase 3 activation. This apparent dissociation between caspase 3 activity and apoptosis has been previously reported in human HCC [25]. Further insight into the molecular mechanisms explaining this observation is warranted.
Previous studies have described a link between nonalcoholic steatohepatitis and HCC [26], and recognized the alterations in hepatic lipid homeostasis to be a key pathway involved in HCC pathogenesis [27,28]. However, the extent of this association remains unclear. We have recently demonstrated that high levels of exogenous SFA promote apoptosis of hepatoma cells, whereas addition of unsaturated FAs or overexpression of SCD prevents the SFA-induced cell death [29]. Our current data extend these observations by demonstrating that inhibition of SCD by either siRNA or a potent pharmacologic inhibitor sensitized HCC cells to chemotherapeutic drugs. The precise mechanisms by which SCD protects HCC cells from chemotherapy-induced apoptosis will require further study. In addition, our study demonstrates dramatic effects on cell proliferation after SCD inhibition, similar to the results depicted for human lung fibroblasts [18]. Based on our observations, it is tempting to speculate that modulation of lipid metabolism by regulating SCD activity may, in turn, influence the cellular proliferation and chemotherapy-induced resistance to apoptosis in human HCC.
Finally, SREBP-1 is a key transcription factor that regulates FA synthesis by upregulating the expression of various lipogenic enzymes including SCD [30]. Potential involvement of SREBP-1 has been implicated in the development of colorectal and breast cancers [31,32], and more recently, it has been shown to mediate an increase in FA synthase promoter activity in hepatoma cells [33]. Our data reveal that the enhanced SCD expression in HCC and chemoresistance was dependent on SREBP-1. We observed a time-dependent SREBP-1 overexpression in HepG2 cells on exposure to chemotherapy, which paralleled that of SCD. Furthermore, our results provide evidence that these changes are dependent on both JNK and PI3K activation but are independent of p38 kinase. In summary, the present study uncovers a link between SCD expression and HCC resistance to currently used chemotherapeutic agents. These concepts have important implications for the pathogenesis and development of novel diagnostic and treatment strategies for human HCC. Recently, specific multikinase inhibitor agents such as sorafenib have shown a survival benefit in advanced HCC [34]. Future in vitro and in vivo studies combining our approach and sorafenib are warranted to explore their potential synergistic therapeutic benefits.
Acknowledgment
Financial support: this work was supported by the National Institutes of Health grant (DK076852) to A.F.
Footnotes
Financial disclosures: the authors have no conflicts to disclose.
REFERENCES
- [1].Baek JY, Hur W, Wang JS, Bae SH, Yoon SK. Selective COX-2 inhibitor, NS-398, suppresses cellular proliferation in human hepatocellular carcinoma cell lines via cell cycle arrest. World J Gastroenterol. 2007;13:1175. doi: 10.3748/wjg.v13.i8.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Caldwell SH, Crespo DM, Kang HS, Al-Osaimi AM. Obesity and hepatocellular carcinoma. Gastroenterology. 2004;127(5 Suppl 1):S97. doi: 10.1053/j.gastro.2004.09.021. [DOI] [PubMed] [Google Scholar]
- [3].Coulouarn C, Gomez-Quiroz LE, Lee JS, et al. Oncogenespecific gene expression signatures at preneoplastic stage in mice define distinct mechanisms of hepatocarcinogenesis. Hepatol (Baltimore, Md) 2006;44:1003. doi: 10.1002/hep.21293. [DOI] [PubMed] [Google Scholar]
- [4].Kanda T, Yokosuka O, Imazeki F, Arai M, Saisho H. Enhanced sensitivity of human hepatoma cells to 5-fluorouracil by small interfering RNA targeting Bcl-2. DNA Cell Biol. 2005;24:805. doi: 10.1089/dna.2005.24.805. [DOI] [PubMed] [Google Scholar]
- [5].Roberts LR, Gores GJ. Hepatocellular carcinoma: molecular pathways and new therapeutic targets. Semin Liver Dis. 2005;25:212. doi: 10.1055/s-2005-871200. [DOI] [PubMed] [Google Scholar]
- [6].Sterpetti P, Marucci L, Candelaresi C, et al. Cell proliferation and drug resistance in hepatocellular carcinoma are modulated by Rho GTPase signals. Am J Physiol. 2006;290:G624. doi: 10.1152/ajpgi.00128.2005. [DOI] [PubMed] [Google Scholar]
- [7].Black PN, Dirusso CC. Transmembrane movement of exogenous long-chain fatty acids: proteins, enzymes, and vectorial esterification. Microbiol Mol Biol Rev. 2003;67:454. doi: 10.1128/MMBR.67.3.454-472.2003. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Li Z, Berk M, Mcintyre TM, Gores GJ, Feldstein AE. The lysosomal-mitochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatol (Baltimore, Md) 2008;47:1495. doi: 10.1002/hep.22183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].De Vries JE, Vork MM, Roemen TH, et al. Saturated but not mono-unsaturated fatty acids induce apoptotic cell death in neonatal rat ventricular myocytes. J Lipid Res. 1997;38:1384. [PubMed] [Google Scholar]
- [10].Ji J, Zhang L, Wang P, et al. Saturated free fatty acid, palmitic acid, induces apoptosis in fetal hepatocytes in culture. Exp Toxicol Pathol. 2005;56:369. doi: 10.1016/j.etp.2005.02.003. [DOI] [PubMed] [Google Scholar]
- [11].Cotrim HP, Parana R, Braga E, Lyra L. Nonalcoholic steatohepatitis and hepatocellular carcinoma: natural history? Am J Gastroenterol. 2000;95:3018. doi: 10.1111/j.1572-0241.2000.03241.x. [DOI] [PubMed] [Google Scholar]
- [12].Moller H, Mellemgaard A, Lindvig K, Olsen JH. Obesity and cancer risk: a Danish record-linkage study. Eur J Cancer. 1994;30A:344. doi: 10.1016/0959-8049(94)90254-2. [DOI] [PubMed] [Google Scholar]
- [13].Pitot HC. Pathways of progression in hepatocarcinogenesis. Lancet. 2001;358:859. doi: 10.1016/S0140-6736(01)06038-X. [DOI] [PubMed] [Google Scholar]
- [14].Cohen P, Ntambi JM, Friedman JM. Stearoyl-CoA desaturase-1 and the metabolic syndrome. Curr Drug Targets. 2003;3:271. doi: 10.2174/1568008033340117. [DOI] [PubMed] [Google Scholar]
- [15].Enoch HG, Catala A, Strittmatter P. Mechanism of rat liver microsomal stearyl-CoA desaturase. Studies of the substrate specificity, enzyme-substrate interactions, and the function of lipid. J Biol Chem. 1976;251:5095. [PubMed] [Google Scholar]
- [16].Miyazaki M, Kim YC, Gray-Keller MP, Attie AD, Ntambi JM. The biosynthesis of hepatic cholesterol esters and triglycerides is impaired in mice with a disruption of the gene for stearoyl-CoA desaturase 1. J Biol Chem. 2000;275:30132. doi: 10.1074/jbc.M005488200. [DOI] [PubMed] [Google Scholar]
- [17].Listenberger LL, Han X, Lewis SE, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci United States America. 2003;100:3077. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Scaglia N, Igal RA. Stearoyl-CoA desaturase is involved in the control of proliferation, anchorage-independent growth, and survival in human transformed cells. J Biol Chem. 2005;280:25339. doi: 10.1074/jbc.M501159200. [DOI] [PubMed] [Google Scholar]
- [19].Yoshikawa T, Shimano H, Amemiya-Kudo M, et al. Identification of liver X receptor-retinoid X receptor as an activator of the sterol regulatory element-binding protein 1c gene promoter. Mol Cell Biol. 2001;21:2991. doi: 10.1128/MCB.21.9.2991-3000.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Busch AK, Gurisik E, Cordery DV, et al. Increased fatty acid desaturation and enhanced expression of stearoyl coenzyme A desaturase protects pancreatic beta-cells from lipoapoptosis. Diabetes. 2005;54:2917. doi: 10.2337/diabetes.54.10.2917. [DOI] [PubMed] [Google Scholar]
- [21].Schulze-Bergkamen H, Fleischer B, Schuchmann M, et al. Suppression of Mcl-1 via RNA interference sensitizes human hepatocellular carcinoma cells towards apoptosis induction. BMC Cancer. 2006;6:232. doi: 10.1186/1471-2407-6-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Takehara T, Liu X, Fujimoto J, Friedman SL, Takahashi H. Expression and role of Bcl-xL in human hepatocellular carcinomas. Hepatol (Baltimore, Md) 2001;34:55. doi: 10.1053/jhep.2001.25387. [DOI] [PubMed] [Google Scholar]
- [23].Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- [24].El-Serag HB. Hepatocellular carcinoma: recent trends in the United States. Gastroenterology. 2004;127(5 Suppl 1):S27. doi: 10.1053/j.gastro.2004.09.013. [DOI] [PubMed] [Google Scholar]
- [25].Persad R, Liu C, Wu TT, et al. Overexpression of caspase-3 in hepatocellular carcinomas. Mod Pathol. 2004;17:861. doi: 10.1038/modpathol.3800146. [DOI] [PubMed] [Google Scholar]
- [26].Brunt EM. Nonalcoholic steatohepatitis. Semin Liver Dis. 2004;24:3. doi: 10.1055/s-2004-823098. [DOI] [PubMed] [Google Scholar]
- [27].Evert M, Schneider-Stock R, Dombrowski F. Overexpression of fatty acid synthase in chemically and hormonally induced hepatocarcinogenesis of the rat. Lab Invest a J Tech Methods Pathol. 2005;85:99. doi: 10.1038/labinvest.3700206. [DOI] [PubMed] [Google Scholar]
- [28].Luedde T, Beraza N, Kotsikoris V, et al. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- [29].Li ZZ, Berk M, Mcintyre TM, Feldstein AE. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: role of stearoyl-CoA desaturase. J Biol Chem. 2009;284:5637. doi: 10.1074/jbc.M807616200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Goodridge AG. Dietary regulation of gene expression: enzymes involved in carbohydrate and lipid metabolism. Annu Rev Nutr. 1987;7:157. doi: 10.1146/annurev.nu.07.070187.001105. [DOI] [PubMed] [Google Scholar]
- [31].Li JN, Mahmoud MA, Han WF, Ripple M, Pizer ES. Sterol regulatory element-binding protein-1 participates in the regulation of fatty acid synthase expression in colorectal neoplasia. Exp Cell Res. 2000;261:159. doi: 10.1006/excr.2000.5054. [DOI] [PubMed] [Google Scholar]
- [32].Yang YA, Morin PJ, Han WF, et al. Regulation of fatty acid synthase expression in breast cancer by sterol regulatory element binding protein-1c. Exp Cell Res. 2003;282:132. doi: 10.1016/s0014-4827(02)00023-x. [DOI] [PubMed] [Google Scholar]
- [33].Roder K, Zhang L, Schweizer M. SREBP-1c mediates the retinoid-dependent increase in fatty acid synthase promoter activity in HepG2. FEBS Lett. 2007;581:2715. doi: 10.1016/j.febslet.2007.05.022. [DOI] [PubMed] [Google Scholar]
- [34].Køstner AH, Sorensen M, Olesen RK, Grønbæk H, Lassen U, Ladekarl M. Sorafenib in advanced hepatocellular carcinoma: a nationwide retrospective study of efficacy and tolerability. Scientific World J. 2013 doi: 10.1155/2013/931972. Article ID 931972. [DOI] [PMC free article] [PubMed] [Google Scholar]