

Abstract
MicroRNAs (miRNAs) are small, regulatory non-coding RNAs that have potent effects on gene expression. Several miRNA are deregulated in cellular processes involved in human liver diseases and regulation of cellular processes. Recent studies have identified the involvement of miR-29 in hepatic fibrosis and carcinogenesis. Although several targets of miR-29 have been identified, there is limited information regarding the cell-type specific roles of miR-29 in the liver, and we sought to evaluate the role of this miRNA in hepatic pathobiology. We report the generation of a tissue–specific knockout mouse to evaluate the role of miR-29 in hepatic fibrosis and carcinogenesis in response to injury. We hypothesized that miR-29 contributes to the hepatocyte driven response to chronic cellular injury that results in fibrosis. In support of this hypothesis, fibrosis and mortality were enhanced in miR29 knockout mice in response to carbon tetrachloride. Genome-wide gene expression analysis identified an over-representation of genes associated with fibrosis. The oncofetal RNA H19 was modulated in a miR-29 dependent manner following exposure to carbon tetrachloride in vivo. The impact of a hepatocyte specific miR-29 knockout on survival following chronic hepatic injury in vivo implicates this miRNA as a potential target for intervention. These results provide evidence of the involvement of miR-29 in chronic hepatic injury, and suggest a role for deregulated hepatocyte expression of miR-29 in the response to hepatic injury, fibrosis and carcinogenesis.
Keywords: fibrosis, liver cancer, transgenic
Introduction
MicroRNAs (miRNAs) are small, regulatory non-coding RNAs that can modulate gene expression by control of translation and transcription. These small RNA molecules exert their effect partly by binding through interactions of the 5′ most nucleotides of the mature miRNA with near perfect complimentarity to the 3′ UTR of the target gene, resulting in either translational repression or enhanced degradation of the mRNA [1–3]. It has been estimated that miRNA may regulate up to one-third of all human genes, and they have been implicated in a diverse number of mammalian cellular processes [4].
Alterations in expression of miRNAs within the liver have been recently shown to critically modulate cellular processes, such as liver carcinogenesis and fibrosis. Amongst the miRNA with deregulated hepatic expression, miR-29 has emerged as an important miRNA capable of modulating both carcinogenesis and fibrosis [5–7]. In addition to the liver effects, miR-29 has also been implicated in several other models of fibrosis, such as systemic sclerosis, pulmonary fibrosis and cardiac fibrosis [8–10]. Several validated targets of miR-29 are involved in cell survival and tumourigenesis. These include oncogenes and apoptosis-related molecules, such as T-cell leukaemia/lymphoma 1, Bcl-2, myeloid cell leukaemia sequence 1 (Mcl-1), cell division cycle 42 and phosphoinositide-3-kinase regulatory subunit 1 [13,11]. Both Bcl-2 and Mcl-1 are direct targets of miR-29, and the mitochondrial pathway is activated in miR-29-promoted apoptosis [5]. The expression of miR-29 can be regulated by c-myc, hedgehog and Toll-like receptor mediated promoter activation [14]. Of note, all collagen-1 and collagen-IV transcripts contain highly conserved binding sites for miR-29 in their 3-untranslated regions, and collagen-IV has been experimentally validated as a miR-29 target [15].
Although several targets of miR-29 have been identified, there is limited information regarding the tissue specific roles in pathobiology. Modulation of miR-29 has been examined in whole livers and experimentally studied in hepatocytes, cholangiocytes and hepatic stellate cells within the liver [6]. As the expression and targets may vary in a cell-type and tissue specific manner, further analysis of hepatic responses requires models that will enable in vivo analysis of cell-type specific miRNA-dependent effects. To address the role of miR-29 in the liver conclusively, during post-natal growth and development as well as in pathophysiological processes, we used the floxed miR29ab1 mice and mice expressing Cre recombinase under albumin promoter and enhancer (Alb-Cre mice) to conditionally knockout miR-29 in hepatocytes. Our goals were to evaluate the role of hepatic expression of miR-29 with a view to elucidating the role of this miRNA in hepatic pathobiology.
Materials and methods
Generation of miR29ab1 conditional knockout mice
Homozygous floxed miR29ab1 mice (C57BL6 strain) were generated as follows. For the targeting construct, two homologous recombination arms were amplified by PCR, on 129 SvJ/X1 genomic DNA, a 5′ one of 4171 bps and a 3′ one of 3857 bps. Also, the genomic fragment to be deleted, of 600 bps containing the miR29a and miR29b1, was amplified the same way and cloned in between two loxP sites, in a pFlox vector. The recombination arms together with the floxed genes were all cloned into Gateway vectors and then assembled together into a destination vector that represented the targeting vector. 129SvJ/X1 ES cells were electroporated with the targeting vector and clones were screened by Southern Blot. DNA was digested with SacI, and labelled with a 3′ probe. Recombinant clones exhibited two bands: a wild-type one of 5.8 kb and a mutant one of 7.4 kb. One positive clone was identified of 336 screened. This one was expanded and the targeting event was confirmed by Southern Blot. The mutant ES cell clone was injected into C57BL/6 blastocysts, and agouti pups were screened by PCR to verify the generation of heterozygous floxed miR29ab1 mice. Albumin-Cre mice (C57BL/6 strain) were obtained from Jackson laboratories, (Bar Harbor, ME, USA) [16]. Homozygous floxed miR29ab1 mice were bred to albumin-Cre mice and the offspring carrying a floxed miR29ab1 allele, and albumin-Cre were again bred to the homozygous floxed miR29ab1 mice. The mating scheme was similar to that described by Brault et al. [17]. This led to a floxed allele and a flox-deleted allele of miR29ab1 and are referred to as miR29ab1flox/flox, Alb-Cre+ or miR29ab1 knockout (miR29 KO). For all studies, miR29ab1wt/twt, Alb-Cre+ were used as controls.
Comparative phenotype analysis
Comprehensive analysis including gross pathology, histopathology, serum chemistry and haematology assessment was performed in knockout mice (Supplemental methods).
Induction of fibrosis
Liver fibrosis was induced with carbon tetrachloride (CCl4; Sigma-Aldrich, St. Louis, MO, USA) intraperitoneally. CCl4 was diluted with olive oil (CCl4:olive oil = 1:7), sterilized with 0.22 μm-filter, and administrated intraperitoneally twice weekly at 4 μl/g body weight for 10 weeks. A total of 36 mice aged 17–19 weeks old (18 miR29ab1 knockout mice and 18 control mice) were randomized into CCl4 treatment group (10 miR29ab1 knockout mice and 9 control mice) or a vehicle treatment group (nine miR29ab1 knockout mice and eight control mice). After 10 weeks treatment, the surviving mice were killed. The liver was removed and was either immediately frozen in liquid nitrogen for storage at −80°C or fixed with 10% neutralized formalin for histological examination. For the quantification of fibrosis, liver tissue was cut in 4 μm-thick sections and was stained with haematoxylin and eosin, and Masson's trichrome. At least five microscopic images of tissue sections with trichrome staining from each mouse were captured randomly by using digital imaging software (NIS-Elements; NIKON, Tokyo, Japan), and the per cent area of fibrosis was analysed and quantified using NIH ImageJ software (National Institute of Health, Bethesda, MD, USA) [18].
RNA isolation
Total RNA was extracted from mouse liver tissue with Trizol reagent (Invitrogen, Carlsbad, CA, USA). RNA concentration was measured using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA), and RNA quality was determined using an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc, Santa Clara, CA, USA). The quality of RNA was considered suitable for array experiments, if samples showed clear peaks of 18S and 28S ribosomal RNA without any DNA contamination sign and had an RNA integrity number >8.0.
Real-time quantitative RT-PCR for miR-29
cDNA was generated using TaqMan microRNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA). The transcribed cDNA was diluted 50 times with DNase-free water and real-time quantitative RT-PCR (qRT-PCR) was performed with TaqMan MiRNA assays (Applied Biosystems) with an Mx3000p System (Stratagene, La Jolla, CA, USA). The determined threshold cycle (CT) was normalized with snoRNA202 as endogenous control, and the relative amount of miR-29 expression in knockout mouse to control mouse was described using the comparative CT method [19]. For studies on H19, pro-collagen I and alpha-smooth muscle actin expression, U6 RNA was used as a control.
Genome-wide expression profiling
Gene expression profiling was performed with total RNA samples from the liver tissues of miR-29ab1 knockout and control mice from both the CCl4 group and vehicle control groups (n = 4 each). The total RNA was hybridized onto Affymetrix GeneChip Mouse Exon ST 1.0 Array according to standard Affymetrix procedures, and data were normalized with full quantile normalization [20] using XRAY software (Biotique System, Reno, NV, USA). Bioinformatics analysis was performed with DAVID [21].
Results
Generation of knockout mouse
Recent studies have identified a role for miR-29 in hepatic survival, fibrosis and carcinogenesis, implicating this miRNA as a key mediator of several pathophysiological processes within the liver [5,6]. To directly evaluate the role of this miRNA within the liver, we generated a liver specific miR29ab1 knockout mouse as outlined in the Methods section (Fig. 1). A reduction in miR-29 was observed within the liver (Fig. 2) but not the spleen. miR-29c expression was not altered (data not shown). Mice with reduced hepatic miR-29ab1 expression were viable and reproduced normally. The behaviour of the knockout mice was not noticeably different from that of controls, and there were no overt lesions noted in the liver of any of the mice. These observations indicate that liver development is not dependent on hepatic expression of miR-29.
Fig. 1.
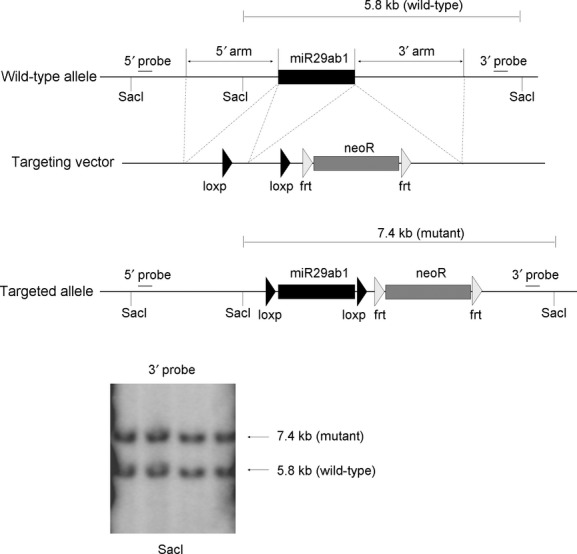
Targeted disruption of the mouse miR29ab1 gene. The structures of the wild-type allele and the disrupted allele are shown. The targeting vector was constructed from two homologous recombination arms, a 5′ one of 4171 bps and a 3′ one of 3857 bps, and a 600 bp genomic fragment containing miR29a and miR29b1 cloned in between two loxP sites. Genomic DNA was used for Southern blot analysis after digestion with SacI and labelled with a 3′ probe. Recombinant clones exhibited two bands: a wild-type one of 5.8 kb and a mutant one of 7.4 kb.
Fig. 2.
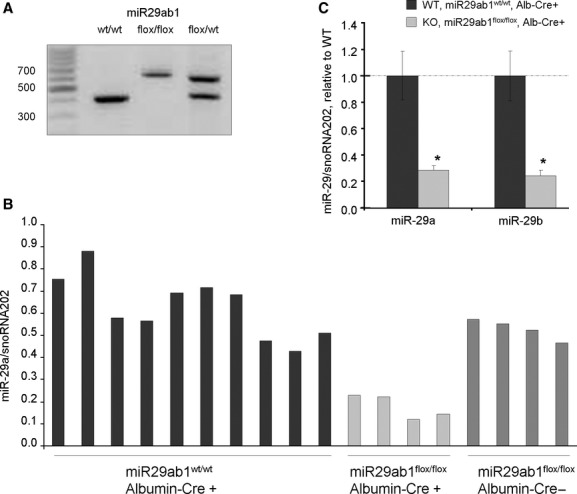
miR-29 expression in vivo. (A) PCR analysis of DNA from mouse tail. Flox sites were inserted upstream and downstream of miR29ab1 site. In presence of Cre, Flox sites could be digested and miR29ab1 site cut-off. Cre was expressed under the albumin promoter and therefore miR-29a and miR-29b was selectively knocked out in hepatocytes. (B) RNA was isolated from liver tissues from wild-type (miR29ab1wt/wt, Cre+), knockout (miR29ab1flox/flox, Cre+) or floxed mice (miR29ab1flox/flox, Cre−), and miR-29a expression was assessed using a Taqman based quantitative RT-PCR assay. Mir-29a expression was selectively reduced in miR29ab1flox/flox, Cre+ animals. (C) The expression of miR-29a and miR-29b was assessed by qRT-PCR and normalized to that of smoRNA202. The bars represent mean and standard error of expression in four mice, relative to wild-type controls (WT). *P < 0.05 relative to controls.
Comparative phenotype analysis
Comparative phenotyping studies did not reveal major morphological or histological differences in major organs between the miR29ab1 knockout mice and control littermate wild-type mice at age 18 weeks. Organ and body weights were similar between the genotypic groups. Haematological analyses did not identify any significant differences in erythrocyte, leucocyte or platelet counts. Biochemical parameters were not overtly different between the two groups except for mild elevations in aspartate aminotransferase and creatine phosphokinase in the knockout mice. Of note, both knockout mice had localized histological lesions in the skeletal muscle and skin that were lacking in wild-type mice. There was evidence of myofiber degeneration, regeneration and fibrosis in skeletal muscle attached to the interscapular brown adipose tissue. Ulcerative dermatitis was also noted in both knockout mice but not in wild-type littermates.
Functional effects on gene expression
To identify the genes that can be modulated in vivo by alterations in miR-29ab1, we analysed gene expression using RNA from livers of 18 week old male mice and their wild-type littermates. A genome-wide expression analysis was performed with the Affymetrix mouse exon 1.0 array gene chip. Compared with wild-type mice, the expression of 116 genes was increased and the expression of 100 genes was decreased by greater than twofold with a P < 0.01 (Supplemental Fig 1). These included alterations in genes that have been implicated in fibrosis, such as PDGF, as well as genes involved in responses to hepatic injury, such as CDKN1A, CYP7A1, MT1F and SSA2. Ingenuity Pathway Analysis of highly deregulated genes (greater than fourfold) revealed that the top two gene networks participated in behaviour, nervous system development and function, cell death (score 48) and organismal injury, cell cycle and lipid metabolism (score 26). The top three canonical pathways were PXR/RXR activation, circadian rhythm signalling and cell cycle: G2M DNA damage checkpoint regulation. Thus, loss of hepatic miR-29 results in deregulation of genes that could modulate several potential pathways and key genes involved in liver injury and fibrosis.
Response to chronic hepatic injury
Hepatic fibrosis is orchestrated by a complex network of signaling pathways, regulating the deposition of extracellular matrix proteins during fibrogenesis [22]. It is believed that miR-29 expression can modulate liver fibrosis [6,7]. The development or use of any hepatic targeting strategy to modulate miRNA in the liver needs to consider the impact of miR-29 knockdown on hepatocytes, the major epithelial cell type. CCl4 results in inflammation and cell injury, which will progress to liver fibrosis and cirrhosis if the repair capacity of the liver is exceeded. To ascertain the involvement of miR-29 in hepatic fibrosis following a hepatotoxic injury, we administered CCl4 to both miR29ab1 knockout and control mice, and assessed the fibrotic responses in these animals. Compared with their wild-type controls an increase in fibrosis was observed in the miR29ab1 knockout mice (Fig. 3). Consistent with these changes, Procollagen 1 expression was increased in miR-29ab knockout mice compared with wild-type controls, with Procollagen1/U6 delta Ct of 7.9 ± 0.3 and 7.2 ± 0.3 respectively (n = 3, P < 0.05). However, a significant change in alpha-smooth muscle actin was not observed [delta Ct of 5.1 ± 1.1 in miR29ab1 knockout compared with 4.8 ± 0.7 in wild-type controls (n = 3, P > 0.05)]. In addition, mortality was increased in the miR29ab1 knockout mice (Fig. 4). These observations indicate that the loss of miR-29ab1 in the liver is pro-fibrogenic and contributes to progressive disease following CCl4.
Fig. 3.
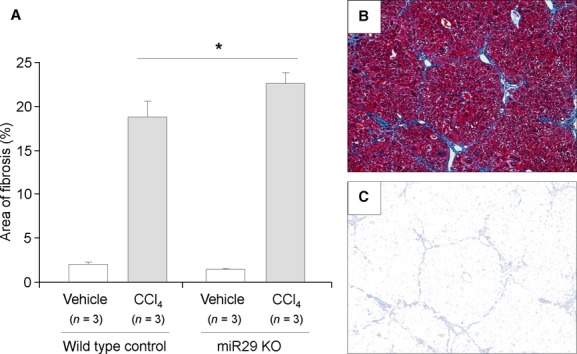
Increased fibrosis in miR29ab1 knockout mice following administration of carbon tetrachloride. CCl4 (0.5 μl/g body weight) or vehicle (olive oil) was administered twice weekly to either miR29ab1 knockout mice or to wild-type mice. The liver was excised after 10 weeks, and the extent of fibrosis assessed by image analysis after staining with Masson's trichrome. Compared with vehicle controls, an increase in fibrosis was noted with CCl4. The extent of fibrosis was greater in miR29ab1 knockout mice than in wild-type mice. (A) bars express a per cent area of fibrosis ± SEM from three mice in each group. *P < 0.05. (B) a representative image of fibrotic liver with trichrome staining. (C) a representative image of the area with extracted fibrosis.
Fig. 4.
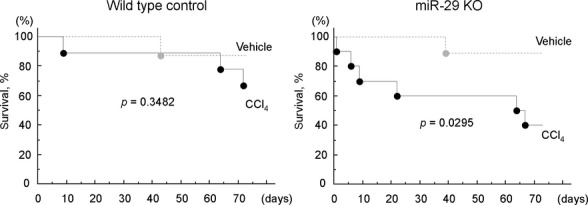
Increased mortality in miR29ab1 knockout mice following carbon tetrachloride (CCl4) administration. CCl4 (0.5 μl/g body weight) or vehicle (olive oil) was administered twice weekly to either miR29ab1 knockout mice or to wild-type control mice. Animals were followed for up to 3 months. Survival curves were generated and analysed by Kaplan–Meier plots. Compared with wild-type control mice, survival was reduced after administration of CCl4 in miR29ab1 knockout mice indicating a greater susceptibility to hepatic injury in these mice.
miR-29 dependent gene expression during chronic hepatic injury
To identify miR-29 dependent genes that contribute to these effects, we examined gene expression in wild-type control mice and miR29ab1 knockout mice treated with CCl4 twice weekly. Whole genome gene expression was examined at 10 weeks using Affymetrix gene chip exon 1.0 ST array. 160 genes were increased in knockout mice but not in controls by CCl4. To understand the functional implications of alterations in these genes, we performed functional bioinformatics analysis using the DAVID programme to identify biological processes altered by miR-29 knockdown in vivo. Gene ontology analysis using DAVID identified regulation of fibroblast proliferation as the most enriched gene ontology classification, with a 24.5-fold enrichment compared with whole genome expression (Table 1). We next sought to identify miR-29 dependent genes modulated during chronic CCl4. In miR29ab1 knockout mice, CCl4 treatment resulted in a decrease in expression of 825 genes (11%) and an increase in 379 genes (5%). Changes in gene expression can arise from loss of miR-29ab1 in hepatocytes, or reflect a fibrotic environment and altered tissue composition with variations in cell populations. To address these changes, we examined genes altered in miR29ab1 knockout or wild-type control mice in response to CCl4. In wild-type mice, CCl4 increased expression of 763/7486 transcripts (10.2%). Amongst these 763 genes, 204 genes were decreased in expression in miR29ab1 knockout mice receiving CCl4 compared with wild-type receiving CCl4 (Table 2). Similarly, CCl4 resulted in decreased transcription of 369/7586 genes (4.9%), and of these, the expression of 24 genes was noted to be increased in the miR29ab1 knockout mice, amongst which, H19 was the most significantly altered in expression. These findings were confirmed using real-time PCR, with delta Ct H19/U6 in miR291ab1 knockout mice of 4.7 ± 0.7 in CCl4 treated mice compared with 8.1 ± 0.5 in untreated controls (n = 3 replicates).
Table 1.
Biological processes altered in miR29ab1 knockout mouse by CCl4 treatment
| Gene ontology term | P-value | Fold enrichment |
|---|---|---|
| Biological processes identified in increased genes | ||
| Regulation of fibroblast proliferation | 0.0005 | 24.5 |
| Regulation of cell proliferation | 0.0014 | 3.0 |
| Positive regulation of macromolecule biosynthetic process | 0.0037 | 2.8 |
| Positive regulation of cellular biosynthetic process | 0.0051 | 2.7 |
| Positive regulation of transcription | 0.0051 | 2.8 |
| Positive regulation of biosynthetic process | 0.0054 | 2.6 |
| Positive regulation of gene expression | 0.0062 | 2.8 |
| Positive regulation of transcription, DNA-dependent | 0.0066 | 2.9 |
| Positive regulation of RNA metabolic process | 0.0069 | 2.9 |
| Positive regulation of cell proliferation | 0.0083 | 3.4 |
| Biological process identified in decreased genes | ||
| RNA processing | 4.08e-08 | 2.9 |
| Translation | 2.06e-07 | 3.2 |
| Protein catabolic process | 5.44e-07 | 2.5 |
| Ribonucleoprotein complex biogenesis | 1.05e-06 | 4.5 |
| Oxidation reduction | 1.23e-06 | 2.3 |
| Ribosome biogenesis | 2.20e-06 | 4.9 |
| Nucleoside bisphosphate metabolic process | 6.04e-06 | 19.9 |
| Macromolecule catabolic process | 9.63e-06 | 2.2 |
| Cellular protein catabolic process | 3.08e-05 | 2.2 |
| Generation of precursor metabolites and energy | 3.44e-05 | 2.9 |
Whole genome gene expression in miR29ab1 knockout mouse liver was examined after CCl4 administration for 10 weeks. Total 160 genes were increased and 555 genes were decreased only in knockout mouse liver but not in control. Functional bioinformatics analysis was performed using the DAVID programme to identify biological processes altered by miR29ab1 knockdown in mice liver. Top 10 biological processes identified in increased and decreased genes are listed.
Table 2.
Genes altered via miR-29ab1 knockdown during CCl4 treatment
| Symbol | Gene description | Fold change |
|---|---|---|
| Genes decreased via miR-29ab1 knockdown | ||
| h19 | H19 foetal liver mRNA | −17.33 |
| ngp | Neutrophilic granule protein | −10.30 |
| retnlg | Resistin like gamma | −9.28 |
| umod | Uromodulin | −6.45 |
| lgals3 | Lectin galactose binding soluble 3 | −5.95 |
| camp | Cathelicidin antimicrobial peptide | −5.46 |
| gpnmb | Glycoprotein (transmembrane) nmb | −5.24 |
| cd24a | CD24a antigen | −5.06 |
| mmp8 | Matrix metallopeptidase 8 | −4.60 |
| sirpb1 | Signal-regulatory protein beta 1 | −4.51 |
| Genes increased via miR-29ab1 knockdown | ||
| retsat | Retinol saturase (all trans retinol 13 14 reductase) | 8.81 |
| cyp4a12b | Cytochrome P450 family 4 subfamily a polypeptide 12B | 6.37 |
| cyp4a12a | Cytochrome P450 family 4 subfamily a polypeptide 12a | 5.93 |
| hsd3b5 | Hydroxy-delta-5-steroid dehydrogenase 3 beta- and steroid delta-isomerase 5 | 3.68 |
| csad | Cysteine sulphinic acid decarboxylase | 3.59 |
| bcl6 | B-cell leukaemia/lymphoma 6 | 3.35 |
| 2810007j24rik | RIKEN cDNA 2810007J24 gene | 3.30 |
| slco1a1 | Solute carrier organic anion transporter family member 1a1 | 3.21 |
| glul | Glutamate-ammonia ligase (glutamine synthetase) | 2.97 |
| camk1d | Calcium/calmodulin-dependent protein kinase ID | 2.95 |
Whole genome gene expression was analysed in control mouse without treatment, control and miR29ab1 knockout mouse with CCl4 treatment. The expression of total 763 genes were increased by CCl4 administration in control mouse, and of these, 204 genes decreased in expression in miR29ab1 knockout mouse receiving CCl4 compared with control mouse receiving CCl4. The expression of total 369 genes were decreased by CCl4 in control mouse, and of these, 24 genes increased in expression in miR29ab1 knockout mouse receiving CCl4 compared with control mouse receiving CCl4. The Top 10 genes with altered expression are listed.
Discussion
Although several experimentally validated targets of miR-29 have been described, the in vivo effects of modulation of this miRNA are not known. Alteration in gene expression can occur as a result of direct targeting of mRNA by miR-29. In addition, several known targets of miR-29 can modulate expression of other genes. The ability of miR-29 to target the DNA methyltransferase DNMT could also influence gene expression through epigenetic modulation of their expression [23,24]. Thus, a global analysis of alterations in gene expression in vivo is necessary to recognize the spectrum of gene changes that are modulated by miR-29.
Recent studies have shown that down-regulation of miR-29 was a frequent event in HCC tissues and an independent prognosis predictor for HCC patients [5]. Furthermore, reintroduction of miR-29 dramatically repressed the tumourigenicity of HCC cells and also sensitized HCC cells to apoptosis triggered by different stimuli [5]. Thus, the miR-29 knockout mouse offers the potential of providing new insights into the fundamental role of miR-29 in hepatocarcinogenesis. The identification of H19 as a miR-29 regulated gene in vivo is noteworthy because this gene was originally identified as a raf regulating gene involved in the expression of αFP in mouse liver and has been implicated in hepatocarcinogenesis [25]. Further studies are warranted to examine the mechanistic link between H19 and miR-29. H19 is an imprinted gene that demonstrates maternal monoallelic expression in some cancers and in foetal tissues. Although H19 expression is reduced post-natally, it can be re-activated during tissue regeneration and can function as an oncogene contributing to tumourigenesis. However, the potential role of H19 in liver fibrosis remains unknown [27,26].
Based on the published experience with the use of the Albumin promoter in cre-lox expression systems, we would not expect deletion in cell-types within the liver that do not express albumin. Cell-type specific modulation of miR-29 also allows dissection of the individual cell types in hepatic fibrosis. Although it is presumed that the main extracellular matrix producing cell type in liver fibrogenesis are the hepatic stellate cells, and alterations in miR-29 expression occurs during myofibroblastic transition of hepatic stellate cells [6], our findings provide evidence that alterations in hepatocyte genes could contribute to experimental fibrosis. However, future studies to examine the contribution of individual cell types would require verification of cell-type specific knockout. To our knowledge, this is the first cell-type specific miRNA knockout mouse of a miRNA implicated in liver fibrosis or carcinogenesis. Using this hepatic specific knockout mouse, we show that genetic knockdown of miR-29ab1 in vivo results in modulation of genes involved in fibrosis, enhanced susceptibility to fibrosis and impaired cell survival following a fibrogenic stimulus. Moreover, the oncofetal RNA H19 was identified as a miR-29 modulated gene following CCl4 exposure. We have not identified any sequence complementarity between miR-29 and H19 that would support direct sequence-based effects on H19. These data identify a critical contribution of miR-29ab1 to fibrosis and carcinogenesis within the liver. The hepatic miR-29 knockout mouse will be a useful tool for such studies to determine the physiological roles of miR-29 in vivo within the liver. Further studies will be required to evaluate the mechanisms by which miR-29ab1 can modulate gene changes that result in HCC during chronic hepatic injury and fibrosis.
Acknowledgments
This study was supported by a grant from the National Institutes of Health, USA (DK069370 to TP). SC and CC generated the knockout mice for the study. TK, IY and CB performed the research. TK and TP designed the research study, analysed the data and wrote the study.
Glossary
- CCl4
carbon tetrachloride
- miRNA
microRNA
- Mcl-1
myeloid cell leukaemia sequence 1
Conflict of interest
There are no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Genome-wide expression analysis of miR29ab1 knockout mouse liver. A genome-wide expression analysis was performed by comparing the expression of genes in miR29ab1 knockout mouse liver to wild-type control mouse liver using the genechip mouse exon 1.0 ST array (affymetrix). Differential expression of genes with P-value <0.05 are plotted [y-axis, log10(P-value); x-axis, log2 (fold change)].
Supplemental methods.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–85. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 3.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 4.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 5.Xiong Y, Fang JH, Yun JP, et al. Effects of microRNA-29 on apoptosis, tumourigenicity, and prognosis of hepatocellular carcinoma. Hepatology. 2010;51:836–45. doi: 10.1002/hep.23380. [DOI] [PubMed] [Google Scholar]
- 6.Roderburg C, Urban GW, Bettermann K, et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology. 2011;53:209–18. doi: 10.1002/hep.23922. [DOI] [PubMed] [Google Scholar]
- 7.Jiang X, Tsitsiou E, Herrick SE, Lindsay MA. MicroRNAs and the regulation of fibrosis. FEBS J. 2010;277:2015–21. doi: 10.1111/j.1742-4658.2010.07632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maurer B, Stanczyk J, Jungel A, et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum. 2010;62:1733–43. doi: 10.1002/art.27443. [DOI] [PubMed] [Google Scholar]
- 9.Cushing L, Kuang PP, Qian J, et al. MIR-29 is a major regulator of genes associated with pulmonary fibrosis. Am J Respir Cell Mol Biol. 2010;45:287–94. doi: 10.1165/rcmb.2010-0323OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Rooij E, Sutherland LB, Thatcher JE, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA. 2008;105:13027–32. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pekarsky Y, Santanam U, Cimmino A, et al. Tcl1 expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Res. 2006;66:11590–3. doi: 10.1158/0008-5472.CAN-06-3613. [DOI] [PubMed] [Google Scholar]
- 12.Mott JL, Kobayashi S, Bronk SF, Gores GJ. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene. 2007;26:6133–40. doi: 10.1038/sj.onc.1210436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park SY, Lee JH, Ha M, et al. miR-29 miRNAs activate p53 by targeting p85 alpha and CDC42. Nat Struct Mol Biol. 2009;16:23–9. doi: 10.1038/nsmb.1533. [DOI] [PubMed] [Google Scholar]
- 14.Mott JL, Kurita S, Cazanave SC, et al. Transcriptional suppression of mir-29b-1/mir-29a promoter by c-Myc, hedgehog, and NF-kappaB. J Cell Biochem. 2010;110:1155–64. doi: 10.1002/jcb.22630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwiecinski M, Noetel A, Elfimova N, et al. Hepatocyte growth factor (HGF) inhibits collagen I and IV synthesis in hepatic stellate cells by miRNA-29 induction. PLoS One. 2011;6:e24568. doi: 10.1371/journal.pone.0024568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Postic C, Shiota M, Niswender KD, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274:305–15. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- 17.Brault V, Moore R, Kutsch S, et al. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–64. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- 18.Zeisberg M, Yang C, Martino M, et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007;282:23337–47. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- 19.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 20.Irizarry RA, Bolstad BM, Collin F, et al. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 22.Friedman SL. Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 2010;7:425–36. doi: 10.1038/nrgastro.2010.97. [DOI] [PubMed] [Google Scholar]
- 23.Fabbri M, Garzon R, Cimmino A, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci USA. 2007;104:15805–10. doi: 10.1073/pnas.0707628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Filkowski JN, Ilnytskyy Y, Tamminga J, et al. Hypomethylation and genome instability in the germline of exposed parents and their progeny is associated with altered miRNA expression. Carcinogenesis. 2010;31:1110–5. doi: 10.1093/carcin/bgp300. [DOI] [PubMed] [Google Scholar]
- 25.Vacher J, Camper SA, Krumlauf R, et al. raf regulates the postnatal repression of the mouse alpha-fetoprotein gene at the posttranscriptional level. Mol Cell Biol. 1992;12:856–64. doi: 10.1128/mcb.12.2.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matouk IJ, DeGroot N, Mezan S, et al. The H19 non-coding RNA is essential for human tumour growth. PLoS One. 2007;2:e845. doi: 10.1371/journal.pone.0000845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamamoto Y, Nishikawa Y, Tokairin T, et al. Increased expression of H19 non-coding mRNA follows hepatocyte proliferation in the rat and mouse. J Hepatol. 2004;40:808–14. doi: 10.1016/j.jhep.2004.01.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genome-wide expression analysis of miR29ab1 knockout mouse liver. A genome-wide expression analysis was performed by comparing the expression of genes in miR29ab1 knockout mouse liver to wild-type control mouse liver using the genechip mouse exon 1.0 ST array (affymetrix). Differential expression of genes with P-value <0.05 are plotted [y-axis, log10(P-value); x-axis, log2 (fold change)].
Supplemental methods.