

The BiP cochaperone ERdj4 removes misfolded proteins from the ER lumen by associating with ERAD machinery. Global deficiency of ERdj4 results in widespread constitutive ER stress, decreased survival, and metabolic derangements in mice. These findings indicate that the chaperone activity of ERdj4 is important for ER homeostasis in vivo.
Abstract
Endoplasmic reticulum–localized DnaJ 4 (ERdj4) is an immunoglobulin-binding protein (BiP) cochaperone and component of the endoplasmic reticulum–associated degradation (ERAD) pathway that functions to remove unfolded/misfolded substrates from the ER lumen under conditions of ER stress. To elucidate the function of ERdj4 in vivo, we disrupted the ERdj4 locus using gene trap (GT) mutagenesis, leading to hypomorphic expression of ERdj4 in mice homozygous for the trapped allele (ERdj4GT/GT). Approximately half of ERdj4GT/GT mice died perinatally associated with fetal growth restriction, reduced hepatic glycogen stores, and hypoglycemia. Surviving adult mice exhibited evidence of constitutive ER stress in multiple cells/tissues, including fibroblasts, lung, kidney, salivary gland, and pancreas. Elevated ER stress in pancreatic β cells of ERdj4GT/GT mice was associated with β cell loss, hypoinsulinemia, and glucose intolerance. Collectively these results suggest an important role for ERdj4 in maintaining ER homeostasis during normal fetal growth and postnatal adaptation to metabolic stress.
INTRODUCTION
Nascent proteins destined for the cell surface, intracellular organelles, or secretion enter the endoplasmic reticulum (ER), where they undergo chaperone-mediated protein folding to achieve a stable conformation. Highly metabolic cells, including pancreatic acinar and β cells, plasma B cells, and serous and mucous cells of the salivary gland, invoke the unfolded protein response (UPR) to increase ER folding capacity and maintain homeostasis in the face of increased protein load and consequent ER stress (Zhang et al., 2002; Iwakoshi et al., 2003; Lee et al., 2005; Iwawaki et al., 2010). The UPR reduces ER burden by attenuating protein translation and up-regulating the machinery involved in protein folding, quality control, and ER-associated degradation (ERAD). The ER transmembrane sensors protein kinase RNA–like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1) detect accumulation of unfolded or misfolded protein in the ER lumen and transduce signals across the ER membrane to alleviate ER stress. PERK activation results in phosphorylation of eukaryotic translation initiation factor 2α, which inhibits translation initiation, thereby reducing the load of newly synthesized proteins within the ER. Activation of ATF6 results in relocation to the Golgi, where proteolytic cleavage releases a cytosolic ATF6 fragment that translocates to the nucleus to stimulate transcription of UPR target genes, including ER chaperones. The activated cytosolic endoribonuclease domain of IRE1 removes an unspliced intron from X-box binding protein 1 (XBP1) mRNA, leading to translation of a transcription factor that up-regulates expression of chaperones involved in protein folding, as well as components of ERAD. Failure of the UPR to resolve ER stress can trigger apoptosis (Schroder and Kaufman, 2005; Ron and Walter, 2007).
Molecular chaperones are critical components of the ER quality control machinery and distinguish among unfolded, correctly folded, and terminally misfolded proteins within the ER. Immunoglobulin-binding protein (BiP)/glucose-regulated protein 78, an ER-localized member of the heat shock protein (HSP) 70 family, is a multifunctional chaperone that assists in the folding of nascent proteins (Lee et al., 1999; Wang et al., 2005), facilitates targeting of misfolded proteins for proteasomal degradation (Nishikawa et al., 2001; Molinari et al., 2002), and plays a key role in activation of UPR sensors in response to ER stress (Bertolotti et al., 2000; Shen et al., 2002a). BiP contains a C-terminal substrate-binding domain (SBD) that binds unfolded substrates with low affinity via interaction with exposed hydrophobic regions. Hydrolysis of ATP bound to the N-terminal nucleotide-binding domain (NBD) of BiP causes high-affinity binding of substrate. Exchange of ADP for ATP releases the substrate from BiP, allowing the substrate to continue folding (Mayer and Bukau, 2005). An arginine residue located on the surface of the NBD in the ATP-bound form is critical for communication with the SBD and interaction with HSP40 cochaperones (Jiang et al., 2005; Awad et al., 2008).
The ER-localized DnaJ (ERdj) family of HSP40 cochaperones induce high-affinity substrate binding by interacting with the NBD of BiP and stimulating ATP hydrolysis. Whereas some ERdjs interact with BiP independently of substrate, others bind substrates directly and recruit BiP (Fewell et al., 2001; Shen and Hendershot, 2005; Jin et al., 2008; Petrova et al., 2008). ERdjs facilitate protein folding and/or degradation of newly translocated (ERdj1–3), unfolded (ERdj3 and ERdj6), or misfolded (ERdj4 and ERdj5) substrates, often by interacting with translocation or ERAD machinery (Otero et al., 2010). ERdj4 is a soluble DnaJ protein that binds BiP through a His-Pro-Asp (HPD) motif in the J domain and likely interacts with unfolded/misfolded substrates through a C-terminal glycine/phenylalanine-rich region (Shen et al., 2002b; Dong et al., 2008; Lai et al., 2012). ERdj4 is ubiquitously expressed at very low levels (Weitzmann et al., 2007; Hageman and Kampinga, 2009) and is up-regulated in response ER stress (Shen et al., 2002b; Dong et al., 2008) by the UPR signal transducer XBP1 (Lee et al., 2003; Kanemoto et al., 2005). ERdj4 binds to unfolded (insulin2) or misfolded (insulin2C96Y, surfactant protein CΔexon4 [SP-CΔexon4], and SP-CL188Q) substrates to facilitate their removal from the ER for degradation by the proteasome (Dong et al., 2008; Lai et al., 2012). Although these studies provided insight into the cellular function of ERdj4, the role of this chaperone in tissue homeostasis is unclear. To address this issue, we generated mice globally deficient in ERdj4 and identified an important role for this chaperone in tissues undergoing physiological stress.
RESULTS
Generation of ERdj4 gene trap mice
Chimeric mice were generated from embryonic stem (ES) cells harboring a gene trap (GT) cassette inserted into the ERdj4 locus (Figure 1A). Offspring carrying the GT allele were backcrossed eight generations to the 129P2 or C57BL/6 strain. Genomic DNA (gDNA) isolated from the progeny of ERdj4+/GT intercrosses was used to confirm the presence of wild-type (WT) and/or GT alleles (Figure 1B). Only one or two copies of the GT cassette were present in ERdj4+/GT or ERdj4GT/GT mice, respectively, confirming that ERdj4 was the only locus that was disrupted (Figure 1C). DNA sequencing confirmed that the GT cassette was inserted into intron 1 of the ERdj4 locus, resulting in splicing of exon 1 with the GT cassette and premature transcript termination (Figure 1A). An increase in β-galactosidase mRNA (Figure 1D) confirmed the presence of the GT cassette, and the virtual absence of ERdj4 mRNA (Figure 1E) confirmed disruption of the ERdj4 locus in mouse embryonic fibroblasts (MEFs) generated from embryonic day 13.5 (E13.5) ERdj4GT/GT mice. Variable expression of ERdj4 mRNA in tissues from adult mice using primers specific for exon 3 (Figure 1A) confirmed that the insertion of the GT cassette resulted in a hypomorphic allele (Figure 1F), a common occurrence in GT mutagenesis (Voss et al., 1998a, b; Voss and Thomas, 2001).
FIGURE 1:

Generation of ERdj4GT/GT mice. (A) The GT allele. The GT cassette was inserted between adenosine 1151 and guanosine 1152 within intron 1 of the ERdj4 locus (NC_000078.6). Quantitative RT (qRT)-PCR was performed using primers specific for β-galactosidase (a, b) and ERdj4 (c, d). β-GEO, β-galactosidase/neomycin resistance fusion gene; IRES, internal ribosome entry site; pA, polyadenylation signal; PLAP, placental alkaline phosphatase; SA, splicing acceptor; TM, transmembrane region. (B) PCR genotyping of the WT and GT alleles in gDNA isolated from the progeny of heterozygous intercrosses. (C) GT copy number was determined in gDNA by the TaqMan Gene Copy Number assay. n = 4 mice/genotype. (D, E) qRT-PCR of β-galactosidase and ERdj4 mRNAs in MEFs. RQ, relative quantitation. n = 3 samples/group. (F) qRT-PCR of ERdj4 mRNA in tissues isolated from 6-wk-old littermates; samples were normalized to β-actin. n = 4 mice/genotype.
Elevated ER stress and increased susceptibility to cell death in ERdj4GT/GT MEFs
To determine whether ERdj4 deficiency enhanced ER stress, we assessed splicing of XBP1 mRNA in control and tunicamycin-treated MEFs generated from ERdj4+/+ and ERdj4GT/GT mice. The spliced form of XBP1 mRNA was elevated at baseline in ERdj4GT/GT MEFs compared with ERdj4+/+ controls (Supplemental Figure S1A, 0 h). After treatment with tunicamycin, which induces ER stress by inhibiting N-linked glycosylation, splicing of XBP1 mRNA was enhanced in both ERdj4+/+ and ERdj4GT/GT MEFs, with more rapid conversion in the latter cells (Supplemental Figure S1A). Consistent with constitutive ER stress in ERdj4GT/GT MEFs, BiP and IRE1α proteins were elevated at baseline (Supplemental Figure S1B, 0 h). Treatment with tunicamycin or the proteasome inhibitor MG-132 increased the expression of BiP and IRE1α in ERdj4GT/GT MEFs. The effect of tunicamycin was less pronounced, and MG-132 had no effect on BiP and IRE1α expression in ERdj4+/+ MEFs. In contrast, the concentration of another ER chaperone, calnexin, was similarly regulated in both groups under normal and stressed conditions (Supplemental Figure S1B). Ultrastructural analyses of ERdj4GT/GT MEFs revealed dilated ER cisternae, providing further evidence of constitutive ER stress (Supplemental Figure S1C). In the absence of exogenous stressors, survival of ERdj4GT/GT MEFs was similar to that of control cells (Supplemental Figure S1D). However, when ERdj4GT/GT MEFs were treated with MG-132, tunicamycin, or thapsigargin (an inhibitor of calcium transport) for 48 h, cell viability was significantly decreased in a dose-dependent manner compared with ERdj4+/+ controls (Supplemental Figure S1D). In keeping with this finding, overexpression of ERdj4 was previously reported to protect against cell death induced by ER stress (Kurisu et al., 2003).
Perinatal lethality associated with growth retardation and hypoglycemia in ERdj4GT/GT mice
Postnatal survival of ERdj4GT/GT progeny, derived from ERdj4+/GT intercrosses, was decreased in a strain-dependent manner. Genotype distribution at E18.5 approximated Mendelian ratios, indicating that lethality resulting from homozygosity of the GT allele occurred perinatally (Table 1). ERdj4GT/GT mice were noticeably smaller than control littermates, with a significant decrease in body weight at E18.5 (Figure 2, A and B). Newborn ERdj4GT/GT mice were severely hypoglycemic, likely contributing to perinatal lethality (Figure 2C). Liver weights were significantly reduced in neonatal ERdj4GT/GT mice, in association with decreased hepatic glycogen stores (Figure 2, D and E). Gluconeogenic enzymes, including glucose-6-phosphatase, phosphoenolpyruvate carboxykinase, and pyruvate carboxylase, were normally expressed in livers of neonatal ERdj4GT/GT mice (Supplemental Figure S2A). Plasma triglycerides and nonesterified fatty acids were also unaffected by ERdj4 deficiency in neonatal mice (Supplemental Figure S2B). Of interest, plasma glucagon was significantly elevated in neonatal ERdj4GT/GT mice in an unsuccessful attempt to correct hypoglycemia (Figure 2F), whereas plasma insulin was unaffected (Supplemental Figure S2C). Collectively these data suggest that impaired glycogen synthesis and/or mobilization resulted in hypoglycemia and subsequent mortality in neonatal ERdj4GT/GT mice.
TABLE 1:
Decreased perinatal survival of ERdj4GT/GT mice.
| Strain | ERdj4+/+ | ERdj4+/GT | ERdj4GT/GT |
|---|---|---|---|
| P21 progenya | |||
| 129P2 × C57BL/6 | 104 | 195 | 56 (15.7%) |
| 129P2 | 47 | 85 | 22 (14.3%) |
| C57BL/6 | 37 | 88 | 16 (11.3%) |
| E18.5 embryosb | |||
| 129P2 × C57BL/6 | 31 | 63 | 28 (23.0%) |
| 129P2 | 14 | 23 | 11 (22.9%) |
| C57BL/6 | 17 | 34 | 22 (30.1%) |
aGenotype distribution of 3-wk-old progeny from heterozygous intercrosses.
bGenotype distribution of E18.5 embryos from heterozygous intercrosses.
FIGURE 2:

Fetal growth retardation and neonatal hypoglycemia in ERdj4GT/GT mice. (A) Size comparison of E18.5 embryos. (B) Body weights of E18.5 embryos. n = 6–14 mice/genotype. (C) Blood glucose levels of vaginally born or cesarean-delivered, nonsuckling neonates. n = 10–11 mice/genotype. (D) Liver weights of neonatal mice. BW, body weight. n = 5–6 mice/genotype. (E) Liver glycogen in neonatal mice. n = 5–6 mice/genotype. (F) Plasma glucagon in neonatal mice. n = 3–5 mice/genotype.
Surviving male and female mice often displayed signs of neurological disorders, including head tilt, abnormal circling behavior, and hyperactivity. Homozygous ERdj4GT/GT intercrosses failed to produce progeny. All subsequent experiments were conducted using littermates derived from heterozygous crosses in the mixed 129P2×C57BL/6 genetic background, unless otherwise indicated.
Constitutive ER stress is associated with histological abnormalities in pancreatic islets of ERdj4GT/GT mice
To determine whether ERdj4 deficiency enhanced ER stress in vivo, we crossed ERdj4+/GT mice to ER stress–activated indicator (ERAI) transgenic mice (Iwawaki et al., 2004). Green fluorescent protein (GFP; resulting from XBP1 splicing and expression of XBP1/GFP fusion protein) was detected in the kidneys and lungs of ERdj4GT/GTERAI mice (Supplemental Figure S3A). Ultrastructural analyses revealed ER distention in renal tubular epithelial cells (Supplemental Figure S3B) and serous cells of the salivary gland (Supplemental Figure S3C). GFP and IRE1α proteins were also increased in pancreatic lysates from ERdj4GT/GTERAI mice compared with ERdj4+/+ERAI controls (Figure 3A). GFP immunofluorescence localized prominently to β cells in islets of pancreatic tissues from both ERdj4+/+ERAI and ERdj4GT/GTERAI mice (Figure 3B), with more GFP-positive cells and increased fluorescence intensity in the latter. Morphometric analyses revealed a 21% decrease in the number of β cells in pancreatic islets of ERdj4GT/GT mice (Table 2). Consistent with this finding, there was a significant increase in the number of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)–positive cells per pancreatic islet in ERdj4GT/GT mice compared with controls (0.29 ± 0.11 vs. 0.06 ± 0.03, p ≤ 0.01, unpublished data). GFP fluorescence was not detected in glucagon-positive α cells in the pancreas of ERdj4+/+ERAI or ERdj4GT/GTERAI mice (Supplemental Figure S4A). Histological analyses of pancreatic sections from ERdj4GT/GT mice identified vacuolated cells in the islets (Figure 3C), and ultrastructural analyses of islets revealed ER distention in both α and β cells of ERdj4GT/GT mice (Figure 3, D and E). Hypomorphic expression of ERdj4 mRNA was confirmed in islets isolated from ERdj4GT/GT mice (Supplemental Figure S4B), linking the loss of ERdj4 in pancreatic islets to elevated ER stress.
FIGURE 3:
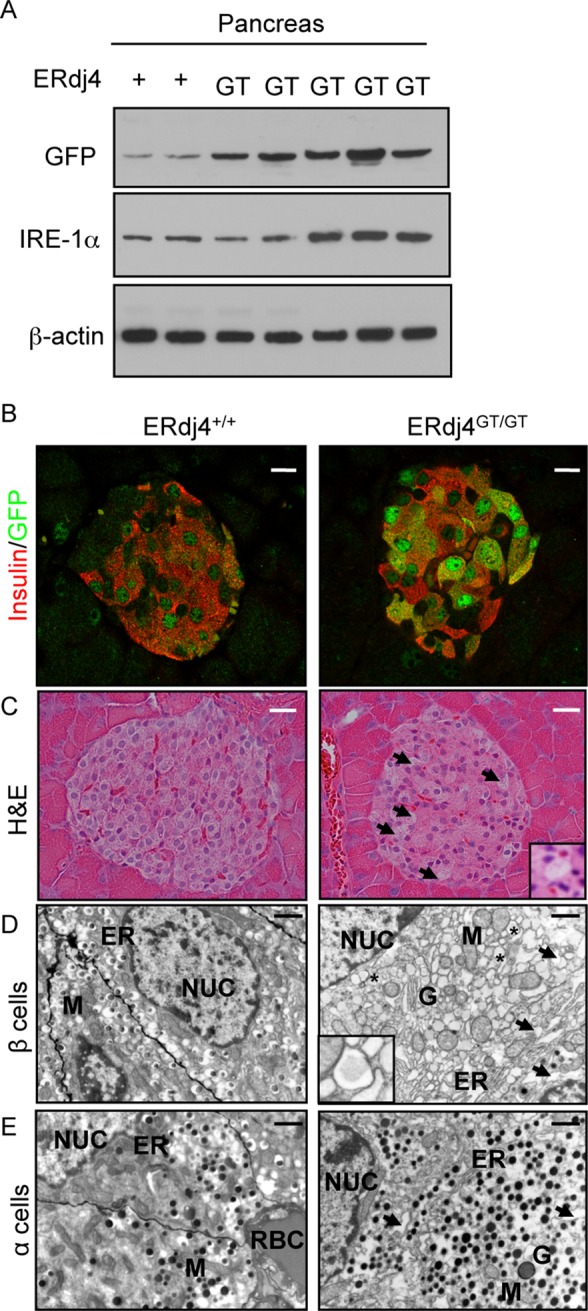
Elevated ER stress in the endocrine pancreas of ERdj4GT/GT mice. (A) Western blot analyses of GFP (reporter for XBP1 splicing), IRE1α, and β-actin (loading control) proteins in pancreatic homogenates from 6-wk-old ERdj4+/+ERAI (n = 2) and ERdj4GT/GTERAI (n = 5) mice. (B) Confocal microscopy of insulin (red) and GFP (green) proteins in pancreatic tissue sections from 8-wk-old ERdj4+/+ERAI (left) and ERdj4GT/GTERAI (right) mice. Scale bars, 10 μm. n = 4–5 mice/genotype. (C) Hematoxylin and eosin (H&E) staining of pancreatic tissue sections from 16- to 20-wk-old mice. Note the vacuolated cells in the islets of ERdj4GT/GT mice (arrows and inset). Scale bars, 10 μm. n = 5 mice/genotype. (D) Electron microscopy of β cells in pancreatic islets of 6-wk-old mice. Note the pronounced ER dilation (arrows) and electron-translucent secretory granules (asterisks and inset) in β cells of ERdj4GT/GT mice. Scale bars, 2 μm. n = 2 mice/genotype. ER, endoplasmic reticulum; G, Golgi complex; M, mitochondria; NUC, nucleus. (E) Electron microscopy of α cells in pancreatic islets of 6-wk-old mice. Note the pronounced ER dilation (arrows) in α cells of ERdj4GT/GT mice. Scale bars, 2 μm. n = 2 mice/genotype. ER, endoplasmic reticulum; G, Golgi complex; M, mitochondria; NUC, nucleus; RBC, red blood cell.
TABLE 2:
Morphometric analyses of pancreatic islets.
| Islet variable | ERdj4+/+ | ERdj4GT/GT |
|---|---|---|
| Number of β cells | 125.76 ± 4.93 | 99.71 ± 4.19† |
| Number of α cells | 35.86 ± 1.62 | 71.96 ± 4.06‡ |
| Ratio of β to α cells | 4.77 ± 0.34 | 1.61 ± 0.07‡ |
| Area of α cells (%) | 17.87 ± 0.74 | 35.38 ± 1.12‡ |
| Total islet area (μm2) | 39,868.67 ± 1647.14 | 43,613.46 ± 1890.15 |
Eight- to 12-wk-old mice.
†p ≤ 0.01.
‡p ≤ 0.001.
Although histology of ERdj4GT/GT acinar cells appeared normal at 8 wk of age (Supplemental Figure S5A), cytoplasmic vacuolation was observed in pancreatic acinar cells from 8-mo-old ERdj4GT/GT mice (Supplemental Figure S5B). Ultrastructural analyses of pancreatic acinar cells revealed dilated ER and multiple immature zymogen granules in ERdj4GT/GT mice (Supplemental Figure S5C), although this did not appear to interfere with acinar cell function, as reflected by similar serum lipase and amylase activity in ERdj4GT/GT and control mice (Supplemental Figure S5D).
Pancreatic α cell hyperplasia and hyperglucagonemia in ERdj4GT/GT mice
Morphometric analyses revealed 101% increase in the number of α cells in pancreatic islets of adult ERdj4GT/GT mice (Table 2). This resulted in 18% increase in α cell area, although total islet area was unchanged (Table 2). Further, glucagon-positive α cells were detected not only in the mantle, but also in the core of islets in ERdj4GT/GT mice (Figure 4A and Supplemental Figure S4A). Ultrastructural analyses also revealed an increase in the number of glucagon granules in α cells of ERdj4GT/GT mice (Figure 3E). Glucagon protein was significantly increased in the pancreas of ERdj4GT/GT mice (Figure 4B), consistent with α cell hyperplasia and increased glucagon granules (Figures 4A and 3E and Table 2). Similar to ERdj4GT/GT neonates (Figure 2F), plasma glucagon was significantly elevated in adult ERdj4GT/GT mice compared with controls (Figure 4C).
FIGURE 4:
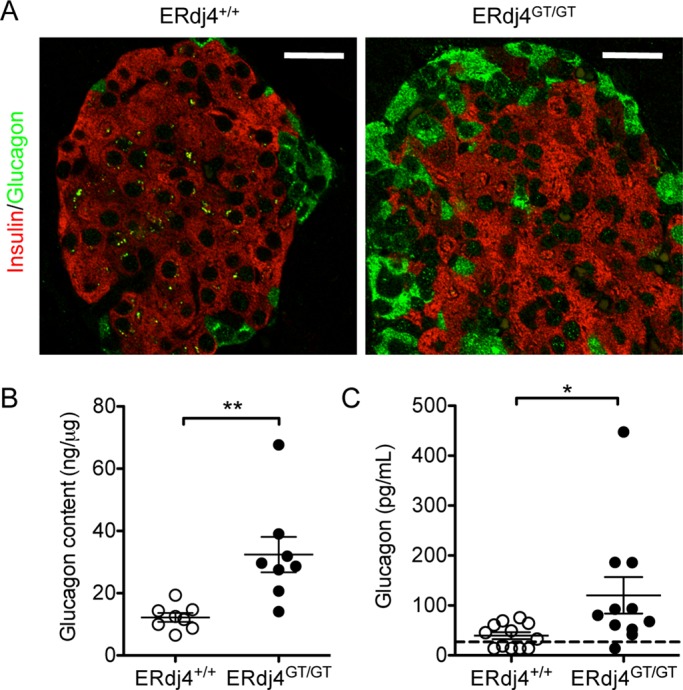
Pancreatic α cell hyperplasia and hyperglucagonemia in ERdj4GT/GT mice. (A) Immunofluorescence of insulin (red) and glucagon (green) in pancreatic tissue sections from 16- to 20-wk-old mice. Scale bars, 10 μm. n = 5 mice/genotype. (B) Glucagon protein in pancreatic extracts of 16- to 20-wk-old mice. n = 8 mice/genotype. (C) Plasma glucagon levels in fasted 8- to 16-wk-old mice. n = 11–12 mice/genotype.
ERdj4 deficiency impairs insulin biosynthesis
Because ERdj4 was previously reported to associate with insulin (Dong et al., 2008), we assessed whether ERdj4 deficiency affected insulin biosynthesis. Although total insulin protein in pancreatic extracts of ERdj4GT/GT mice was similar to that in ERdj4+/+ mice (Supplemental Figure S6), colocalization of proinsulin and BiP was enhanced in pancreatic β cells, indicating accumulation of proinsulin in the ER (Figure 5A). Metabolic labeling of an equal number of islets, followed by immunoprecipitation, identified a modest but significant increase in proinsulin protein in ERdj4GT/GT mice compared with controls (Figure 5B); however, proinsulin was likely underrepresented in this experiment due to β cell loss in ERdj4GT/GT islets (Table 2). Ultrastructural analyses revealed an increase in translucent (immature) insulin granules in β cells of ERdj4GT/GT mice (Figure 3D, inset), and plasma proinsulin:insulin ratios were significantly increased in ERdj4GT/GT mice (Figure 5C), suggesting a disruption in proinsulin maturation. Total plasma insulin was significantly lower at baseline in fasted ERdj4GT/GT mice (Figure 5D, 0 min) and remained significantly lower than controls after glucose administration (Figure 5D, 15 and 30 min).
FIGURE 5:

ERdj4 deficiency impairs insulin biosynthesis. (A) Immunofluorescence of insulin (red) and BiP (green) in pancreatic tissue sections from 16- to 20-wk-old mice. Scale bars, 5 μm. n = 3 mice/genotype. (B) Metabolic labeling of islets stimulated with 16.7 mM glucose from 6-wk-old C57BL/6 littermates (n = 3 mice/genotype). Equal numbers of trichloroacetic-precipitable counts per minute were immunoprecipitated with insulin antibody, and the immunoprecipitates were separated by SDS–PAGE. Proinsulin signal was visualized by autoradiography and quantitated by Multi Gauge software. The gel is a representative image of an experiment performed in triplicate. (C) Plasma proinsulin/insulin ratio in 12-wk-old, fasted mice. n = 8 mice/genotype. (D) Plasma insulin levels before and after glucose administration to fasted 12-wk-old mice. n = 8–9 mice/genotype.
Hypoinsulinemia causes glucose intolerance in ERdj4GT/GT mice
Blood glucose was normal in fasted ERdj4GT/GT mice (Figure 6A) despite reduced plasma insulin (Figure 5D, 0 min). However, after glucose administration, blood glucose remained significantly elevated in ERdj4GT/GT mice compared with controls (Figure 6B). Of importance, administration of insulin effectively lowered blood glucose in ERdj4GT/GT mice, confirming that glucose intolerance in ERdj4GT/GT mice was the result of hypoinsulinemia rather than a defect in the insulin signaling pathway (Figure 6C).
FIGURE 6:
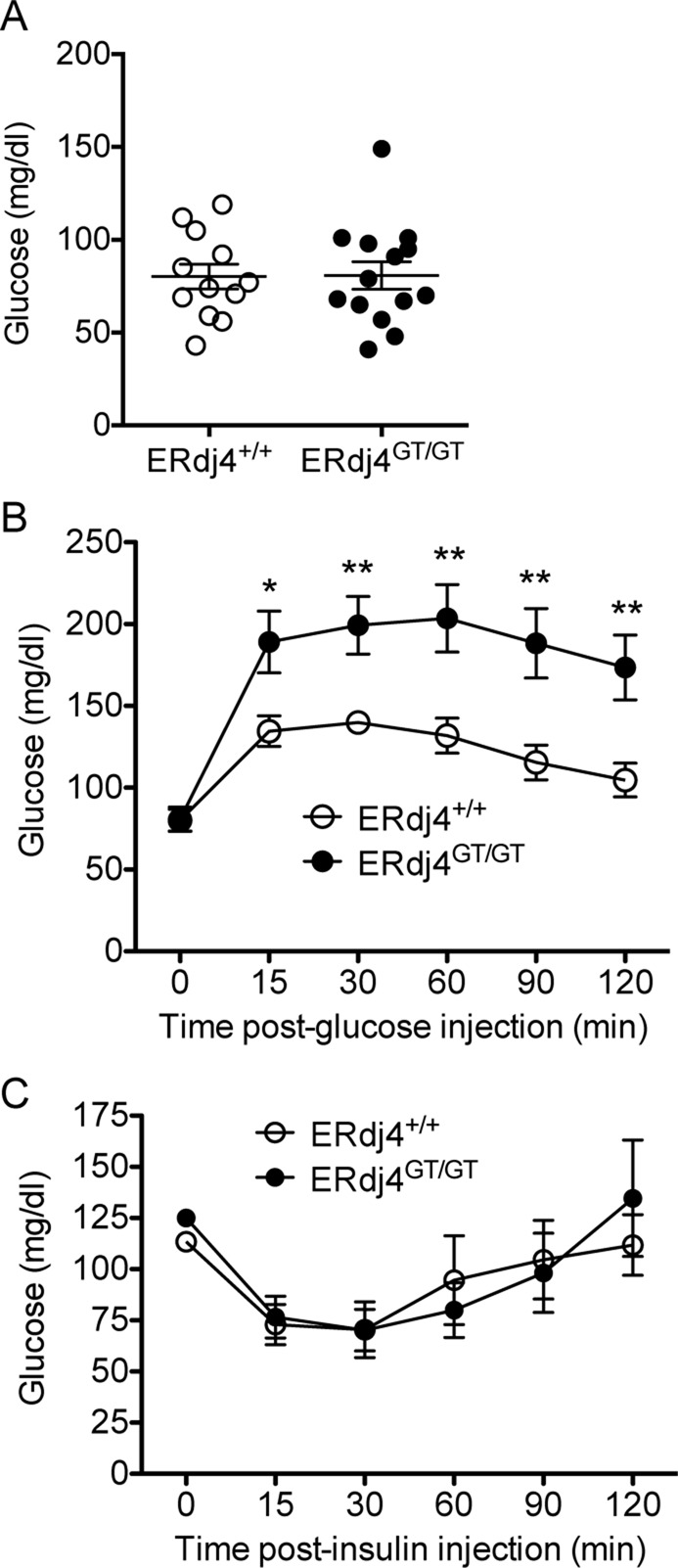
Hypoinsulinemia causes glucose intolerance in ERdj4GT/GT mice. (A) Fasting blood glucose levels in 16- to 20-wk-old mice. n = 12–14 mice/genotype. (B) Blood glucose levels over time after administration of glucose to fasted 12- to 16-wk-old mice. n = 12–14 mice/genotype. (C) Blood glucose levels over time after administration of human insulin to fasted 16- to 20-wk-old mice. n = 5 mice/genotype.
DISCUSSION
ERdj4 is a soluble ER chaperone that can directly bind client proteins (Dong et al., 2008; Lai et al., 2012) and recruit BiP to promote substrate solubilization and/or folding (Shen et al., 2002b). Several lines of evidence previously suggested that the chaperone activity of ERdj4 was largely restricted to the ERAD pathway. First, expression of ERdj4 is normally low and rapidly up-regulated in response to ER stress (Shen et al., 2002b; Kurisu et al., 2003), including stress resulting from expression of terminally misfolded SP-C (Dong et al., 2008). Second, ERdj4 exhibited prolonged association with misfolded SP-C but not with the corresponding wild-type protein (Dong et al., 2008). Third, misfolded SP-C coprecipitated in a complex containing ERdj4 and the cytosolic ATPase p97/VCP (Dong et al., 2008). Finally, ERdj4 transiently associated with ERAD-specific derlin-1 (Lai et al., 2012). Taken together, these results suggest that ERdj4 may function in cooperation with the retrotranslocation machinery late in the ERAD pathway. The results of the present study, demonstrating perinatal lethality and metabolic derangements in ERdj4GT/GT mice, suggest an expanded role for ERdj4 in adaptation to ER stress associated with normal growth and metabolism. One possible explanation for these findings is that ERdj4 associates with proteins that are abnormally prone to misfolding and/or fold more slowly than other substrates, leading to prolonged ERdj4/client interaction and elimination via ERAD under conditions of stress. Although direct evidence is lacking, it is also possible that ERdj4 is required for productive folding of some wild-type substrates, in addition to selective elimination of terminally misfolded substrates from the ER lumen.
Hypomorphic expression of ERdj4 resulted in normal Mendelian distribution of genotypes at E18.5; in contrast, survival of newborn ERdj4GT/GT pups was significantly decreased in a strain-dependent manner. Neonatal lethality can arise from defects in respiration, suckling, or metabolism (Turgeon et al., 2009). ERdj4GT/GT pups did not display overt signs of respiratory distress and contained milk in their stomachs, consistent with normal respiration and suckling. However, newborn ERdj4GT/GT pups were severely hypoglycemic, with significantly reduced hepatic glycogen stores, consistent with impaired glycogenesis and/or glycogenolysis. Plasma glucagon was elevated in neonatal ERdj4GT/GT mice, likely in attempt to increase glucose levels by stimulating glycogen breakdown. Hyperglucagonemia persisted in adulthood and was associated with α cell hyperplasia; in contrast, plasma insulin was normal in newborn mice but declined in adult animals in association with β cell loss (discussed later). Collectively these results suggest that loss of ERdj4 expression perturbs glycogen metabolism during the critical transition from placental nutrition to suckling. Consistent with this conclusion, lower hepatic glycogen stores were linked to fetal growth restriction (Gruppuso and Brautigan, 1989; Lanoue et al., 1999), a prominent phenotype in ERdj4GT/GT mice. ERdj4 is highly expressed in the placenta compared with other tissues (Hageman and Kampinga, 2009), and IRE1α, a signaling molecule upstream of ERdj4, is essential for development of the placental labyrinth layer (Iwawaki et al., 2009), where nutrients and gas are exchanged between mother and fetus. Thus, although the precise molecular pathway(s) underlying the perinatal function of ERdj4 remains uncertain, expression of this chaperone is important for normal fetal development and adaptation to increased metabolic demand in the immediate postnatal period.
In adult mice, ERdj4 deficiency resulted in β cell pathology associated with hypoinsulinemia and glucose intolerance. These findings are consistent with the concept that components of the UPR, including ERdj4, play an important role in cells that synthesize large amounts of protein in response to metabolic cues. Previous studies linked diabetes pathogenesis to altered expression of the ER stress sensors PERK, ATF6, and IRE1α/XBP1. Mutations in PERK and ATF6α were associated with disrupted glucose homeostasis and diabetes in humans (Delepine et al., 2000; Allotey et al., 2004; Senee et al., 2004; Thameem et al., 2006; Meex et al., 2007). Deficiency of PERK, XBP1, or ERdj6/p58IPK (an XBP1 target gene and ERdj/HSP40 family member) resulted in spontaneous diabetes in mice due to a failure in development and/or function of β cells (Harding et al., 2001; Scheuner et al., 2001; Zhang et al., 2002; Ladiges et al., 2005; Lee et al., 2011). Further, the finding of increased expression of XBP1 in β cells of ERdj4GT/GT mice is similar to that in pancreatic islets of diabetic mouse models and type 2 diabetic patients (Huang et al., 2007; Laybutt et al., 2007; Marchetti et al., 2007). Overall these results support the concept that loss of ERdj4 expression exacerbates ER stress associated with increased metabolic demand in β cells. This finding may be generalizable, as increased ER stress was detected in a variety of cells/tissues from ERdj4GT/GT mice.
Elevated ER stress was associated with retention/accumulation of proinsulin in the ER of β cells in ERdj4GT/GT mice. Several possibilities could explain this finding, including increased synthesis, impaired maturation, and/or decreased ERAD of unfolded/misfolded proinsulin. BiP associates with proinsulin in the ER to promote folding and prevent aggregation of hydrophobic domains (Schmitz et al., 1995; Scheuner et al., 2005). Under conditions of metabolic stress and increased insulin biosynthesis, ERdj4 may initiate the interaction between proinsulin and BiP and/or promote proinsulin folding by stimulating ATPase activity. Alternatively, given the substantial role of ERdj4 in trafficking unfolded/misfolded proteins to the proteasome (Dong et al., 2008), an increase in proinsulin in the ER may result from reduced degradation. ERdj4GT/GT mice also showed evidence of impaired proinsulin processing, including an abundance of immature secretory granules and an increased plasma proinsulin:insulin ratio. Thus it is possible that the chaperone activity of ERdj4 may also be involved in maturation of the insulin processing enzymes Pcsk1, Pcsk2, and/or CPE. Overall ERdj4 deficiency resulted in a defect in insulin biosynthesis that likely contributed to hypoinsulinemia.
Hypoinsulinemia in ERdj4GT/GT mice was also associated with β cell loss. The UPR activates cell death pathways in response to protein accumulation that cannot be resolved through attenuation of protein translation or up-regulation of protein folding/degradation machinery (Back and Kaufman, 2012). Although it occurred at a relatively low frequency, cell death was significantly increased in pancreatic islets of ERdj4GT/GT mice. In support of this finding, several mouse models with genetic alterations in UPR components exhibited increased β cell death in association with ER stress (Zhang et al., 2002; Ladiges et al., 2005). Alternatively, β cell loss can be the result of dedifferentiation. A recent study (Talchai et al., 2012) demonstrated that increased metabolic stress caused β cells to revert to a progenitor phenotype that no longer produced insulin: progenitor cells can redifferentiate to α cells, resulting in hyperglucagonemia. Because α cell hyperplasia and hyperglucagonemia are characteristic of ERdj4GT/GT mice, it is possible that increased β cell stress not only resulted in death, but also caused reprogramming.
ERdj4 was previously identified as a BiP cochaperone that is up-regulated in response to ER stress to facilitate the removal of unfolded/misfolded proteins from the ER lumen. In this study, hypomorphic expression of ERdj4 in mice decreased perinatal survival in association with fetal growth restriction, reduced hepatic glycogen stores, and hypoglycemia. Adult ERdj4GT/GT mice exhibited elevated ER stress in pancreatic β cells associated with hypoinsulinemia and glucose intolerance. Given the evidence of constitutive ER stress in multiple organs of ERdj4GT/GT mice, the chaperone activity of ERdj4 may extend to a broad range of protein substrates. Overall this work reveals a potential role for ERdj4 in adaptation to ER stress associated with increased metabolism.
MATERIALS AND METHODS
Mice
ES cells harboring a GT cassette inserted into the ERdj4 locus (Bay Genomics, ES cell line KST256) were purchased from the Mutant Mouse Regional Resource Center. GT ES cells were injected into blastocysts to generate chimeras. Animals were backcrossed eight generations onto the 129P2 or C57BL/6 genetic background. With the exception of perinatal lethality analyses (Figure 2, C57BL/6), all experiments in this study used 129P2×C57BL/6 littermates. The presence of the GT cassette was confirmed by PCR using primers designed for WT and GT alleles (WT, forward, 5′-AAG CCC AGT AAT AAC CCA ACT TAT C-3′; GT, forward, 5′-TGC TGG AGT CTA GCT ACT TAT CCA C-3′; WT/GT, reverse, 5′-CTG TCT TTT CTG TGT TTG GCT AGA A-3′). ERAI mice (Iwawaki et al., 2004) were purchased from RIKEN BRC, and the presence of the ERAI transgene was confirmed by PCR using primers designed by the supplier (forward, 5′-GAA CCA GGA GTT AAG ACA GC-3′; reverse, 5′-GAA CAG CTC CTC GCC CTT GC-3′). All mice were housed in a pathogen-free barrier facility with free access to food and water. Animal procedures were performed under protocols approved by Cincinnati Children's Hospital Medical Center's Institutional Animal Care and Use Committee.
RNA isolation and real-time PCR
Total RNA was isolated from cells and tissues using the RNeasy Plus Mini Kit (Qiagen, Venlo, Netherlands), and cDNA was synthesized using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). XBP1 was detected in MEFs by standard real-time (RT) PCR using XBP1-specific primers (forward, 5′-GTG GTT GAG AAC CAG GAG TTA AGA-3′; reverse, 5′-AGA ATC TGA AGA GGC AAC AGT GTC-3′). PCR products were separated on 4% agarose gels and visualized by ethidium bromide staining. Quantitative RT-PCR was performed with 25 ng of cDNA per reaction on the ABI 7300 system with TaqMan Assays (Applied Biosystems, Foster City, CA) for mouse ERdj4 (exon 3, Mm01622956_s1), a custom-designed β-galactosidase/neomycin resistance region of the GT cassette (forward, 5′-CTG TTC CGT CAT AGC GAT AAC GA-3′; reverse, 5′-TTG CCA GCG GCT TAC CAT-3′; reverse, 5′-FAM-CCA GCG CCA CCA TC-NFQ-3′), and mouse β-actin (endogenous control, Mm00607939_s1). Relative quantitation was assessed using SDS software (Applied Biosystems).
Gene copy-number assay
Tail gDNA (20 ng) was prepared in quadruplicate as recommended by Applied Biosystems. The reaction mix included a custom-designed TaqMan Copy Number Assay targeted for the β-galactosidase region of the GT cassette and a TaqMan Copy Number Reference Assay specific for the mouse transferrin receptor gene (Applied Biosystems). Absolute quantitation was performed with the ABI 7300 system, followed by gene-copy-number analysis with SDS and CopyCaller software (Applied Biosystems).
Immunoblot analyses
Tissue lysate preparation and immunoblot analyses were performed as previously described (Bridges et al., 2003). Briefly, tissue was harvested and homogenized in RIPA buffer (Teknova) containing a mammalian protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). Homogenates were centrifuged at 5000 × g for 10 min to clear tissue debris, and protein concentration in the supernatants was assessed using the Micro BCA Kit (Pierce Biotechnology, Rockford, IL). Lysates were analyzed by SDS-polyacrylamide gels under reducing conditions, transferred to nitrocellulose membranes, and immunoblotted with antibodies specific for IRE1α (Cell Signaling Technology, Danvers, MA), GFP (Invitrogen), BiP (Cell Signaling Technology), calnexin (Cell Signaling Technology), or β-actin (Seven Hills Bioreagents, Cincinnati, OH).
Cell culture
MEFs were isolated from ERdj4+/+ and ERdj4GT/GT embryos on day 13.5 as described by Conner (2001). Cells were cultured in DMEM with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 200 mM l-glutamine and passaged at least three times before use in experiments. Cell viability was assessed (CellTiter 96 AQueous One Solution Cell Proliferation Assay; Promega, Fitchburg, WI) after treatment with tunicamycin (EMD Millipore, Billerica, MA), thapsigargin (EMD Millipore), or MG-132 (EMD Millipore) for 48 h at the indicated concentrations.
Histology and electron microscopy
Tissues were fixed in 4% paraformaldehyde, embedded in paraffin, and cut at 5 μm for hematoxylin and eosin staining and immunostaining, as previously described (Bridges et al., 2003). For immunofluorescence, slides underwent antigen retrieval by boiling in 10 mM citrate buffer (pH 6.0) for 20 min, followed by blocking in normal donkey serum (Jackson ImmunoResearch, West Grove, PA) for 2 h. Sections were stained with primary antibodies specific for insulin (ab7842; Abcam, Cambridge, England), glucagon (ab932; EMD Millipore), BiP (G9043; Sigma-Aldrich), and GFP (ab13970; Abcam), followed by application of secondary antibodies conjugated to Alexa Fluor 488, Alexa Fluor 555, or Alexa Fluor 594 (Invitrogen). Image stacks were acquired by 0.25-μm step distance using a Nikon C1 Si Confocal Imaging System and processed for deconvolution to remove image blurring using Autoquant X 2.2 (Media Cybernetics, Sarasota, FL). For TUNEL analyses, paraffin-embedded tissue sections were stained using the ApopTag Peroxidase In Situ Apoptosis Detection Kit per manufacturers’ instructions (EMD Millipore). TUNEL-positive cells in pancreatic islets of ERdj4+/+ (n = 5 mice, 54 islets) and ERdj4GT/GT (n = 4 mice, 24 islets) mice were visualized and manually counted using the Axio Imager.A2 microscope (Zeiss, Oberkochen, Germany) under a 20× objective lens.
For electron microscopy, cells and tissues were processed as previously described (Ridsdale et al., 2011). Briefly, samples were fixed in 2% paraformaldehyde (Electron Microscopy Sciences [EMS], Hatfield, PA), 2% glutaraldehyde (EMS), and 0.1% calcium chloride in 0.1 M sodium cacodylate buffer (pH 7.3) for 30 min, followed by transfer to fresh fixative at 4°C overnight. Fixed cells and tissues were cut into 1- to 2-mm blocks, incubated with 1% osmium tetroxide (EMS) and 1.5% potassium ferrocyanide (Sigma-Aldrich) in 0.1 M sodium cacodylate buffer (pH 7.3) for 2 h, stained en bloc with 4% aqueous uranyl acetate (EMS) at 4°C overnight, dehydrated with a graded series of alcohol, and embedded in expoxy resin EMbed 812 (EMS) or Quetol 651 (EMS). Ultrathin 90-nm sections were cut using a Reichert Ultracut E ultramicrotome (Leica Microsystems, Wetzlar, Germany). Electron micrographs were collected using a Hitachi H-7650 TEM (Hitachi High Technologies America, Dallas, TX) equipped with an AMT transmission electron microscope charge-coupled device camera (Advanced Microscopy Techniques, Woburn, MA).
Morphometric analyses of pancreatic islets
Pancreatic tissue was harvested from 8- to 12-wk-old ERdj4+/+ (n = 8) and ERdj4GT/GT (n = 5) mice, fixed in 4% paraformaldehyde, and embedded in paraffin. Tissues were sectioned at 5 μM using a Leica RM2235 microtome, and one section was collected every 100 μM. After immunofluorescence for insulin and glucagon (described earlier), images were acquired using the Axioplan 2 microscope (Zeiss) equipped with an AxioCam MRm camera and analyzed using Imaris, version 7.6.4 (Bitplane, Belfast, United Kingdom). The β and α cell numbers were obtained by counting DAPI-positive nuclei in insulin- and glucagon-positive sections. Twenty islets (≥30 cells/islet) were analyzed per mouse.
Islet isolation and metabolic labeling
Pancreatic islets were isolated from 6- to 8-wk-old mice as previously described (Zmuda et al., 2011). Briefly, the pancreas was perfused with liberase (0.5 mg/ml; Roche Applied Science, Penzberg, Upper Bavaria, Germany) and removed for digestion in a 37°C water bath for 20 min. After washing and filtering the digest, islets were separated from exocrine cells by density gradient centrifugation using Histopaque 1077 (Sigma-Aldrich). Islets were collected from the Histopaque/media interface and washed several times before culturing overnight in CRML 1066 (Cellgro, Manassas, VA) containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. For metabolic labeling experiments, 100 islets were hand-picked from each genotype and repeatedly washed in Krebs–Ringer bicarbonate buffer (KRB) containing 1% bovine serum albumin. To stimulate insulin biosynthesis, islets were incubated in KRB containing 2.8 mM glucose for 1 h at 37°C and then resuspended in KRB containing 16.7 mM glucose and incubated for another hour (Welsh et al., 1986; Skelly et al., 1998). During the last 20 min of incubation, islets were labeled with 0.5 mCi/ml [35S]methionine/cysteine (MP Biomedicals, Santa Ana, CA) for 60 min. Cells were lysed and immunoprecipitated with guinea pig anti-insulin antibody (ab7842; Abcam) and protein G plus agarose beads (Santa Cruz Biotechnology, Dallas, TX) at 4°C overnight. Immunoprecipitates were analyzed by SDS–PAGE under reducing conditions and visualized by autoradiography, and the proinsulin signal was quantitated using Multi Gauge software, version 3 (Fugifilm, Tokyo, Japan).
Serum lipase and amylase activity
Blood was collected from mice through the retroorbital sinus into serum separator tubes (BD Biosciences, Franklin Lakes, NJ). Serum was submitted to the Clinical Laboratory at Cincinnati Children's Hospital for enzyme analyses.
Triglyceride and nonesterified fatty acid analyses
Blood was collected from neonatal mice through decapitation into microtubes containing lithium heparin (Sarstedt, Nuembrecht, Germany). Plasma was submitted to the Cincinnati Mouse Metabolic Phenotyping Center for lipid analyses.
Insulin and glucagon assays
Insulin and glucagon were extracted from the pancreas by repeated homogenization in acid-ethanol (0.18 M HCl in 70% ethanol; Sjoholm et al., 2001). Mouse insulin (Crystal Chem, Downers Grove, IL) and glucagon (R&D Systems, Minneapolis, MN) were assessed by enzyme-linked immunosorbent assay (ELISA), and the results were normalized to total protein in pancreatic extracts. Plasma proinsulin (ALPCO Diagnostics, Salem, NH) and insulin were determined by ELISA in 6-h-fasted mice at baseline or after intraperitoneal injection with 10% glucose. Plasma glucagon in neonatal and adult mice was analyzed by the Cincinnati Mouse Metabolic Phenotyping Center.
Glycogen assay
Neonatal livers were harvested, weighed, and flash frozen in liquid nitrogen until assayed. Liver tissue was homogenized in 200 μl of distilled water by successive passage through 22 and 25G needles. Homogenates were boiled at 100°C for 5 min and then centrifuged at 15,300 × g for 5 min. The supernatant was assayed for glycogen using the Glycogen Colorimetric/Fluorometric Assay Kit (Biovision, San Francisco, CA). Total glycogen content per liver was normalized to liver weight.
Glucose and insulin tolerance tests
Mice were fasted for 16 h with free access to water. Blood glucose levels were determined by collecting blood from the tail vein at baseline using an ACCU-CHEK Aviva glucose monitor (Roche Applied Science) before intraperitoneal injection of 10% glucose (1 unit/g of body weight). Blood glucose levels were monitored over time after injection. Insulin tolerance tests were performed on 6-h-fasted mice by intraperitoneal injection of human insulin (0.75 U/kg; Eli Lilly, Indianapolis, IN), followed by blood glucose measurements as described.
Statistical analyses
All data represent mean ± SEM with a significance difference reported as *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001. Two-way comparisons were analyzed by two-tailed, unpaired Student's t test using Prism software (GraphPad, La Jolla, CA).
Supplementary Material
Acknowledgments
We thank Eileen Elfers and Jonathan Katz for assistance with islet isolation and Mary Falconieri for metabolic labeling experiments. This study was supported by National Institute of Health Grants HL103923 (T.E.W.) and HL086492 (T.E.W.).
Abbreviations used:
- ATF6
activating transcription factor 6
- BiP
immunoglobulin-binding protein
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- ERAI
ER stress–activated indicator
- ERdj
ER-localized DnaJ
- GFP
green fluorescent protein
- GT
gene trap
- HSP
heat shock protein
- IRE1
inositol-requiring enzyme 1
- MEF
mouse embryonic fibroblast
- PERK
protein kinase RNA–like ER kinase
- SBD
substrate-binding domain
- SP-C
surfactant protein C
- UPR
unfolded protein response
- WT
wild type
- XBP1
X-box binding protein 1
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-06-0319) on December 11, 2013.
REFERENCES
- Allotey RA, et al. The EIF2AK3 gene region and type I diabetes in subjects from South India. Genes Immun. 2004;5:648–652. doi: 10.1038/sj.gene.6364139. [DOI] [PubMed] [Google Scholar]
- Awad W, Estrada I, Shen Y, Hendershot LM. BiP mutants that are unable to interact with endoplasmic reticulum DnaJ proteins provide insights into interdomain interactions in BiP. Proc Natl Acad Sci USA. 2008;105:1164–1169. doi: 10.1073/pnas.0702132105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem. 2012;81:767–793. doi: 10.1146/annurev-biochem-072909-095555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- Bridges JP, Wert SE, Nogee LM, Weaver TE. Expression of a human surfactant protein C mutation associated with interstitial lung disease disrupts lung development in transgenic mice. J Biol Chem. 2003;278:52739–52746. doi: 10.1074/jbc.M309599200. [DOI] [PubMed] [Google Scholar]
- Conner DA. Mouse embryo fibroblast (MEF) feeder cell preparation. Curr Protoc Mol Biol. 2001. Chapter 23, Unit 23.2. [DOI] [PubMed]
- Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet. 2000;25:406–409. doi: 10.1038/78085. [DOI] [PubMed] [Google Scholar]
- Dong M, Bridges JP, Apsley K, Xu Y, Weaver TE. ERdj4 and ERdj5 are required for endoplasmic reticulum-associated protein degradation of misfolded surfactant protein C. Mol Biol Cell. 2008;19:2620–2630. doi: 10.1091/mbc.E07-07-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fewell SW, Travers KJ, Weissman JS, Brodsky JL. The action of molecular chaperones in the early secretory pathway. Annu Rev Genet. 2001;35:149–191. doi: 10.1146/annurev.genet.35.102401.090313. [DOI] [PubMed] [Google Scholar]
- Gruppuso PA, Brautigan DL. Induction of hepatic glycogenesis in the fetal rat. Am J Physiol. 1989;256:E49–54. doi: 10.1152/ajpendo.1989.256.1.E49. [DOI] [PubMed] [Google Scholar]
- Hageman J, Kampinga HH. Computational analysis of the human HSPH/HSPA/DNAJ family and cloning of a human HSPH/HSPA/DNAJ expression library. Cell Stress Chaperones. 2009;14:1–21. doi: 10.1007/s12192-008-0060-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7:1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- Huang CJ, Lin CY, Haataja L, Gurlo T, Butler AE, Rizza RA, Butler PC. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes. 2007;56:2016–2027. doi: 10.2337/db07-0197. [DOI] [PubMed] [Google Scholar]
- Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4:321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- Iwawaki T, Akai R, Kohno K. IRE1alpha disruption causes histological abnormality of exocrine tissues, increase of blood glucose level, and decrease of serum immunoglobulin level. PLoS One. 2010;5:e13052. doi: 10.1371/journal.pone.0013052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwawaki T, Akai R, Kohno K, Miura M. A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat Med. 2004;10:98–102. doi: 10.1038/nm970. [DOI] [PubMed] [Google Scholar]
- Iwawaki T, Akai R, Yamanaka S, Kohno K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc Natl Acad Sci USA. 2009;106:16657–16662. doi: 10.1073/pnas.0903775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Prasad K, Lafer EM, Sousa R. Structural basis of interdomain communication in the Hsc70 chaperone. Mol Cell. 2005;20:513–524. doi: 10.1016/j.molcel.2005.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Awad W, Petrova K, Hendershot LM. Regulated release of ERdj3 from unfolded proteins by BiP. EMBO J. 2008;27:2873–2882. doi: 10.1038/emboj.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemoto S, Kondo S, Ogata M, Murakami T, Urano F, Imaizumi K. XBP1 activates the transcription of its target genes via an ACGT core sequence under ER stress. Biochem Biophys Res Commun. 2005;331:1146–1153. doi: 10.1016/j.bbrc.2005.04.039. [DOI] [PubMed] [Google Scholar]
- Kurisu J, Honma A, Miyajima H, Kondo S, Okumura M, Imaizumi K. MDG1/ERdj4, an ER-resident DnaJ family member, suppresses cell death induced by ER stress. Genes Cells. 2003;8:189–202. doi: 10.1046/j.1365-2443.2003.00625.x. [DOI] [PubMed] [Google Scholar]
- Ladiges WC, Knoblaugh SE, Morton JF, Korth MJ, Sopher BL, Baskin CR, MacAuley A, Goodman AG, LeBoeuf RC, Katze MG. Pancreatic beta-cell failure and diabetes in mice with a deletion mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes. 2005;54:1074–1081. doi: 10.2337/diabetes.54.4.1074. [DOI] [PubMed] [Google Scholar]
- Lai CW, Otero JH, Hendershot LM, Snapp E. ERdj4 protein is a soluble endoplasmic reticulum (ER) DnaJ family protein that interacts with ER-associated degradation machinery. J Biol Chem. 2012;287:7969–7978. doi: 10.1074/jbc.M111.311290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanoue L, Liu XJ, Koski KG. Postnatal profiles of glycogenolysis and gluconeogenesis are modified in rat pups by maternal dietary glucose restriction. J Nutr. 1999;129:820–827. doi: 10.1093/jn/129.4.820. [DOI] [PubMed] [Google Scholar]
- Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, Biankin AV, Biden TJ. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia. 2007;50:752–763. doi: 10.1007/s00125-006-0590-z. [DOI] [PubMed] [Google Scholar]
- Lee YK, Brewer JW, Hellman R, Hendershot LM. BiP and immunoglobulin light chain cooperate to control the folding of heavy chain and ensure the fidelity of immunoglobulin assembly. Mol Biol Cell. 1999;10:2209–2219. doi: 10.1091/mbc.10.7.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Chu GC, Iwakoshi NN, Glimcher LH. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 2005;24:4368–4380. doi: 10.1038/sj.emboj.7600903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Heidtman K, Hotamisligil GS, Glimcher LH. Dual and opposing roles of the unfolded protein response regulated by IRE1alpha and XBP1 in proinsulin processing and insulin secretion. Proc Natl Acad Sci USA. 2011;108:8885–8890. doi: 10.1073/pnas.1105564108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti P, Bugliani M, Lupi R, Marselli L, Masini M, Boggi U, Filipponi F, Weir GC, Eizirik DL, Cnop M. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia. 2007;50:2486–2494. doi: 10.1007/s00125-007-0816-8. [DOI] [PubMed] [Google Scholar]
- Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meex SJ, et al. Activating transcription factor 6 polymorphisms and haplotypes are associated with impaired glucose homeostasis and type 2 diabetes in Dutch Caucasians. J Clin Endocrinol Metab. 2007;92:2720–2725. doi: 10.1210/jc.2006-2280. [DOI] [PubMed] [Google Scholar]
- Molinari M, Galli C, Piccaluga V, Pieren M, Paganetti P. Sequential assistance of molecular chaperones and transient formation of covalent complexes during protein degradation from the ER. J Cell Biol. 2002;158:247–257. doi: 10.1083/jcb.200204122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa SI, Fewell SW, Kato Y, Brodsky JL, Endo T. Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J Cell Biol. 2001;153:1061–1070. doi: 10.1083/jcb.153.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero JH, Lizak B, Hendershot LM. Life and death of a BiP substrate. Semin Cell Dev Biol. 2010;21:472–478. doi: 10.1016/j.semcdb.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova K, Oyadomari S, Hendershot LM, Ron D. Regulated association of misfolded endoplasmic reticulum lumenal proteins with P58/DNAJc3. EMBO J. 2008;27:2862–2872. doi: 10.1038/emboj.2008.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridsdale R, Na CL, Xu Y, Greis KD, Weaver T. Comparative proteomic analysis of lung lamellar bodies and lysosome-related organelles. PLoS One. 2011;6:e16482. doi: 10.1371/journal.pone.0016482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Vander Mierde D, Song B, Flamez D, Creemers JWM, Tsukamoto K, Ribick M, Schuit FC, Kaufman RJ. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat Med. 2005;11:757–764. doi: 10.1038/nm1259. [DOI] [PubMed] [Google Scholar]
- Schmitz A, Maintz M, Kehle T, Herzog V. In vivo iodination of a misfolded proinsulin reveals co-localized signals for BiP binding and for degradation in the ER. EMBO J. 1995;14:1091–1098. doi: 10.1002/j.1460-2075.1995.tb07092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Senee V, et al. Wolcott-Rallison syndrome: clinical, genetic, and functional study of EIF2AK3 mutations and suggestion of genetic heterogeneity. Diabetes. 2004;53:1876–1883. doi: 10.2337/diabetes.53.7.1876. [DOI] [PubMed] [Google Scholar]
- Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002a;3:99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- Shen Y, Hendershot LM. ERdj3, a stress-inducible endoplasmic reticulum DnaJ homologue, serves as a cofactor for BiP's interactions with unfolded substrates. Mol Biol Cell. 2005;16:40–50. doi: 10.1091/mbc.E04-05-0434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Meunier L, Hendershot LM. Identification and characterization of a novel endoplasmic reticulum (ER) DnaJ homologue, which stimulates ATPase activity of BiP in vitro and is induced by ER stress. J Biol Chem. 2002b;277:15947–15956. doi: 10.1074/jbc.M112214200. [DOI] [PubMed] [Google Scholar]
- Sjoholm A, Arkhammar P, Berggren PO, Andersson A. Polyamines in pancreatic islets of obese-hyperglycemic (ob/ob) mice of different ages. Am J Physiol Cell Physiol. 2001;280:C317–C323. doi: 10.1152/ajpcell.2001.280.2.C317. [DOI] [PubMed] [Google Scholar]
- Skelly RH, Bollheimer LC, Wicksteed BL, Corkey BE, Rhodes CJ. A distinct difference in the metabolic stimulus-response coupling pathways for regulating proinsulin biosynthesis and insulin secretion that lies at the level of a requirement for fatty acyl moieties. Biochem J. 1998;331:553–561. doi: 10.1042/bj3310553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell. 2012;150:1223–1234. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thameem F, Farook VS, Bogardus C, Prochazka M. Association of amino acid variants in the activating transcription factor 6 gene (ATF6) on 1q21-q23 with type 2 diabetes in Pima Indians. Diabetes. 2006;55:839–842. doi: 10.2337/diabetes.55.03.06.db05-1002. [DOI] [PubMed] [Google Scholar]
- Turgeon B, Meloche S, De Recherche I. Interpreting neonatal lethal phenotypes in mouse mutants: insights into gene function and human diseases. Physiol Rev. 2009:89, 1–26. doi: 10.1152/physrev.00040.2007. [DOI] [PubMed] [Google Scholar]
- Voss AK, Thomas T. Identification of novel genes by gene trap mutagenesis. Methods Mol Biol. 2001;175:377–396. doi: 10.1385/1-59259-235-X:377. [DOI] [PubMed] [Google Scholar]
- Voss AK, Thomas T, Gruss P. Compensation for a gene trap mutation in the murine microtubule-associated protein 4 locus by alternative polyadenylation and alternative splicing. Dev Dyn. 1998a;212:258–266. doi: 10.1002/(SICI)1097-0177(199806)212:2<258::AID-AJA10>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Voss AK, Thomas T, Gruss P. Efficiency assessment of the gene trap approach. Dev Dyn. 1998b;212:171–180. doi: 10.1002/(SICI)1097-0177(199806)212:2<171::AID-AJA3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Wang N, Daniels R, Hebert DN. The cotranslational maturation of the type I membrane glycoprotein tyrosinase: the heat shock protein 70 system hands off to the lectin-based chaperone system. Mol Biol Cell. 2005;16:3740–3752. doi: 10.1091/mbc.E05-05-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzmann A, Baldes C, Dudek J, Zimmermann R. The heat shock protein 70 molecular chaperone network in the pancreatic endoplasmic reticulum—a quantitative approach. FEBS J. 2007;274:5175–5187. doi: 10.1111/j.1742-4658.2007.06039.x. [DOI] [PubMed] [Google Scholar]
- Welsh M, Hammer RE, Brinster RL, Steiner DF. Stimulation of growth hormone synthesis by glucose in islets of Langerhans isolated from transgenic mice. J Biol Chem. 1986;261:12915–12917. [PubMed] [Google Scholar]
- Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol. 2002;22:3864–3874. doi: 10.1128/MCB.22.11.3864-3874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmuda EJ, Powell CA, Hai T. A method for murine islet isolation and subcapsular kidney transplantation. J Vis Exp. 2011 doi: 10.3791/2096. doi:10.3791/2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.