

Abstract
Malignant peripheral nerve sheath tumors (MPNSTs) develop sporadically or in the context of neurofibromatosis type 1 (NF1). EGFR overexpression has been implicated in MPNST formation, but its precise role and relevant signaling pathways remain unknown. We found that EGFR overexpression promotes mouse neurofibroma transformation to aggressive MPNST (GEM-PNST). Immunohistochemistry demonstrated phosphorylated STAT3 (Tyr705) in both human MPNST and mouse GEM-PNST. A specific JAK2/STAT3 inhibitor FLLL32 delayed MPNST formation in an MPNST xenograft nude mouse model. STAT3 knockdown by shRNA prevented MPNST formation in vivo. Finally, reducing EGFR activity strongly reduced pSTAT3 in vivo. Thus, an EGFR-STAT3 pathway is necessary for MPNST transformation and establishment of MPNST xenografts growth but not for tumor maintenance. Efficacy of the FLLL32 pharmacological inhibitor in delaying MPNST growth suggests that combination therapies targeting JAK/STAT3 might be useful therapeutics.
Keywords: EGFR, STAT3, Sarcoma, MPNST, NF1
Introduction
Malignant peripheral nerve sheath tumors (MPNSTs) are aggressive incurable sarcomas that arise in 0.001% of the general population. However, the incidence of MPNST increases to 8-13% in neurofibromatosis type 1 (NF1) patients (1-4). In NF1, benign peripheral nerve plexiform and subcutaneous neurofibromas can undergo malignant transformation to MPNSTs (5, 6). MPNSTs are a major source of mortality for NF1 patients because only complete surgical resection is curative and complete resection is often not feasible due to tumor location or metastasis. MPNST patients are often not sensitive to chemotherapy or radiation therapy (1, 7).
MPNSTs derive from either neural crest cells or glial cells that dedifferentiate to a neural crest-like cell (8-11). Hyperactive Ras, resulting from the loss function of neurofibromin, is a major contributor of Schwann cells malignant transformation to MPNST. Indeed, mass spectroscopy-based analysis of an MPNST cell line failed to detect mutations in 19 commonly activated oncogenes (12).
Epidermal growth factor receptor (EGFR or erbB1) is a 170 kDa transmembrane tyrosine kinase, which triggers signaling processes that promote cell proliferation, migration, adhesion, and angiogenesis, and inhibits apoptosis (13). While peripheral nerve Schwann cells that might be cells of origin of MPNST do not express EGFR, Schwann cell progenitors and MPNST cells do (DeClue et al 2000, Li et al 2002). Thus EGFR is expressed in 80% of human MPNSTs, and most human and mouse MPNST cell lines (14-18).
EGFR is deregulated in a variety of solid malignant tumors by overexpression of EGFR protein, activating point mutations in the tyrosine kinase domain, or other mutations. In NF1, EGFR is over-expressed – not activated by mutation (15, 18). In a compound cis-heterozygous Nf1 and p53 (NPcis) mouse sarcoma model (9, 10), loss of EGFR function by breeding the NPcis mouse to a mouse carrying an EGFR hypomorph in mutation Wa2 (Wa2+) (19) resulted in delayed sarcoma formation (20). Taken together the data support a role for EGFR in MPNSTs. It is unclear, however, whether EGFR is sufficient or necessary for MPNST malignant transformation and/or tumor maintenance.
Signal transducer and activator of transcription 3 (STAT3) is a latent transcription factor that participates in transcriptional activation of apoptosis and cell-cycle progression, and has been implicated as an oncogene and therapeutic target in neoplastic disease (21). STAT3 phosphorylation at (Tyr705) is essential for STAT3 dimerization that, in turn, is required for STAT3 binding to DNA-promoter regions and, thus, transcriptional activation (Battle and Frank, 2002). The EGFR-src-STAT3 oncogenic pathway is involved in cell transformation and tumor escape from genotoxic treatment (22). STAT3 also regulates self-renewal and growth of glioblastoma stem cells (23).
STAT3 is hyperactivated in Nf1−/− primary astrocytes and STAT3 inhibition by cucurbitacin-I blocked human MPNST growth in vivo when delivered for 7 days at onset of cell xenograft (24). Whether EGFR-dependent STAT3 activation plays a role in MPNST transformation is unknown. Here we report that EGFR overexpression in a neurofibroma mouse model is sufficient to promote neurofibroma transformation into mouse MPNST (GEM-PNST). STAT3 (Tyr705) was hyper-phosphorylated in mouse GEM-PNSTs and human MPNSTs, and inhibition of STAT3 with the specific JAK2/STAT3 inhibitor FLLL32, which blocks STAT3 activity both by blocking STAT3 dimerization and JAK2 interaction with STAT3 (25), delayed MPNST xenograft formation in nude mice. Short-heparin RNA targeting STAT3 (shSTAT3) prevented MPNST xenograft tumor formation in vivo. These data support a role for the EGFR-STAT3 pathway in MPNST transformation, but not in tumor maintenance.
Results
Overexpression of CNP-hEGFR is sufficient to drive aggressive sarcoma formation in the Nf1fl/fl;DhhCre neurofibroma mouse
We used a neurofibroma mouse model in which 100% of mice develop benign neurofibromas, but there is no GEM-PNST formation (26). To test whether increased EGFR expression is sufficient to drive GEM-PNST formation, we bred Nf1fl/+;DhhCre;CNP-hEGFR and Nf1fl/fl mice – obtaining Nf1fl/fl;DhhCre;CNP-hEGFR and Nf1fl/fl;DhhCre mice in the expected Mendelian ratios. All fifteen Nf1fl/fl;DhhCre;CNP-hEGFR mice developed neurofibromas. Surprisingly, five mice (33.3%) developed visible tumors at 4-8 months of age. These tumors were rapidly growing and either under or protruding through the skin; the protruding tumors sometimes exhibited surface ulceration (Figure 1A). The histological and immunohistochemical features of the rapidly growing tumors are consistent with GEM-grade III PNST (27). In one tumor we detected a transition zone of neurofibroma [low to moderate cellularity and nuclear atypia of rare cells, scattered S100β+ cells (Figure 1C, 1D, 1F) ] to densely cellular tumor GEM-PNST characterized by spindle-shaped cells with numerous mitotic figures and foci of necrosis (Figure 1C, 1E). In addition, all GEM-PNSTs showed association between nerves and scattered S100β+ cells (Figure 1G), typical of GEM-grade III PNSTs. All GEM-grade III PNST tumors expressed the human EGFR transgene (6 of 6) (Figure 1H). The results suggest that GEM-grade III PNSTs developed from pre-existing neurofibromas.
Figure 1. Overexpression of CNP-hEGFR promotes GEM-PNST formation in Nf1fl/fl;DhhCre mice.
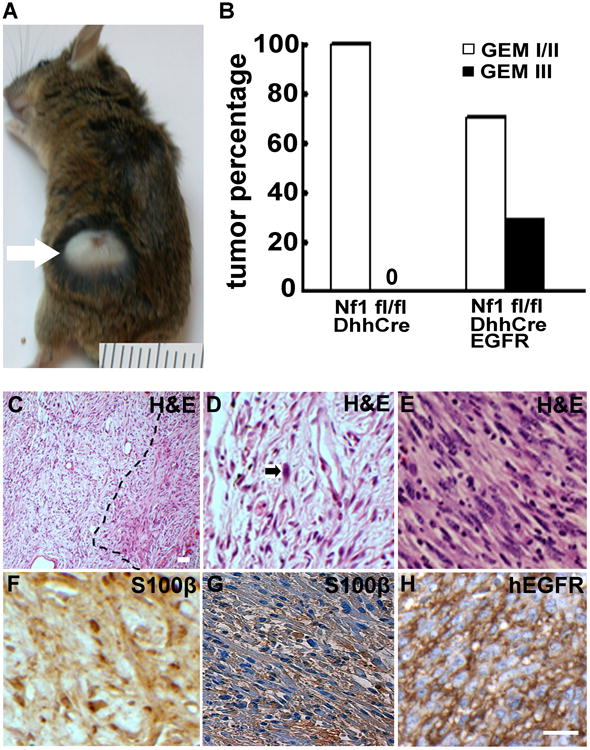
(a) Representative photograph of a rapidly enlarging tumor, with surface ulceration, in an Nf1fl/fl;DhhCre;CNP-hEGFR mouse. A ruler showing 1 mm markings is included. (b) Grading of tumors. The y-axis shows percentages of GEM-I/II (white bars), and GEM-III PNSTs (black bar) in Nf1fl/fl;DhhCre, Nf1fl/fl; DhhCre;CNP-hEGFR, and Nf1fl/fl;DhhCre;Wa2 mice. (c-h) Neurofibroma and GEM-III PNST histology and immunostaining in paraffin-embedded sections (c) Low magnification shows a neurofibroma and GEM-PNST transition zone within a tumor (dot line). (d) H&E shows low to moderate cellularity and nuclear atypia of rare cells (black arrow) in neurofibroma. (e) H&E staining showing densely cellular tumors composed of spindle-shaped cells with numerous mitotic figures, foci of necrosis, large fascicles cells with large pleomorphic nuclei in GEM-PNST. (f) S100β staining shows some S100β+ cells (brown) in neurofibroma. (g) S100β staining shows S100β+ nerve bundles entrapped in mostly S100β− tumor cells in GEM-PNST. (h) immunostaining of EGFR+ cells visualized with DAB (brown) – confirming that tumor cells are derived from CNP-hEGFR transgene-expressing cells. Bar = 20 μm.
STAT3 is activated in human MPNSTs and mouse GEM-PNSTs
If pathways upstream of STAT3 regulate MPNST growth, pSTAT3 should be detected in these peripheral nerve tumors. We therefore labeled paraffin sections of human MPNSTs and mouse GEM-PNSTs with antibodies that recognize the major STAT3-phosphorylation sites. Tyrosine 705 is necessary for STAT3 dimerization and translocation to the nucleus. Immunopositive staining for pSTAT3 (Tyr705) was detected in all human MPNSTs (n = 15) and mouse GEM-PNSTs (n = 6) (Figure 2A and 2B). The percentage of cells expressing pSTAT3 (Tyr705) varied among tumors (from less than 25% to more than 75%, Figure 2C). In contrast, detectable levels of pSTAT3 (Ser727) were found in only one of four mouse GEM-PNSTs, and only two of four human MPNSTs (data not shown).
Figure 2. STAT3 is activated in both mouse GEM-PNSTs and human MPNSTs.
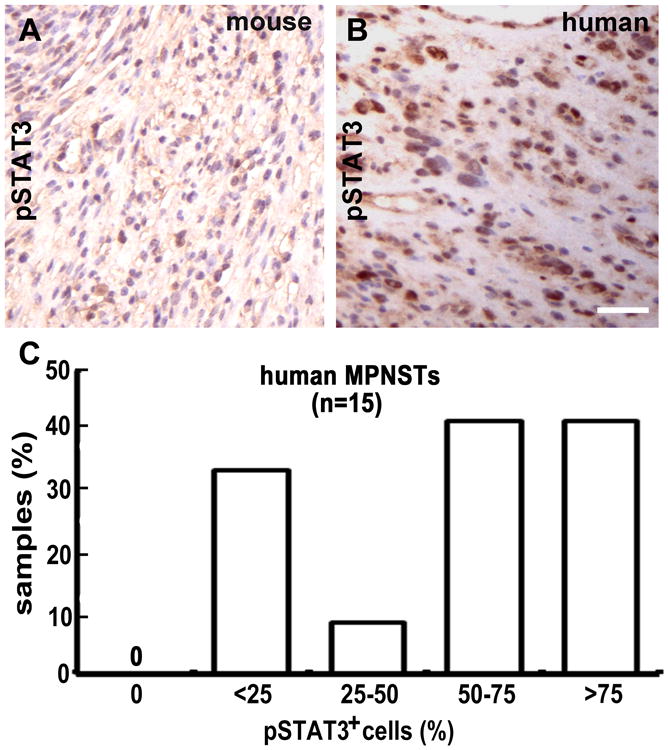
(a-b) pSTAT3 immunostaining in both (a) mouse GEM-PNST and (b) human MPNST, visualized with DAB (brown). (c) Quantification of pSTAT3-expressing cells in human MPNSTs.
JAK2/STAT3 inhibition prevents MPNST cells growth in vitro
To test if STAT3 or the Stat kinases JAK 1, 2, or 3 is relevant to MPNST cell growth, we screened JAK and JAK2/STAT3 inhibitors. We conducted a dose-response analysis of FLLL32, a specific JAK2/STAT3 inhibitor (25) on five MPNST cell lines, four of which were from NF1 patients. After four days of treatment, FLLL32 decreased cell survival in 5/5 tested cell lines. The average IC50 was around 200 nM (Figure 3C). In contrast, treatment of normal human Schwann cells with FLLL32 (0.5μM) did not affect viability of normal human Schwann cells (Supplemental figure 1A).
Figure 3. Inhibition of JAK2/STAT3 decreases MPNST cell growth in vitro.
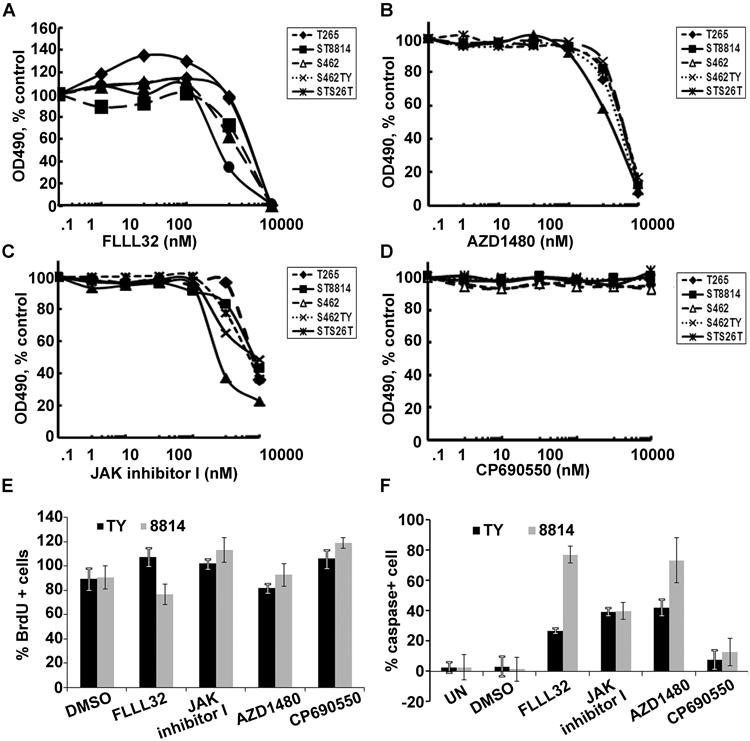
MTS assay showing decreased living cells in response to (a) FLLL32, (b) AZD1480, (c) JAK inhibitor I, and (d) CP-690550 in five human MPNST cell lines. (e) BrdU incorporation assay of FLLL32, JAK inhibitor I, AZD1480, CP690550, and DMSO control on S462TY (black) and 8814(grey) cells. (f) apoptosis assay of FLLL32, JAK inhibitor I, AZD1480, CP690550, and DMSO (0.1%) control on S462TY (black) and 8814 (grey) cells.
We then tested effects of JAK inhibitors on the same human MPNST cell lines. The JAK1/AK2 inhibitor AZD1480 (Figure 3B) and JAK 1, 2, and 3 inhibitor JAK inhibitor I (Figure 3C) had similar effects as FLLL32, decreasing human MPNST cell survival. The JAK3 inhibitor CP6690550 did not affect cell viability (Figure 3D). These inhibitors had no significant effects on cell proliferation as evidenced by BrdU incorporation (Figure 3E), but FLLL32, JAK inhibitor I, and AZD1480 induced cell apoptosis (Figure 3F). These inhibitors did not affect viability of normal human Schwann cells (Supplemental figure 1A). These human MPNST cell lines exhibited very similar dose kinetics in response to JAK inhibitors treatment because they have similar level of pSTAT3 (Tyr 705) and EGFR (Supplemental figure 1A). We performed western blots on lysates from inhibitor treated 8814 and S462TY cells. All the inhibitors inhibited pSTAT3 and pJAK2, but none of them affected pERK except CP690550 (Supplemental figure 1C, 1D). These results suggest that JAK1/2 mediated STAT3 signaling contributes to MPNST cell survival. Due to the unavailability of a specific JAK1 inhibitor, we could not differentiate if the survival effect was caused by JAK1 or JAK2, or both. However, together with the JAK2/STAT3 inhibitor FLLL32 results, we concluded that JAK2/STAT3 inhibition can prevent MPNST cells growth.
Pharmacological inhibition of STAT3 delays MPNST growth in vivo
We tested whether blocking the JAK2/STAT3 pathway might affect growth in an MPNST xenograft model. We treated nude mice with S462TY MPNST tumors >250mm3 (n = 10) with FLLL32 (200mg/kg) daily intraperitoneal injection. We sacrificed the treated mice by 23 consecutive days of treatment when the tumor size reached >10% of the mouse body weight, and did not detect significant changes in tumor size compared to vehicle treated controls (Figure 4A). However, when we treated the nude mice with S462TY MPNST tumors beginning when tumors were small (average 50mm3) (n = 10) with FLLL32 (200mg/kg) daily, tumor formation was significantly delayed (∼2 weeks) compared to vehicle treated controls (n=12, Figure 4B, p<0.0001). These results suggest that STAT3 affects the early stage of MPNST tumor growth.
Figure 4. JAK2/STAT3 inhibitor FLLL32 delays mouse MPNST growth in vivo.
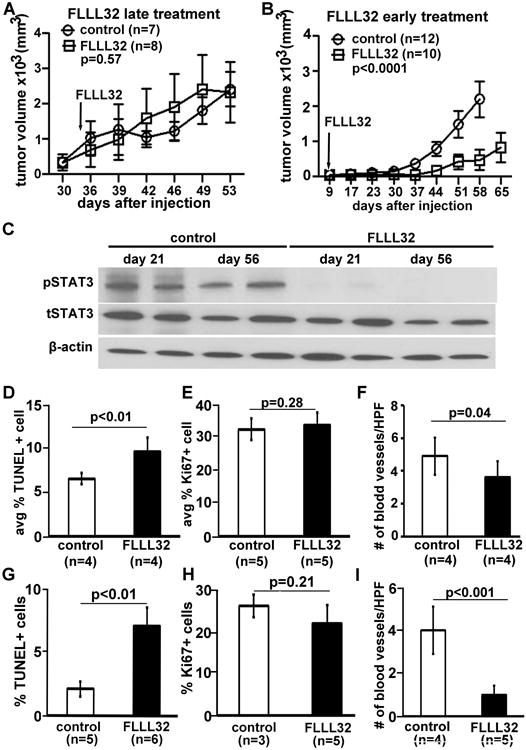
(a-b) Differential inhibition effect of FLLL32 on S462TY mouse MPNST growth on nude mice. (a) There is no significant different change of tumor size when the treatment started with tumor >250mm3 (n=8) compared to vehicle treated controls (n = 7) with FLLL32 (200mg/kg) daily (p=0.57). (b) There is significant different change of tumor size when the treatment started with tumor ∼50mm3 (n=10) compared to vehicle treated controls (n = 10) with FLLL32 (200mg/kg) daily (p<0.0001). (c) Western blot of FLLL32 inhibition effects on MPNST xenografts. β-action was used as loading control. (d, g) FLLL32 promotes S462 TY MPNST cell death at both early (d) or later (g) stage. (e, h) FLLL32 has no effect on inhibiting s S462 TY MPNST cell proliferation at both early (e) or late (h) stage. (f, i) FLLL32 has no significant effect on blood vessel at early stage (f), but has significant difference in blood vessels at later stage (i) compared to vehicle controls.
To determine how FLLL32 delays MPNST growth, mice treated with 200mg/kg/day FLLL32 or vehicle control for 14 days (early group) when FLLL32 is effective or 56 days (late group) when tumors were resistant to FLLL32 were sacrificed one hour after the final dose. We performed western blot to determine pSTAT3 levels at day 21 (2 weeks after FLLL32 treatment) and day 56 (at the end of experiment) with or without FLLL32 treatment. pSTAT3 was low at both time points, indicated that the drug is effective at both time points (Figure 4C). We then analyzed cell death, cell proliferation, and blood vessels in paraffin sections of excised MPNSTs by TUNEL and Ki67 staining, respectively. In the early group, the average percentage of TUNEL+, dying cells was 9.6±1.6% in FLLL32-treated MPNSTs vs 6.6±0.6% in control (p<0.01, Figure 4D). There was no significant difference in the percentage of Ki67+, proliferating cells in FLLL32-treated MPNSTs (33.7±3.7%, n=5) compared to vehicle control (32.4±3.2%, n=5) (p=0.28, Figure 4E). In the late group, the average percentage of TUNEL+, dying cells was 7.1±1.4% in FLLL32-treated MPNSTs vs 2.1±0.6% in control (p<0.01, Figure 4G). There was no significant difference in the percentage of Ki67+, proliferating cells in FLLL32-treated MPNSTs (21.9±4.3%, n=5) compared to vehicle control (26±2.7%, n=3) (p=0.21, Figure 4H). We also analyzed the number of blood vessels per high power field (HPF) by staining these tumors with the endothelial cell marker MECA 32. Interestingly, there was a decrease in blood vessels per HPF in the early stage of treatment group (p=0.04, Figure 4F), and more effect in the late stage of treatment group (p<0.001, Figure 4I), suggesting that decreased vasculature may also be involved in the drug treatment induced tumor cell death.
The efficacy of STAT3 only in early but not late stage is likely to involved factors other than proliferation, death or change in vasculature, such as initial establishment of tumors, change tumor cytokines, or other factors
STAT3 deficiency abrogates MPNST formation in vivo
With any chemical inhibitor, there is the potential for lack of specificity. Therefore, to confirm the role of STAT3 in MPNST, we transduced NF1 patient-derived S462TY MPNST cells with a lentivirus encoding shRNA targeting STAT3; a non-targeting shRNA also expressing YFP served as a control. Western blot analysis confirmed that STAT3 level was decreased after infection of MPNST cells with this vector, which did not change pAKT or pERK levels, indicating that other signaling pathways remain intact in shSTAT3 treated MPNST cells (Figure 5A). MTS assay of shSTAT3 expressing cells revealed decreased numbers of viable cells, but in contrast to cells exposed to JAK/STAT3 inhibitors cleaved caspase-3 was not detectable, suggesting that these cells were not dying by apoptosis (data not shown). It is possible that target JAK as well as STAT with inhibitors cause a shift to apoptosis. We injected sh-non-target controls or shSTAT3 cells subcutaneously into nu/nu (nude) mice (n = 6). Tumor growth was not detectable on the right sides of animals, injected with STAT3 shRNA-expressing MPNST cells (Figure 5B, arrowhead). However, the left sides, which received the same number of non-target shRNA-expressing MPNST cells, exhibited significant tumor growth (Figure 5B, arrow). Mice were sacrificed at day 64, when volumes of control non-target tumors reached allowable limits (Figure 5C). These results confirm a key role for STAT3 in MPNST tumor formation in xenografts.
Figure 5. Genetic inhibition of STAT3 prevents growth of S462TY xenograft in nude mice.
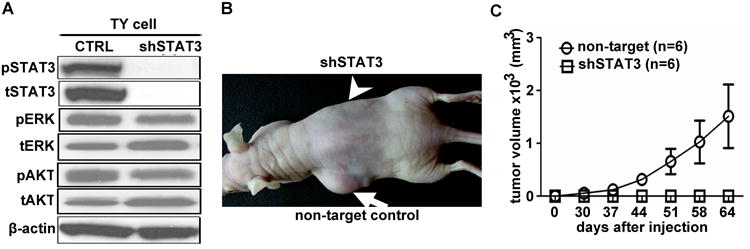
(a) Western blot confirming knock down of both pSTAT3 and total STAT3 (tSTAT3) in NF1 patient-derived S462TY MPNST cells infected with shRNA-STAT3 lentivirus; control is non-target YFP-shRNA. A and B designated different shRNA. (b) Athymic nude mice were injected subcutaneously with shRNA STAT3 (shRNA A)-expressing S462TY MPNST cells in one flank (arrow head), and non-target shRNA-expressing cells in the other flank (arrow). Tumor size was photographed at day 64. Tumor growth was only detectable on the control side (arrow). (c) Tumor volume was measured twice weekly, and mice sacrificed at day 64. Tumor volume calculations demonstrate that shSTAT3 blocks growth of MPNST S462TY xenografts (n=6) compared to non-target (n=6) (p<0.01).
EGFR/JAK2/STAT3 activation contributes to MPNST transformation
To determine if EGFR expression contributed to STAT3 (Tyr705) phosphorylation in vivo, we monitored STAT3 (Tyr705) phosphorylation in the NPcis mouse GEM-PNST compared to NPcis;Wa2/+ mouse GEM-PNSTs (20). pSTAT3 decreased significantly in NPcis;Wa2+; mouse GEM-PNSTs (n=4, Figure 6A), compared to NPcis mouse GEM-PNSTs (n=4, Figure 6B, 6C, p<0.001). Western blots confirmed decreased pJAK2, total JAK2 (tJAK2) and pSTAT3 in NPcis;Wa2/+ mouse GEM-PNST tumors compared to the NPcis GEM-PNSTs (Figure 6D), suggesting that the decrease of pSTAT3 (Tyr 705) is attributed at least in part JAK2, but we can not exclude the possibility that the STAT3 is directly phosphorylated and activated by EGFR. To determine if EGFR expression correlates with STAT3 (Tyr705) phosphorylation, we stimulated serum starved 8814 cells with EGF (20ng/mL) for 10 minutes and monitored pEGFR and pSTAT3 (Tyr705) levels. Both EGFR and STAT3 (Tyr705) were robustly phosphorylated in the EGF stimulated cells (Figure 6E). In 8814 cells treated with the EGFR tyrosine kinase inhibitor OSI-774, pSTAT3 (Tyr705) deceased correlated with pEGFR level (Figure 6F). These gain and loss of function of EGFR results demonstrate that EGFR is a significant activator of STAT3 in MPNST cells.
Figure 6. EGFR/STAT3 activation contributes to MPNST transformation.

(a) Immunofluorescence data showing that pSTAT3 is detected in mouse GEM-PNST (A; NPcis) and decreased in NPcis;Wa2/+ mouse GEM-PNSTs (b). (c) Quantification of the percentage of pSTAT3+ cells shows that the decrease is significant (p<0.001). (d) Western blots showed decreased pJAK2, tJAK2 and pSTAT3 in NPcis;Wa2/+ mouse PNST tumors compared to the PNSTs. Number 1 and 2 indicated tumors. Β-actin was used as control. The number indicated the pJAK2/JAK2 ratio. (e) Western blots showed EGF stimulation increased pEGFR and pSTAT3 levels. (f) Western blots showed EGFR inhibition decreased pEGFR and pSTAT3 levels. For all of the Western bots, β-action was used as loading control
Discussion
We found that increasing EGF receptor levels in Nf1 peripheral nerve glial cells is sufficient to promote the transformation of benign neurofibroma to aggressive GEM-PNST, the counterpart of human MPNST. Activated STAT3 was prominent in human MPNSTs and mouse GEM-PNSTs, and a JAK2/STAT3 inhibitor delayed the growth of MPNST in nude mice. Furthermore, EGFR provided a major input to STAT3 activation in vitro and in vivo. The data support the hypotheses that STAT3 is sufficient for MPNST transformation but in spite of effects vasculature and tumor cell survival, is not necessary for tumor growth in established MPNST.
One third of Nf1fl/fl;DhhCre CNP-hEGFR mice developed GEM-PNST. As all Schwann cell in the CNP-hEGFR mouse nerve express EGFR (20) and all the GEM-PNST tumor cells express hEGFR (Figure 1H), therefore we infer co-expression of Nf1 and EGFR in the target cells. It is likely that additional events must occur in these cells to promote transformation to GEM-PNST, accounting for the low penetrance of aggressive tumors in this model. This is consistent with the numerous genetic changes in human MPNSTs (28). Our data support a role for EGFR expression levels early in sarcoma formation. This is consistent with our findings that 1) while shSTAT3 prevented xenograft formation, early treatment of tumors delayed MPNST formation while treatment of established tumors with STAT3 inhibitor did not. And 2) reduced EGFR activity delayed but did not prevent MPSNT growth (Ling et al., 2005). A role for EGFR expression levels in early sarcoma formation could explain why EGFR expression levels in resected MPNST are not prognostic (18). Our gain-of-function data in genetically engineered mice also suggests that increased levels of EGFR might serve as a modifier predisposing to MPNST formation. Studies searching for modifiers of Nf1 GEM-PNST identified a locus on mouse chromosome 11, close to Egfr (29-31). However, modifier effects might also derive from increased expression of EGFR ligands or EGFR ligand proteases, and/or increased expression of other pathway genes.
STAT3 is known to act as an oncogene, initiating tumor formation and maintaining tumor cell proliferation in other tumor systems (21). Consistent with these studies, our data support a key role for STAT3 in transformation of MPNST. In tissue culture, MPNST cells remained responsive to JAK and JAK2/STAT3 inhibitors, as they do to EGFR inhibitors (1, 7), (32). Ligand-bound, phosphorylated EGFR physically interacts with the SH2 domain of STAT3 – leading to phosphorylation of STAT3 at Tyr705, STAT3 dimerization, translocation to the nucleus, and STAT3-activated gene transcription (33). EGFR and phosphorylated STAT3 (Tyr705) are co-expressed in many cancers (34-37). In human MPNST and mouse GEM-PNSTs, STAT3 was phosphorylated at Tyr705, and STAT3 phosphorylation was robust in all MPNST tissue sections. In contrast, STAT3 was phosphorylated at Ser727 in only a low percentage of cells, and in less than 20% of tested human and mouse tumors. A recent in vitro study found STAT3 phosphorylated on Ser727 in an MPNST cell line (24). Thus, while Ser727 may be relevant in some rare tumors, STAT3 (Tyr705) is more consistently phosphorylated in and relevant to MPNSTs. EGFR signaling pathways induce epithelial-mesenchymal transition via STAT3-mediated TWIST gene expression in breast cancer (38). Therefore, it is perhaps significant that TWIST is frequently over-expressed in MPNST (28, 39). STAT3 is a potential therapeutic target, since blocking it with shRNA or the non-specific inhibitor curcumin inhibits growth in several cancer models (24, 34-37). Use of the specific JAK2/STAT3 inhibitor, FLLL32, and/or its derivatives, should enable future clinical testing.
In summary, EGFR promotes MPNST formation via JAK2/STAT3 activation. Pharmacologic inhibition of JAK2/STAT3 using FLLL32 delays MPNST growth, and shSTAT3 eliminates MPNST formation. These data suggest that the EGFR-STAT3 axis contributes to MPNST initiation. However, once tumors become established, they no longer respond to EGFR or STAT3 inhibition.
Materials and methods
Animals
Mice were housed in temperature- and humidity-controlled facilities on a 12-hour dark-light cycle with free access to food and water. The animal care and use committees of Cincinnati Children's Hospital Medical Center or University of Minnesota approved all animal procedures. C57Bl/6 CNP-hEGFR transgenic mice (CNP-hEGFR/+) (20) were interbred to obtain homozygous mice (CNP-hEGFR/CNP-hEGFR). Nf1fl/fl;DhhCre mice on a 129/Bl/6 mixed background (26). DhhCre mice were bred onto the Nf1fl/fl background to obtain an F1 generation (Nf1fl/+;DhhCre/+); F1 and CNP-hEGFR/CNP-hEGFR mice were bred to obtain F2 Nf1fl/+;DhhCre/+;CNP-hEGFR+, and F2 and Nf1fl/fl mice were bred to obtain Nf1fl/fl; DhhCre, CNP-hEGFR+. Littermates served as controls.
Cell culture
MPNST cell lines included T265, ST8814, S462, S462TY [derived from the S462 cell line (40) by two rounds of in vivo growth as xenografts], and STS26T were maintained in DMEM/10% FBS/1% penicillin/streptomycin. For drug treatment, inhibitors were added two hours after plating of cells. MTS assays were performed at day 4 as described (39). For EGF stimulation assays, 80% confluent of ST8814 cells were serum starved overnight, EGF (20ng/mL) was added for 10 minutes and cells were collected for western blot analysis. For OSI-774 inhibition assays, ST8814 cells were treated with OSI-774 (3.3μM) for 48 hours and then collected for western blot analysis. DMSO (0.1%) was used as control. Primary human Schwann cells were the gift of Patrick Wood Miami Project to the Paralysis, and were cultured as described (41).
In vivo drug treatment
We dissolved FLLL32 in 12.5% alcohol + 12.5% cremaphor by boiling for 15 minutes. Drug was prepared fresh every other day. We administered FLLL32 ( 200mg/kg) or vehicle (12.5% alcohol + 12.5% cremaphor) to tumor bearing nude mice daily for 2 or 7 weeks by intraperitoneal injection in 200 – 300ul based on mouse weight. Mice were weighed twice a week.
Immunohistochemistry and histology
Tissue was embedded in paraffin, and blocks were cut to generate 6-μm sections. Deparaffinized sections were stained with either H&E or incubated overnight at 4°C with antibodies: anti-S100β (Dako, Carpinteria, CA), anti-human EGFR (Invitrogen, Grand Island, NY), Ki67 (Novacastra Leica Microsystems, Buffalo Grove, IL) or anti-pSTAT3 (Tyr705) (Abcam, Cambridge, MA). The next day, sections were incubated in HRP-labeled secondary antibody and staining was detected by DAB (DAKO). For immunefluoresence staining, sections were incubated with species specific fluorophores (Jackson Laboratory, Bar Harbor, ME). For the TUNEL staining, we used acidification as recommended (Roche, Indianapolis, IN). Sections were viewed with a microscope (Carl Zeiss, Thornwood, NY) equipped with a digital imaging system.
Lentiviral infection
We infected S462TY cells at 60% confluence with shSTAT3 or non-target control (Sigma). We incubated lentiviral particles with MPNST cells in the presence of polybrene (8μg/ml; Sigma-Aldrich) daily for 3 days, followed by selection in puromycin (2.5μg/ml; Sigma-Aldrich). Surviving cells were collected for xenograft injection.
Mouse xenografts
We injected 2.3×106 S462TY MPNST cells in a total volume of 150 μl of 30% Matrigel (BD Biosciences, San Jose, CA) into flanks of 5- to 6-week-old female athymic nude (nu/nu) mice (Harlan, IN); right flanks were injected with shSTAT3 cells, and left with non-target control cells. Tumor volume was calculated as L * W2 (π/6), where L is the longest diameter and W is the width Tumors were dissected one hour after last dose and immediately flash frozen in liquid nitrogen for biochemistry or fixed in 4% paraformaldehyde for histology.
Western blots
Tumor proteins were extracted using extraction buffer (20 mMNaPO4, 150 mM NaCl, 2 mMMgCl2, 0.1% Nonidet P-40, 10% glycerol, 10 mM sodium fluoride, 0.1 mM sodium orthovanadate, 10 mM sodium pyrophosphate, 100 μM phenylasine oxide, 10 nM okadaic acid, 1 mM dithiothreitol, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 10 μg/ml pepstatin, 10 μg/ml tosyl-l-phenylalanine chloromethyl ketone, and 10 μg/ml N α-tosyl-l-lysine chloromethyl ketone). Protein concentration was estimated using Coomassie® Plus Protein Assay Reagent. Proteins (40 μg/lane) were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis on 4–20% tris-glycine gel (Invitrogen, Carlsbad, CA) and electrotransferred to polyvinylidene diflouride membrane. Membranes were blocked with 5% nonfat milk + 0.1% TBST to minimize nonspecific binding. Antibodies recognizing EGFR, pJAK2, JAK2, pSTAT3, STAT3, pERK, ERK, and β-actin (Cell Signaling, Danvers, MA) were used. Antibody binding to the membrane was visualized using a chemiluminescent detection system (ECL, Amersham, Arlington Heights, IL). The bands obtained were quantified by Gel-pro analyzer (Media Cybernetics, Inc, Rockville, MD). Anti-β-actin was used as a loading control. At least three different tumor lysates were analyzed for each antigen.
Supplementary Material
{kind=link}
Acknowledgments
We thank Dr. Andre Bernards (Massachusetts General Hospital) and Drs. Jiayuh Lin, Pui-Kai Li, and Gregory B. Lesinski (Ohio State University) for helpful discussions. This work was supported by the National Institutes of Health (R01 NS28840 and P50 NS057531 to N.R.), and Department of Defense New Investigator Award (NF100053) and an Ohio State University Comprehensive Cancer Center Pelotonia Idea Grant (to J.W.). The American Cancer Society (IRG-67-003-44) supported J.R.F.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
References
- 1.Widemann BC. Current status of sporadic and neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Curr Oncol Rep. 2009 Jul;11(4):322–8. doi: 10.1007/s11912-009-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Evans D, Baser M, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39(5):311–4. doi: 10.1136/jmg.39.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002 Mar 1;62(5):1573–7. [PubMed] [Google Scholar]
- 4.Tucker T, Wolkenstein P, Revuz J, Zeller J, Friedman JM. Association between benign and malignant peripheral nerve sheath tumors in NF1. Neurology. 2005 Jul 26;65(2):205–11. doi: 10.1212/01.wnl.0000168830.79997.13. [DOI] [PubMed] [Google Scholar]
- 5.Ferner RE. Neurofibromatosis 1. Eur J Hum Genet. 2007 Feb;15(2):131–8. doi: 10.1038/sj.ejhg.5201676. [DOI] [PubMed] [Google Scholar]
- 6.Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1? Glia. 2008 Nov 1;56(14):1590–605. doi: 10.1002/glia.20776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katz D, Lazar A, Lev D. Malignant peripheral nerve sheath tumour (MPNST): the clinical implications of cellular signalling pathways. Expert Rev Mol Med. 2009;11:e30. doi: 10.1017/S1462399409001227. [DOI] [PubMed] [Google Scholar]
- 8.Miller SJ, Jessen WJ, Mehta T, Hardiman A, Sites E, Kaiser S, et al. Integrative genomic analyses of neurofibromatosis tumours identify SOX9 as a biomarker and survival gene. EMBO Mol Med. 2009 Jul;1(4):236–48. doi: 10.1002/emmm.200900027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cichowski K, Shih TS, Schmitt E, Santiago S, Reilly K, McLaughlin ME, et al. Mouse models of tumor development in neurofibromatosis type 1. Science. 1999;286:2172–6. doi: 10.1126/science.286.5447.2172. [DOI] [PubMed] [Google Scholar]
- 10.Vogel KS, Klesse LJ, Velasco-Miguel S, Meyers K, Rushing EJ, Parada LF. Mouse tumor model for neurofibromatosis type 1. Science. 1999;286:2176–9. doi: 10.1126/science.286.5447.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joseph NM, Mosher JT, Buchstaller J, Snider P, McKeever PE, Lim M, et al. The loss of Nf1 transiently promotes self-renewal but not tumorigenesis by neural crest stem cells. Cancer Cell. 2008 Feb;13(2):129–40. doi: 10.1016/j.ccr.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun D, Tainsky MA, Haddad R. Oncogene Mutation Survey in MPNST Cell Lines Enhances the Dominant Role of Hyperactive Ras in NF1 Associated Pro-Survival and Malignancy. Transl Oncogenomics. 2012;5:1–7. doi: 10.4137/TOG.S8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yarden Y. The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2001;37(Suppl):S3–8. doi: 10.1016/s0959-8049(01)00230-1. [DOI] [PubMed] [Google Scholar]
- 14.Li H, Velasco-Miguel S, Vass W, Parada L, DeClue J. Epidermal growth factor receptor signaling pathways are associated with tumorigenesis in the Nf1:p53 mouse tumor model. Cancer Res. 2002;62(15):4507–13. [PubMed] [Google Scholar]
- 15.Perry A, Kunz S, Fuller C, Banerjee R, Marley E, Liapis H, et al. Differential NF1, p16, and EGFR patterns by interphase cytogenetics (FISH) in malignant peripheral nerve sheath tumor (MPNST) and morphologically similar spindle cell neoplasms. J Neuropathol Exp Neurol. 2002;61(8):702–9. doi: 10.1093/jnen/61.8.702. [DOI] [PubMed] [Google Scholar]
- 16.DeClue JE, Heffelfinger S, Benvenuto G, Ling B, Li S, Rui W, et al. Epidermal growth factor receptor expression in neurofibromatosis type-1 related tumors and NF1 animal models. J Clin Invest. 2000;105(9):1–10. doi: 10.1172/JCI7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frohnert PW, Stonecypher MS, Carroll SL. Constitutive activation of the neuregulin-1/ErbB receptor signaling pathway is essential for the proliferation of a neoplastic Schwann cell line. Glia. 2003 Aug;43(2):104–18. doi: 10.1002/glia.10232. [DOI] [PubMed] [Google Scholar]
- 18.Keizman D, Issakov J, Meller I, Maimon N, Ish-Shalom M, Sher O, et al. Expression and significance of EGFR in malignant peripheral nerve sheath tumor. J Neurooncol. 2009 Sep;94(3):383–8. doi: 10.1007/s11060-009-9862-z. [DOI] [PubMed] [Google Scholar]
- 19.Luetteke N, Phillips H, Qiu T, Copeland N, Earp H, Jenkins N, et al. The mouse waved-2 phenotype results from a point mutation in the EGF receptor tyrosine kinase. Genes Dev. 1994 Feb 15;8(4):399–413. doi: 10.1101/gad.8.4.399. [DOI] [PubMed] [Google Scholar]
- 20.Ling B, Wu J, Miller S, Monk K, Shamekh R, Rizvi T, et al. Role for the epidermal growth factor receptor in neurofibromatosis-related peripheral nerve tumorigenesis. Cancer Cell. 2005;7(1):65–75. doi: 10.1016/j.ccr.2004.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009 Nov;9(11):798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vigneron A, Gamelin E, Coqueret O. The EGFR-STAT3 oncogenic pathway up-regulates the Eme1 endonuclease to reduce DNA damage after topoisomerase I inhibition. Cancer Res. 2008 Feb 1;68(3):815–25. doi: 10.1158/0008-5472.CAN-07-5115. [DOI] [PubMed] [Google Scholar]
- 23.Sherry MM, Reeves A, Wu JK, Cochran BH. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells. 2009 Oct;27(10):2383–92. doi: 10.1002/stem.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banerjee S, Byrd JN, Gianino SM, Harpstrite SE, Rodriguez FJ, Tuskan RG, et al. The neurofibromatosis type 1 tumor suppressor controls cell growth by regulating signal transducer and activator of transcription-3 activity in vitro and in vivo. Cancer Res. 2010 Feb 15;70(4):1356–66. doi: 10.1158/0008-5472.CAN-09-2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin L, Hutzen B, Zuo M, Ball S, Deangelis S, Foust E, et al. Novel STAT3 phosphorylation inhibitors exhibit potent growth-suppressive activity in pancreatic and breast cancer cells. Cancer Res. 2010;70(6):2445–54. doi: 10.1158/0008-5472.CAN-09-2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu J, Williams JP, Rizvi TA, Kordich JJ, Witte D, Meijer D, et al. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell. 2008 Feb;13(2):105–16. doi: 10.1016/j.ccr.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stemmer-Rachamimov A, Louis D, Nielsen G, Antonescu C, Borowsky A, Bronson R, et al. Comparative pathology of nerve sheath tumors in mouse models and humans. Cancer Res. 2004;64(10):3718–24. doi: 10.1158/0008-5472.CAN-03-4079. [DOI] [PubMed] [Google Scholar]
- 28.Beert E, Brems H, Daniels B, De Wever I, Van Calenbergh F, Schoenaers J, et al. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer. 2011 Dec;50(12):1021–32. doi: 10.1002/gcc.20921. [DOI] [PubMed] [Google Scholar]
- 29.Reilly KM, Broman KW, Bronson RT, Tsang S, Loisel DA, Christy ES, et al. An imprinted locus epistatically influences Nstr1 and Nstr2 to control resistance to nerve sheath tumors in a neurofibromatosis type 1 mouse model. Cancer Res. 2006 Jan 1;66(1):62–8. doi: 10.1158/0008-5472.CAN-05-1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reilly KM, Tuskan RG, Christy E, Loisel DA, Ledger J, Bronson RT, et al. Susceptibility to astrocytoma in mice mutant for Nf1 and Trp53 is linked to chromosome 11 and subject to epigenetic effects. Proc Natl Acad Sci U S A. 2004 Aug 31;101(35):13008–13. doi: 10.1073/pnas.0401236101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walrath JC, Fox K, Truffer E, Gregory Alvord W, Quinones OA, Reilly KM. Chr 19(A/J) modifies tumor resistance in a sex- and parent-of-origin-specific manner. Mamm Genome. 2009 Apr;20(4):214–23. doi: 10.1007/s00335-009-9179-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Torres KE, Zhu QS, Bill K, Lopez G, Ghadimi MP, Xie X, et al. Activated MET is a molecular prognosticator and potential therapeutic target for malignant peripheral nerve sheath tumors. Clin Cancer Res. 2011 Jun 15;17(12):3943–55. doi: 10.1158/1078-0432.CCR-11-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shao H, Cheng HY, Cook RG, Tweardy DJ. Identification and characterization of signal transducer and activator of transcription 3 recruitment sites within the epidermal growth factor receptor. Cancer Res. 2003 Jul 15;63(14):3923–30. [PubMed] [Google Scholar]
- 34.Fukuda A, Wang S, Morris Jt, Folias A, Liou A, Kim G, et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell. 2011;19(4):441–55. doi: 10.1016/j.ccr.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lesina M, Kurkowski M, Ludes K, Rose-John S, Treiber M, Klöppel G, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19(4):456–69. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 36.Guryanova O, Wu Q, Cheng L, Lathia J, Huang Z, Yang J, et al. Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell. 2011;19(4):498–511. doi: 10.1016/j.ccr.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marotta L, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker S, Bloushtain-Qimron N, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+CD24− stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121(7):2723–35. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei Y, et al. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007 Oct 1;67(19):9066–76. doi: 10.1158/0008-5472.CAN-07-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller SJ, Rangwala F, Williams J, Ackerman P, Kong S, Jegga AG, et al. Large-scale molecular comparison of human schwann cells to malignant peripheral nerve sheath tumor cell lines and tissues. Cancer Res. 2006 Mar 1;66(5):2584–91. doi: 10.1158/0008-5472.CAN-05-3330. [DOI] [PubMed] [Google Scholar]
- 40.Frahm S, Mautner VF, Brems H, Legius E, Debiec-Rychter M, Friedrich RE, et al. Genetic and phenotypic characterization of tumor cells derived from malignant peripheral nerve sheath tumors of neurofibromatosis type 1 patients. Neurobiol Dis. 2004 Jun;16(1):85–91. doi: 10.1016/j.nbd.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 41.Rosenbaum T, Rosenbaum C, Winner U, Muller HW, Lenard HG, Hanemann CO. Long-term culture and characterization of human neurofibroma-derived Schwann cells. J Neurosci Res. 2000 Sep 1;61(5):524–32. doi: 10.1002/1097-4547(20000901)61:5<524::AID-JNR7>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.