

Premature S-phase entry due to Cdh1 ablation results from premature loss of the CDK inhibitor p27 and a reduced requirement for cyclin E1. This prolonged S phase coincides with slowed replication fork elongation and fewer replication terminations, both of which could contribute to genome instability.
Abstract
Cdh1, a coactivator of the anaphase-promoting complex (APC), is a potential tumor suppressor. Cdh1 ablation promotes precocious S-phase entry, but it was unclear how this affects DNA replication dynamics while contributing to genomic instability and tumorigenesis. We find that Cdh1 depletion causes early S-phase onset in conjunction with increase in Rb/E2F1-mediated cyclin E1 expression, but reduced levels of cyclin E1 protein promote this transition. We hypothesize that this is due to a weakened cyclin-dependent kinase inhibitor (CKI)–cyclin-dependent kinase 2 positive-feedback loop, normally generated by APC-Cdh1–mediated proteolysis of Skp2. Indeed, Cdh1 depletion increases Skp2 abundance while diminishing levels of the CKI p27. This lowers the level of cyclin E1 needed for S-phase entry and delays cyclin E1 proteolysis during S-phase progression while corresponding to slowed replication fork movement and reduced frequency of termination events. In summary, using both experimental and computational approaches, we show that APC-Cdh1 establishes a stimulus–response relationship that promotes S phase by ensuring that proper levels of p27 accumulate during G1 phase, and defects in its activation accelerate the timing of S-phase onset while prolonging its progression.
INTRODUCTION
Ubiquitin-mediated proteolysis plays essential roles in regulating eukaryotic cell cycle progression. The transitions between and maintenance of each cell cycle phase are controlled by the activation of specific cyclin-dependent kinase (CDK)/cyclin complexes. Once these transitions occur, however, ubiquitin-mediated degradation of cyclins and other cell cycle regulators inactivates CDK and resets the cell to prepare it for the next division cycle. From late mitosis to G1 phase, proteolysis of mitotic cyclins and other mitotic regulators depends on the activity of the anaphase-promoting complex (APC), an E3 ubiquitin ligase (Kraft et al., 2003; Castro et al., 2005; Peters, 2006). APC is activated by CDK1/cyclin B phosphorylation, and it catalyzes the polyubiquitylation of cyclin B (King et al., 1995; Lahav-Baratz et al., 1995; Kraft et al., 2003). This negative-feedback loop results in the degradation of cyclin B by the 26S proteosome and leads to the inactivation of CDK1. Cdc20 and Cdh1 are two adaptor proteins required to temporally control APC activity, targeting it to different substrates during the cell cycle (Kramer et al., 1998; Castro et al., 2005). The APC-Cdc20 complex facilitates the metaphase-to-anaphase transition, whereas in late mitosis and the subsequent G1 phase, its Cdc20 cofactor is replaced by dephosphorylated Cdh1 to form the APC-Cdh1 complex (Kramer et al., 2000; Peters, 2006).
From yeast to humans, Cdh1 plays a conserved role in eukaryotic cell cycle regulation. Many APC-Cdh1 targets, including Skp2, cyclin A, Polo-like kinase 1, and the Aurora kinases, are potential oncoproteins that are overexpressed in various cancers (Waesch et al., 2010). Use of mouse genetic models showed that Cdh1 functions as a haploinsufficient tumor suppressor (Garci-Higuera et al., 2008). Whereas many APC-Cdh1 substrates are mitotic regulators, Cdh1-deficient cell lines are capable of proliferating and show no significant defects at mitotic exit (Sigl et al., 2009). Conversely, Cdh1 depletion caused precocious and prolonged S-phase periods (Engelbert et al., 2007; Garci-Higuera et al., 2008; Gao et al., 2009). What remained unknown, however, is which specific CDK/cyclin complex stimulates this premature G1/S transition, and the fact that redundancy exists between the CDKs and cyclins that drive cell cycle progression was a potential confounding factor in this problem (Aleem et al., 2005; Santamaria et al., 2007). Cyclin A was reported to accumulate prematurely in Cdh1-depleted cells and had been implicated as the premature activator of CDK2 during early S-phase onset, but the involvement of other cyclins had not been described (Sigl et al., 2009). Additional work showed that Cdh1 was necessary to maintain G0/G1 phases by degrading not only cyclins, but also Skp2, which is an F-box protein of the SKP1-CUL1-F-box (SCF) complex that targets CDK inhibitors (CKIs) for degradation (Carrano et al., 1999; Bornstein et al., 2003; Kamura et al., 2003). Attenuated accumulation of the CKIs (p27 and p21) in Cdh1-depleted cells occurred due to Skp2 stabilization, and early SCF activation was implicated in permitting premature S-phase entry (Bashir et al., 2004; Wei et al., 2004). Together these previous findings provided initial insight into how reduced APC-Cdh1 activity could lead to cell cycle deregulation and genome instability, potentially culminating with tumorigenesis. However, direct links had not been established between Cdh1 loss, its subsequent effect on the abundance and activities of S-phase-promoting and -inhibiting factors, and how these changes alter S-phase dynamics at the molecular level.
In the present study, we show that Cdh1 ablation not only accelerates S-phase entry, but also lowers its requirement for cyclin E1, despite increases in its Rb-E2F1–mediated expression. We describe how APC-Cdh1 initiates a series of inhibitory steps that culminate in a positive-feedback loop essential to generate the mammalian S-phase switch. We propose that Cdh1 ablation lessens the potential strength of the double-negative (positive) feedback loop generated between CDK2 and the CKI p27. This in turn causes the response of CDK2 to cyclin E1 stimulus level to become more linear, altering the dynamics of S-phase entry and progression. This was indeed the case, as p27 depletion mirrored the lowered requirement for cyclin E1 levels at S-phase entry—like those ablated for Cdh1—and cells entered S-phase prematurely. Cdh1 knockdown not only caused DNA replication to initiate hours earlier at lower cyclin E1 levels, but it also significantly prolonged S phase due to a reduced rate of replication fork movement, corresponding to delayed destruction of cyclin E1. Together these changes in the response to and removal of cyclin E1 stimulus implied an alteration in the switching of CDK2 at the G1/S transition. Cdh1 depletion also significantly reduced the frequency of replication termination events, and these effects combined could lead to replication stress and increase the potential for genome instability. Using ordinary differential equation (ODE) modeling, we then demonstrated qualitatively how loss of Cdh1—through increase in Skp2 levels—could permit an earlier but blunted activation of CDK2 as cyclin E1 accumulates; a model lacking p27 exhibited similar behavior. This supported the idea that reduction in Cdh1 and subsequent loss of p27 would permit premature activation of CDK2 and early initiation of DNA replication but that its response would be less abrupt. In summary, by combining single-cell experimental approaches and modeling, we reveal for the first time on the systems-level how loss of Cdh1 function leads to corresponding alterations in the dynamics of S-phase onset and progression, where the onset of replication errors and genome instability could occur.
RESULTS
Cdh1 ablation induces early S-phase entry and enrichment of S-phase cells in culture
Previous studies using flow cytometry and bromodeoxyuridine (BrdU)-labeling methods revealed that Cdh1 ablation leads to early onset of DNA replication (Engelbert et al., 2007; Gao et al., 2009; Sigl et al., 2009). To measure precisely this shortening of G1 phase in cells depleted for Cdh1, we transfected asynchronous HeLa cells with small interfering RNAs (siRNAs) directed against Cdh1, its earlier APC-activating counterpart, Cdc20, or the firefly luciferase, GL2, along with an S-phase biosensor, proliferating cell nuclear antigen (PCNA)–yellow fluorescent protein (YFP; Hahn et al., 2009). We then imaged cells live by time-lapse microscopy. siRNA treatment reduced Cdh1 and Cdc20 levels efficiently in HeLa cells (Figure 1A), and examination of their G1-phase duration—from cell division to the formation of PCNA punctum during DNA synthesis—revealed that Cdh1 depletion induced S-phase entry nearly 40% earlier (3.4 h) than for control cells (p < 0.001; Figure 1, B and C). Ablation of Cdc20, however, caused a subtle change in G1 duration (0.4 h earlier; p = 0.023; Figure 1B), supporting that G1-phase maintenance is executed by APC-Cdh1.
FIGURE 1:

Depletion of Cdh1 shortens G1-phase duration in HeLa cells. (A) Immunoblots for indicated proteins in HeLa cell lysates (30 μg of protein) at 54 and 70 h post siRNA transfection. (B) Average G1 duration times for GL2, Cdh1, and Cdc20 siRNA–transfected cells (N = 3; nGL2 = 474, nCdh1 = 608, and nCdc20 = 223 cells). Error bars represent a confidence interval of 95% (95% CI). (C) Montages of live-cell images showing representative GL2 siRNA–transfected cell (top) and Cdh1 siRNA–transfected cell (bottom). White arrowheads indicate PCNA punctum formation and S-phase entry. (D) Immunoblots for indicated proteins in HeLa cell lysates (30 μg of total protein) after siRNA transfection with or without a Cdh1-rescue plasmid. Double asterisks indicate endogenous Cdh1; single asterisk indicates ectopic pECFP-mutCdh1. (E) Cell cycle distributions of cells in D, determined by EdU labeling and PI staining, followed by flow cytometry. Data are plotted as means ± SEM (N = 3).
Consistent with prior reports, flow cytometry corroborated that S phase was prematurely initiated and lengthened in Cdh1-ablated cells, with a 20% decrease in the G1-phase population and a 20% increase in the S-phase population (Figure 1E). Cells cotransfected with Cdh1 siRNA and an siRNA-insensitive Cdh1 mutant, pECFP-mutCdh1, recovered nearly 70% of the G1-phase cell population that had been shifted to S phase in the Cdh1 knockdown (Figure 1, D and E). This G1-phase recovery by the siRNA-resistant plasmid was not due to nonspecific effects of its overexpression, since transfection of the pECFP-mutCdh1 plasmid alone caused minimal changes in the G1 (∼10% reduction) and S-phase (∼10% increase) populations relative to the GL2 control (Supplemental Figure S1).
Removal of Cdh1 drives premature accumulation of cyclin B and E proteins through different mechanisms
Prior studies reported premature cyclin B accumulation in Cdh1-depleted cells (Engelbert et al., 2007; Sigl et al., 2009). We also observed increased cyclin B protein content as Cdh1-depleted cells transited through G1 and into S phase, along with other APC-Cdh1 targets, such as AurA and Polo-like kinase 1 (Plk1; Figure 2, A and B). Surprisingly, cyclin E1, which is not an APC-Cdh1 substrate, also accumulated prematurely in Cdh1-depleted cells (Figure 2, A and B). To determine whether precocious accumulation of these cyclins occurred due to early increases in Rb/E2F-mediated transcription, we performed real-time quantitative reverse transcription PCR (qRT-PCR) assays against several E2F1 targets in Cdh1-ablated cells, including cyclin A2, cyclin B1, cyclin E1, and PCNA. Compared to GL2 siRNA–transfected cells, Cdh1-depleted cells had more transcripts from all four of these E2F1-transcribed genes as they progressed from G1 into S phase, but the increase in cyclin E1 mRNA was the most significant (Figure 2, C and D, and Supplemental Figure S2). At 18 h after release from triple-thymidine block, Cdh1-depleted cells contained 2–2.5 times as much cyclin E1 mRNA, and this exceeded the maximum cyclin E1 mRNA abundance observed at S-phase entry in control cells (26–28 h) (Figure 2, C and D, and Supplemental Figure S2). In contrast to cyclin E1, levels of cyclin A2 and cyclin B1 transcript increased only nominally: 18 h after thymidine release, there was 0.1- to 0.2-fold more cyclin A2 mRNA and 0.2- to 0.5-fold more cyclin B1 mRNA in Cdh1 siRNA–transfected cells (Figure 2D). This implied that the increase in cyclin B1 protein might have resulted from a lack of APC-Cdh1–directed proteolysis rather than a significant gain in cyclin B1 transcription (Figure 2B). Consistent with increases in transcription of other E2F1-transcribed genes, however, Cdh1-depleted cells also contained more PCNA mRNA (Figure 2D). Thus, despite a shortened G1 phase, Cdh1 depletion increased the accumulation of certain S-phase transcripts and proteins on the population level relative to GL2 siRNA–transfected control cells.
FIGURE 2:

Cdh1 knockdown in HeLa cells alters the kinetics of cyclin B1 and E1 accumulation and causes early transcription of certain E2F1 targets. (A) Immunoblots for indicated proteins in HeLa cell lysates (35 μg of protein) prepared at time points after GL2 and Cdh1 siRNA transfection with triple-thymidine block and release. (B) Quantitation of cyclin A2, B1, and E1 proteins was performed using ImageJ; values were normalized to tubulin and then plotted as means ± SD (N = 3). (C) Percentage cells in S phase from transfections in A were determined by PI staining and flow cytometry. Data are plotted as means ± SD (N = 3). (D) Relative transcript levels of cyclin A2 (top left), cyclin B1 (top right), cyclin E1 (bottom left), and PCNA (bottom right) determined by qRT-PCR using RNA prepared from GL2 and Cdh1 siRNA–transfected cells at time points after release from triple-thymidine block. Relative mRNA levels were determined using the ΔΔCt method and normalized to GAPDH expression, then converted to fold increase over signal detected in GL2 siRNA–transfected cells (18 h). Data are plotted as means ± SEM (N = 3).
Cyclin E alone stimulates early S-phase entry in cells lacking Cdh1
A prior study linked premature accumulation of cyclin A to early S-phase onset in Cdh1-ablated cells (Sigl et al., 2009). Our finding that cyclin A levels did not change during our time courses, as well the possibility of redundancy between mitotic and S-phase cyclins in promoting S phase, motivated us to test the S-phase-promoting function of different cyclins in cells lacking Cdh1. To identify which cyclin triggers early S-phase onset in Cdh1-depleted cells, we cotransfected siRNAs directed against cyclin A2, B1, or E1 into asynchronous HeLa cells along with Cdh1 siRNA and PCNA-YFP (Figure 3A) and determined their G1 duration by live-cell imaging. Cdh1 knockdown induced PCNA punctum formation and S-phase entry ∼33% (3.1 h) earlier in these experiments (p < 0.001), and double knockdown of cyclin E1 and Cdh1 recovered more than half of this G1-phase duration (p < 0.001; Figure 3B). Ablation of both cyclin A2 and Cdh1, however, did not prolong G1 phase (p = 0.146; Figure 3B). These data indicated that cyclin E1 was involved in regulating premature S-phase onset in Cdh1-depleted cells and that cyclin A2 was not, in contrast with previous findings (Sigl et al., 2009). The early increase in cyclin A–associated kinase activity in this prior study may have been a consequence of early S-phase entry being stimulated by cyclin E1. We also found that a subtle recovery of G1 phase, ∼10% (36 min), occurred by ablating cyclin B and Cdh1 (p = 0.011; Figure 3B), but the removal of Cdh1, cyclin B1, and cyclin E1 together did not significantly alter G1 timing relative to Cdh1-depleted cells without cyclin E1 (p = 0.059; Supplemental Figure S3). Consistent with results of these single-cell analyses, flow cytometry revealed that knocking down only cyclin E1 recovered the G1 population in asynchronous Cdh1-depleted cells (Figure 3C and Supplemental Figure S4A). Indeed, the role of cyclin E1 in promoting S-phase entry in Cdh1-depleted cells followed suit with cells lacking cyclin E1 alone: removal of cyclin E1 from asynchronous HeLa cells prolongs G1 phase relative to control cells, whereas removal of cyclin E1 from cells ablated for Cdh1 lengthens G1 phase relative to cells only lacking Cdh1 (Supplemental Figure S4, B and C). In summary, whereas cyclin B1 protein accumulated significantly in the absence of Cdh1, it did not stimulate premature S-phase entry. In contrast, elevated levels of cyclin E1 corresponded to premature S-phase onset in Cdh1-ablated cells. However, it was unclear whether this increase in cyclin E1 occurred before or during this precocious S phase.
FIGURE 3:

Precocious cyclin E1 accumulation corresponds to early S-phase onset in Cdh1-depleted cells. (A) Immunoblots for indicated proteins in HeLa cell lysates (30 μg of protein) after indicated siRNA transfection for live-cell imaging. (B) Average G1 duration times for GL2, Cdh1, Cdh1/cyclin A2, Cdh1/cyclin B1, and Cdh1/cyclin E1 siRNA–transfected cells (N = 3; nGL2 = 92, nCdh1 = 217, nCdh1/CycA2 = 165, nCdh1/CycB1 = 185, nCdh1/CycE1 = 109 cells). Error bars represent 95% CI. (C) Cell cycle distribution of cells in A was determined by EdU labeling, PI staining, and flow cytometry. Data are plotted as means ± SEM (N = 3).
APCCdh1/SCFSkp2/p27 functions in control of S-phase entry timing
qRT-PCR results reflected increased activation of the Rb/E2F1 pathway that corresponded to early S-phase onset, and this presumably elevated cyclin E1 protein levels in Cdh1-ablated cells. Because cyclin E1 transcription is initiated by the action of CDK4/CDK6/cyclin D in the Rb/E2F1 pathway, and cyclin D proteolysis coincides with S-phase entry (Alao, 2007; Kanie et al., 2011), we were curious about whether Cdh1 depletion would prematurely reduce cyclin D abundance. Indeed, immunoblotting revealed a decrease in cyclin D1 protein as cells progressed from G1 into early S phase after Cdh1 depletion, possibly due to early destruction of cyclin D1 (Figure 4, A and B). This raised a question: how does this reduction in cyclin D1 connect to the removal of Cdh1? Earlier studies showed that Cdh1 knockdown stabilized the F-box protein Skp2 and led to an attenuated accumulation of one of the SCFSkp2 substrates, p27 (Bashir et al., 2004; Wei et al., 2004); our findings were consistent with this prior report (Figure 4, A and B). p27 is a well-studied CDK2 inhibitor, and its ubiquitylation and degradation during late G1 phase requires CDK2/cyclin E phosphorylation of Thr-187 (Carrano et al., 1999). This topology generates a potential double-negative (positive) feedback loop that had been proposed earlier by computational biologists to contribute to the biochemical switch controlling the G1/S transition in mammalian cells: first, by p27 inhibiting CDK2/cyclin E activation, and second, with the subsequent proteolysis of p27 by SCF through CDK2/cyclin E activation and phosphorylation of p27 (Figure 4C; Thron, 1999; Qu et al., 2003). We hypothesized that not merely did loss of p27 in Cdh1-ablated cells prevent the inhibition of CDK2/cyclin E, but that less of this inhibitor might also alter how CDK2 responds to cyclin E stimulus at the G1/S boundary. Therefore we first tested the hypothesis that increasing p27 expression in Cdh1-depleted cells would prolong G1 phase. Indeed, ectopic expression of p27 in asynchronous Cdh1-ablated cells recovered ∼33% of G1 phase compared with control cells (p < 0.001; Figure 4, D and E) and enriched the G1 population (Figure 4F and Supplemental Figure S5A). This increase in G1-phase cells relative to the control population occurred when pECFP-p27 was expressed alone, as well as in cells ablated for Cdh1 (Supplemental Figure S5, B and C). Together these data corroborated that inhibition of CDK2 in Cdh1-depleted cells is less effective due to lower p27 levels. Corresponding to a reduction in p27, cyclin D1 abundance was also diminished, suggesting that accelerated cyclin E1 expression and subsequent S-phase entry promoted premature cyclin D1 proteolysis. In summary, the activity of Cdh1 is clearly essential to prevent premature accumulation of Skp2 and its early activation of SCFSkp2, permitting p27 accumulation and preventing early CDK2/cyclin E1 activation.
FIGURE 4:

Overexpressing p27 recovers G1-phase duration in Cdh1-depleted cells. (A) Immunoblots for indicated proteins in HeLa cell lysates (35 μg of protein) prepared at indicated time points after GL2 and Cdh1 siRNA transfection with triple-thymidine block and release. (B) Quantitation of cyclin D1, p27, and Skp2 proteins was done using ImageJ, and values are normalized to tubulin and plotted as means ± SD (N = 3). (C) Schematic of positive-feedback motifs in CDK2/cyclin E activation. These include a double-negative-feedback loop of CDK2/cyclin E inhibiting its inhibitor p27, and a positive-feedback loop of CDK2/cyclin E promoting cyclin E transcription. (D) Immunoblots for Cdh1, cyclin E1, p27, Skp2, and tubulin in HeLa cell lysates (30 μg of protein) after siRNA transfection with or without pECFP-p27 in cells before and after imaging. Double asterisks indicate endogenous p27; single asterisk indicates ectopic pECFP-p27. (E) Average G1 duration times for GL2 and Cdh1 siRNA– and Cdh1 siRNA + pECFP-p27–transfected HeLa cells (N = 3; nGL2 = 302, nCdh1 = 394, nCdh1/p27 = 230 cells). Error bars represent 95% CI. (F) Cell cycle distribution of cells in B was determined by EdU labeling, PI staining, and flow cytometry. Data are plotted as means ± SEM (N = 3).
Reduction of p27 in Cdh1-depleted cells lessens cyclin E1 threshold for S-phase entry
Although increased levels of cyclin E1 corresponded to premature S-phase entry in cells lacking Cdh1, it was unknown whether the latter was a cause or an effect of the former. To test whether Cdh1 knockdown altered the activation of CDK2—potentially affecting how it switches—measurement of CDK2 activity over short time intervals would be required. Unfortunately, attempts at showing precise changes in the rate of CDK2 activation in cell populations ablated for Cdh1 were unsuccessful. This was attributed in part to the fact that the synchrony of cell populations was lost as a result of changes in cell cycle timing due to Cdh1 knockdown. This made measuring CDK2 activity on a bulk level over small changes in time unfeasible. As surrogates to direct CDK2 activity assays, we implemented two different approaches. First, we repeated the Cdh1 ablation in thymidine-synchronized cell populations, and after their release, probed lysates for a CDK2-specific phosphorylation of Rb (pT821; Zarkowska and Mittnacht, 1997). Indeed, Rb phosphorylation at this site was prematurely elevated in cells lacking Cdh1 (Supplemental Figure S6). We then measured differences in the levels of cyclin E1 protein before and during S phase at the single-cell level. If CDK2 activation were altered, its requirement for cyclin E1 at the G1/S transition, as well as the removal of cyclin E1 during S phase, would serve as an indirect but sensitive readout of these changes. HeLa cells were synchronized, transfected with GL2 and Cdh1 siRNAs, and released (Figure 5A). At 18 h postrelease, samples were collected at 2-h intervals, fixed, probed for DNA and cyclin E1 contents, and then analyzed by flow cytometry. GL2-transfected cells remained predominantly in G1 phase from 18 through 22 h postrelease, and by 24 h, the initial entry of cells into S phase coincided with elevated cyclin E1 protein (Figure 5, A and B, GL2 siRNA). Cdh1-ablated cells, however, exhibited some notable differences when compared with their control counterparts (Figure 5, A and B, Cdh1 siRNA). By comparing the relative abundance of cyclin E1 in gated G1 cells just before their disappearance into S phase—with the GL2 siRNA-transfected population trailing roughly 4 h behind the accelerated Cdh1-ablated population (Figure 5B; compare GL2: 24 h to Cdh1: 20 h)—we discovered that G1 cells lacking Cdh1 contained less cyclin E1 on average (Figure 5C). This revealed that Cdh1 knockdown did not increase but actually reduced the content of cyclin E1 in cells before S-phase entry, presumably corresponding to a lowered threshold for CDK2 activation. Not only did Cdh1 removal change how S-phase entry was stimulated, but it also markedly altered cyclin E1 removal during S-phase progression. At 28 h, a snapshot of cyclin E1 abundance over the entire S-phase control-cell population revealed its plateau in the first 10–20% of S phase, followed by its relatively uniform reduction as cells progressed through its later stages (Figure 5, B and D, GL2 siRNA). Cdh1 knockdown, however, appeared to cause two major differences (Figure 5, B and D, Cdh1 siRNA). First, cyclin E1 protein accumulated in these cells up to the first 30–40% of S phase, suggesting that translation of cyclin E1 continued for ∼3–4 h after S-phase entry. This is consistent with an increasing trend in cyclin E1 transcription that occurred up to 24 h during the time course in Cdh1-ablated cell populations (Figure 2D). Second, after cyclin E1 levels plateaued, they dropped slowly during the final half of S phase (Figure 5D); this also occurred concomitant with a decreasing trend of cyclin E1 mRNA. Of interest, although cyclin E1 protein abundance throughout S phase was different between the control and Cdh1-ablated cells, the trend in changes in its mRNA levels between these two treatments was similar (Figure 2D). Whereas differences in cyclin E1 abundance were apparent before and during S phase in Cdh1-ablated cells, there were no marked differences in cyclin A2 content (Supplemental Figure S7A), and cyclin B1 accumulation was more substantial during mid to late S phase (Supplemental Figure S7B).
FIGURE 5:

Cdh1 depletion alters the pattern of cyclin E accumulation in HeLa cells progressing from G1 to S phase. (A) Immunoblots for indicated proteins in HeLa cell lysates (35 μg of protein) prepared at time points after GL2 and Cdh1 siRNA transfection of HeLa cells with triple-thymidine block and release. (B) Representative zebra plots of cyclin E1 protein abundance vs. DNA content in cells transfected with siRNAs directed against GL2 or Cdh1, corresponding to time points in A. (C) Probability densities of G1-phase cyclin E1 abundance in GL2 and Cdh1 siRNA–transfected cells shown in B at 24 and 20 h, respectively. (D) Mean cyclin E1 protein content in 154,751 GL2 and 282,860 Cdh1 siRNA–transfected HeLa cells as they progress through successive intervals of S phase (by increasing bins of DNA content), plotted as means ± SEM (N = 3; some data points mask error bars).
The timing of S-phase entry is dictated at least in part by the CDK2 inhibitor p27, and this also raised the question of whether a reduction in its levels due to the absence of Cdh1 is a direct contributing factor: does loss of p27 in Cdh1-ablated cells reduce the potential for positive feedback that underlies CDK2/cyclin E1 activation? We found that Cdh1 depletion could, in theory, reduce the strength of this feedback loop indirectly by up-regulating Skp2 and decreasing p27 levels. Thus we hypothesized that lowering p27 levels directly would also decrease the cyclin E1 abundance needed for CDK2 activation, causing it to respond linearly to cyclin E1 and subsequently accelerate S-phase entry. To test this hypothesis, we transfected HeLa cells with siRNAs directed toward GL2, Cdh1, or p27 and then thymidine synchronized and released them (Figure 6A). Cells were probed for p27 and cyclin E1 and gated (Supplemental Figure S8A), and then DNA contents were plotted for cells staining p27+/cyclin E1+ and p27−/cyclin E1+ (Figure 6B). The GL2-transfected population gained p27+/cyclin E1+ cells through 24 h postrelease (Figure 6B, top, black trace), and p27-/cyclin E1+ cells then began to transit into S phase (Figure 6B, top, shaded gray). Like the Cdh1-ablated cells, p27 knockdown strongly reduced the number of p27+ cells, and cyclin E1+ cells entered S phase ∼4 h earlier (Figure 6B, middle and bottom). Both Cdh1 and p27 ablation permitted S-phase entry with diminished cyclin E1 (Figure 6, C and D), but cyclin E1 abundance in the latter eventually matched that of the control during S-phase progression (Figure 6, C and E). There had been no demonstration of a “hard” cyclin E1 threshold for S phase in mammalian cells before this study, but a bivariate plot of p27 versus cyclin E1 revealed how important p27 is to accumulate proper levels of cyclin E1 during G1 phase and before S-phase entry (Supplemental Figure S8). GL2 siRNA–transfected cells showed three pronounced populations of cells with distinct p27 and cyclin E1 contents as they transited through G1: p27+/cycE1−, p27+/cycE1+, and p27−/cycE1+ (Supplemental Figure S8B and inset). As described earlier, once p27−/cycE1+ cells accumulate, they remain in G1 phase for a period before S-phase entry. However, once Cdh1- and p27-ablated cells gain cyclin E1, they immediately pass from G1 into S phase (Figure 6B and Supplemental Figure S8B). These data demonstrate not only that APC-Cdh1 activity is necessary to provide the proper gating for S-phase entry through stabilizing p27, but also that this stabilization of p27 ensures that cyclin E1 stimulus is generated properly.
FIGURE 6:

Partial knockdown of p27 mirrors the acceleration of S-phase onset caused by Cdh1 loss. (A) Immunoblots for indicated proteins in HeLa cell lysates (30 μg of protein) prepared at time points after GL2, Cdh1, and p27 siRNA transfection of HeLa cells with triple-thymidine block and release. (B) DNA histograms of cell populations in A that have been gated for the following p27 and cyclin E1 contents: p27+/cycE1+ (black trace) and p27−/cycE+ (shaded gray). Inset, histograms indicating cell cycle distribution of all 30,000 cells that were analyzed at each time point. (C) Representative zebra plots of cyclin E1 protein abundance vs. DNA content in cells transfected with siRNAs directed against GL2, Cdh1, or p27, corresponding to time points in A. (D) Probability densities of G1-phase cyclin E1 abundance in GL2 and Cdh1 siRNA– and p27 siRNA–transfected cells shown in C at 26 and 22 h, respectively. (E) Mean cyclin E1 protein content in GL2 and Cdh1 siRNA–transfected HeLa cells as they progress through successive intervals of S phase (by increasing bins of DNA content), plotted as means ± SEM (N = 3; some data points mask error bars).
Together these data reveal for the first time that the S-phase requirement for cyclin E1 protein changes in Cdh1-ablated cells, as well as the effectiveness of its removal during S phase. This is the first demonstration that APC-Cdh1 function is essential to establish the proper relationship between cyclin E1 and S-phase entry and that its stabilization of p27 is essential for this switching mechanism to operate properly. It is therefore change in how this switch responds to cyclin E1 that drives the accelerated S-phase entry in Cdh1-ablated cells, and not a rapid increase in cyclin E1 synthesis per se or the premature synthesis of any other G1-phase or M-phase-promoting cyclin.
Cdh1 ablation prolongs S phase
Our analysis of cells lacking Cdh1 revealed that less cyclin E1 could stimulate S-phase entry and that its turnover was delayed during S-phase progression. This implied that a change occurred in the response of its cognate kinase—CDK2—apparently reducing its threshold and possibly causing it to turn on more gradually. This type of alteration in CDK2 activation might increase the window of time when CDK2 activity sits at an intermediate level, causing premature DNA replication, while potentially leading to defects in S-phase onset and progression. In comparison, past studies showed that cyclin E1 overexpression also accelerated S-phase entry (Resnitzky et al., 1994; Keck et al., 2007). However, because cyclin E1 overexpression alone should not impede the function of any positive-feedback loops that drive CDK2 activation and the G1/S switch, we hypothesized that it might only cause relatively modest effects on S-phase dynamics: CDK2 activation may occur earlier due to an increase in stimulus, but it should not perturb S-phase progression as drastically as cells lacking Cdh1.
To test this hypothesis, we measured the duration of S phase by in vivo labeling of replication factories using a PCNA-YFP expression construct. To test whether removal of Cdh1 changes how cells respond to either normal or overexpressed levels of cyclin E, we cotransfected a pECFP-cyclin E1 overexpression construct into HeLa cells along with either GL2 or Cdh1 siRNA (Figure 7A). Consistent with earlier findings, either Cdh1 depletion or cyclin E1 overexpression alone shortened the duration of G1 phase by ∼25% (p < 0.001; Figure 7B). Overexpressing cyclin E1 in Cdh1-depleted cells further shortened G1 phase, demonstrating that more of this CDK2 stimulus could further accelerate S-phase entry (Figure 7B). Next we set out to determine how the duration of S phase might be affected due to these perturbations. Earlier separate studies using flow cytometry showed that either knocking down Cdh1 or overexpressing cyclin E lengthened the period of DNA replication, due to an enrichment of S-phase cells. In our analysis, compared with a 50-min-prolonged S phase (8% longer) in cells overexpressing cyclin E1 alone (p < 0.05), Cdh1-depleted cells took ∼20% longer (130 min) than their GL2 siRNA–treated counterparts (p < 0.001), and overexpressing cyclin E1 in Cdh1-ablated cells did not further prolong S phase (Figure 7C). Together these findings led us to hypothesize that Cdh1 is essential to build the dynamics necessary to confer to cells a switch-like and complete transition into, and proper progression through, S phase in response to cyclin E1.
FIGURE 7:
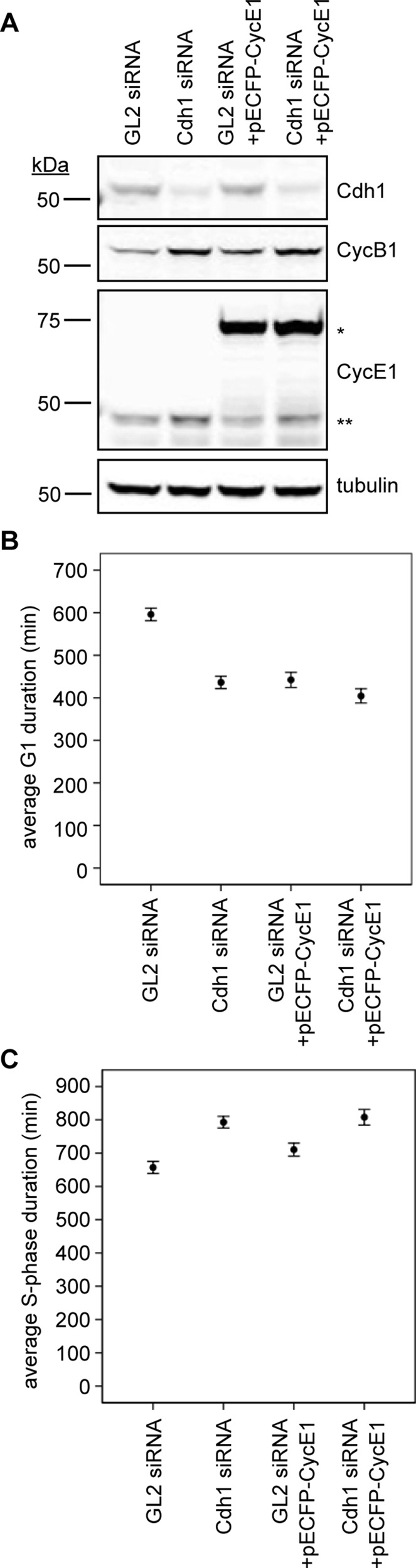
Cdh1 ablation accelerates S-phase entry onset and prolongs its duration. (A) Immunoblots for indicated proteins in HeLa cell lysates (30 μg of protein) after siRNA transfection with or without a pECFP-cyclin E1. Single asterisk indicates ectopic pECFP-cyclin E1; double asterisks indicate endogenous cyclin E1. (B) Plot of average G1 duration times for GL2 and Cdh1 siRNA–, GL2 siRNA + pECFP-cyclin E1–, and Cdh1 siRNA + pECFP-cyclin E1–transfected cells (N = 3; nGL2 = nCdh1 = nGL2/CycE1 = nCdh1/CycE1 = 250 cells). (C) Average S-phase durations for GL2 and Cdh1 siRNA–, GL2 siRNA + pECFP-cyclin E1–, and Cdh1 siRNA + pECFP-cyclin E1–transfected cells (N = 3; nGL2 = 77, nCdh1 = 94, nGL2/CycE1 = 84, nCdh1/CycE1 = 72 cells). Error bars represent 95% CI.
Cdh1 knockdown reduces rate of replication fork movement and frequency of replication terminations
Cdh1 loss induces a premature and prolonged S phase, corresponding to a lowered requirement for cyclin E1 at the G1/S transition and an increased expression of cyclin E1 during S-phase progression. To determine how replication dynamics is affected by Cdh1 knockdown, we applied a DNA fiber-spreading method (Chastain et al., 2006) to asynchronous HeLa cells treated with either Cdh1 or GL2 siRNA (Figure 8A). Images of labeled DNA fibers were analyzed using the Computer Aided Scoring and Analysis (CASA) software package (Yaping et al., 2011), and the average rate of replication fork elongation was determined, as well as the number of active replication forks and the frequencies of replication origins and termination events. We found that Cdh1 depletion reduced the fork progression rate by ∼20% compared with control cells (p = 0.004; Figure 8, B and C), accounting for the prolonged S-phase duration described earlier (Figure 7C). Of interest, there was no decrease in the number of replication forks in the former (p = 0.002; Figure 8D), and the frequency of replication origins was not significantly different from the control (p = 0.517; Figure 8, B and E). Surprisingly, there were >2.5-fold fewer termination events (p = 0.005; Figure 8, B and F). The fact that the average number of replication origins that fired was not diminished in the Cdh1-ablated cells indicated that they did not have a diminished capacity to initiate DNA replication (i.e., these cells apparently had no fewer origins licensed than control cells). Despite the fiber analyses having been performed in asynchronous cells, it is plausible that the shortened G1 in Cdh1-depleted cells leads to their enrichment during a slowed S-phase progression and insufficient time for additional terminations to be observed in our assays. Taken together, the reduction in replication elongation rates and termination frequency may have resulted from defective replisome function and/or stalled replication forks and could be a contributing factor to genome instability in the absence of Cdh1. This will be examined in future studies.
FIGURE 8:

Cdh1 loss slows the rate of replication fork elongation and decreases the frequency of termination events. (A) Immunoblots for indicated proteins in HeLa cell lysates (30 μg of protein) after siRNA transfection. (B) DNA fibers pulse labeled with IdU (red) and CldU (green) from cells transfected with indicated siRNAs. (C) Histogram of average fork elongation rate in GL2 and Cdh1 siRNA–transfected cells. Data are plotted as means ± SEM (N = 3, nGL2 = 143, nCdh1 = 170). (D) Histogram of the number of replication forks per megabase of DNA in GL2 and Cdh1 siRNA–transfected cells. Means of the two treatments from triplicate experiments were summed, and individual values from each experiment were normalized. Adjusted data are pooled and plotted as means ± SEM (N = 3, nGL2 = 26, nCdh1 = 32). (E) Histogram of frequencies of replication origins detected in GL2 and Cdh1 siRNA–transfected cells (N = 3, nGL2 = 481, nCdh1 = 677). (F) Histogram of frequencies of replication termination events detected in GL2 and Cdh1 siRNA–transfected cells. Data are plotted as means ± SEM (N = 3, nGL2 = 481, nCdh1 = 677).
It was possible that the 20% reduction in replication rate occurred due to activation of DNA damage response pathways, so we assayed Cdh1- and p27-depleted cells for detectable changes in three DNA damage markers: p-Chk1(S345), p-Chk2(T68), and p-H2AX(S139) (Supplemental Figure S9). Treatment of asynchronous HeLa cells with either hydroxyurea (HU; from 0.5–5 mM) or aphidicolin (10–50 μM) for 2 h caused easily detectable increases in all three markers (Supplemental Figure S9A). Whereas treatment of cells with HU or aphidicolin increased these signals, elimination of Cdh1 from an asynchronous HeLa cell population did not produce any detectable increase, either at 2 or 5 d posttransfection (Supplemental Figure S9, B and C). Ablating either Cdh1 or p27 from a second cell type—osteosarcoma cells (U2OS)—mirrored these results, and neither immunoblotting/flow cytometry probing for various DNA damage markers (Supplemental Figure S10) nor immunofluorescence for p-H2AX(S139) (Supplemental Figure S11) revealed any substantial DNA damage response. Hence, the reduction in the rate of fork movement in Cdh1-depleted cells occurs due to factors other than DNA-damage-induced, Chk-mediated signaling or a response to double-stranded DNA breaks.
In summary, our findings support that S-phase onset can occur without the replication machinery being incapacitated due to a lack of replication initiation factors. However, slowed fork movement and a precipitous drop in terminations implied an incidence of defects in replication that could be a result of changes in the dynamics of CDK2 activation at the G1/S boundary and possibly in its activity during S phase. Given the results, we propose that a positive-feedback-driven switch into S phase mediated by p27 and CDK2/cyclin E1 could be key to driving the firing of replication origins and properly sustaining S-phase progression. It is through APC-Cdh1 function that this p27-mediated feedback loop would be generated.
DISCUSSION
In this study, we sought to determine how an M-phase-initiated negative-feedback loop mediated by APC-Cdh1 sets the biochemical stage for S-phase entry. We found that Cdh1 knockdown increased cyclin E1 protein abundance in a population of cells as they progressed prematurely into S phase. Removal of cyclin E1, but neither cyclin A nor B, consequently prolonged the shortened G1 phase in Cdh1-depleted cells. This provided evidence that cyclin E1 and its cognate kinase, CDK2, stimulate precocious S-phase initiation in HeLa cells devoid of Cdh1.
CDK2/cyclin E is the central regulator of the G1/S transition, where its substrates are involved in transcriptional control and the initiation of DNA replication. The Rb/E2F1 pathway regulates expression of many S-phase genes and is sequentially activated, first through phosphorylation by CDK4/6/cyclin D and then by CDK2/cyclin E (Harbour et al., 1999; Harbour and Dean, 2000). Cyclin E transcription is first stimulated by E2F1 and increases CDK2 activity, initiating a positive-feedback loop that further amplifies its expression (Ohtani et al., 1995). In Cdh1-depleted cell populations, increased cyclin E1 protein corresponded to increase in cyclin E1 mRNA, but, surprisingly, they had less cyclin E1 than control cells as they transited into S phase. This was the first evidence that Cdh1 loss did not induce premature S-phase entry by overexpressing cyclin E1 or others but was due to changes in their response to cyclin E1.
Given that cyclin E1 levels in Cdh1-ablated cells were lessened at G1/S, this raised a question: what drives premature S-phase entry? We hypothesized that reduced CKI levels weakened the inhibitory leg of the S-phase-promoting positive-feedback loop, decreasing the nonlinearity of its response. One potential source of double-negative feedback in S-phase entry mirrors the topology of the CDK1 switch at mitotic entry, where it is linked to inhibitors, Wee1 and Myt1 (Pomerening et al., 2005, 2008). A similar scheme potentially generates positive feedback in CDK2 activation and involves at least one CKI, p27 (Figure 9A). p27 is a CDK inhibitor of the Cip/Kip family, and it peaks during early G1 phase (Bloom and Pagano, 2003; Chu et al., 2008). Nuclear export and Kip1 ubiquitylation-promoting complex–mediated degradation slowly remove p27 and permit basal activation of CDK2/cyclin E during early G1 (Bloom and Pagano, 2003; Chu et al., 2008). Basal turnover of p27 is also mediated by Skp2-dependent/Thr-187 phosphorylation–independent degradation, followed by its proteolysis at G1/S due to CDK2 phosphorylation and SCFSkp2-mediated ubiquitylation (Figure 9A, left) (Malek et al., 2001; Chu et al., 2008). Thus p27 loss may permit early CDK2 activation with less cyclin E1, and this could hinder accumulation of sufficient cyclin E1/inactive CDK2 needed to properly drive S phase (Figure 9A, right). Indeed, Cdh1 depletion reduced p27 coincident with elevated Skp2, and less cyclin E1 drove premature S-phase entry. Based on the use of cyclin E1 protein abundance as a CDK2 activity surrogate, its increase and plateau after S-phase initiation differed substantially from those in control cells, where cyclin E1 diminished upon S-phase entry. This cyclin E1 accumulation coincided with continued cyclin E1 transcription, suggesting that inactivation of its transcription, and possibly its proteolysis, were not stimulated properly in Cdh1-null cells.
FIGURE 9:

p27 and CDK2 generate a double-negative (positive) feedback loop indirectly as a result of APC-Cdh1 activity. (A) Wiring diagram of the double-negative (positive) feedback loop between p27 and CDK2/cyclin E (left) and its weakening by Cdh1 loss (right). Dotted line extending from active CDK2 depicts phosphorylation of inactive p27/CDK2; lightened species indicate their reduction (right). (B–D) Effect of Skp2 variation; increase in Skp2 represents decrease in APC-Cdh1 activity. (B) Temporal response of p27 abundance from an ODE model represented in A as Skp2 is increased from 1 (black), to 1.5 (red), or to 3 (blue). (C) Temporal responses of cyclin E1 (cycE) abundance (solid) and CDK2 activity (dotted) as Skp2 is increased. (D) Steady-state responses of CDK2/cyclin E as Skp2 is increased. (E–G) Effect of p27 knockdown, represented by decrease in rate of p27 synthesis (ksyn_p27), whose behavior is similar to that of Skp2 increase.
How might CDK2 activation vary between cells with or without Cdh1? We explored this by constructing a phenomenological ODE model of a minimal CDK2/cyclin E switch (Figure 9, B–D). It includes simple chemical kinetic terms for generating p27 and cyclin E, as well as double-negative (positive) feedback between active CDK2 and the inactive, CDK2-bound p27 that culminates in p27 destruction (Figure 9A; see the Supplementary Material [including Supplemental Tables S1 and S2] for model). Two features of this model depend on p27 input and the positive feedback it generates. First, its accumulation produces a delayed and steepened switch-like temporal activation of CDK2 that could be anticipated at G1/S (Figure 9C; dotted black). Second, its generation of positive feedback is evidenced by bistability in CDK2 activation at steady state: the thresholds to switching CDK2 on and off are far apart, making this system robust (Figure 9D; black). Our model also revealed that its dynamics could change due to Cdh1 depletion by altering the parameter Skp2: increasing Skp2 reflected a progressive loss of Cdh1. As Skp2 levels were increased, numerous features supported our experimental findings, including p27 loss (Figure 9B; red and blue). First, CDK2 activated early, suggesting a premature S phase; second, decreased steepness in CDK2 activation occurred, indicating that cells would possess intermediate CDK2 activity for a prolonged period (Figure 9C; dotted black, red, and blue). Finally, steady-state analysis showed a loss of positive feedback and nonlinearity (hysteresis curve) as Skp2 levels increased (Figure 9D), along with CDK2 activation—and possibly the G1/S transition itself—occurring at lower cyclin E1 levels (Figure 9, C and D; solid black, red, and blue). In all, this simple model simulates Cdh1 knockdown qualitatively and suggests that relaxing the positive-feedback-driven switch between active CDK2 and CDK2-bound p27 would cause an early but less abrupt increase in CDK2 activity at G1/S. In addition, p27 ablation was represented in the model by reducing its synthesis rate, ksyn_p27. On this change, a phenotype similar to Cdh1 knockdown occurred (Figure 9, E–G). Like our experimental results, either Cdh1 or p27 ablation caused early CDK2 activation at reduced cyclin E1 levels, suggesting that their dynamics could be similar. Our model underscores the critical role of Cdh1 in preserving this positive feedback by stabilizing p27, and in turn, governing CDK2 activation at S-phase onset.
Extending from past studies involving loss of Cdh1 function, we determined that this perturbation changes the dynamics of S-phase promotion. First, less cyclin E1 stimulated S-phase entry; second, dramatic changes occurred in its accumulation and loss as cells progressed through a prolonged S phase. Changes in CDK2 activation at the G1/S transition could pose risks to DNA replication fidelity, but it was unclear how the dynamics of replication might be affected and lead to error commitment. Alterations in CDK activity can affect the number of replication factories and replication rates (Thomson et al., 2010). Using DNA fiber analysis, we indeed found that Cdh1 depletion slowed replication fork progression and reduced the frequency of termination events. This demonstrated that loss of Cdh1—and potentially deregulation of CDK2 activation—could lead to inefficient phosphorylation of downstream targets and impaired fork progression. Slow-moving or stalled replication forks are often associated with double-stranded DNA breaks and a DNA damage response. These latter defects were detected previously in Cdh1-depleted cells and could interfere with genome integrity (Engelbert et al., 2007; Garci-Higuera et al., 2008; Sigl et al., 2009). However, we found no detectable damage checkpoint responses in Cdh1-ablated HeLa or U2OS cells. Delayed and/or fewer terminations might increase the incidence of these defects and, combined with intermediate CDK2 activities, could lead to genomic instability by inefficient inhibition of rereplication. Future work will be directed at understanding how altering termination events affects DNA replication and genome stability and determining whether subtle reductions in APC-Cdh1 activity promote transformation.
In summary, Cdh1 is a critical part of the pathway that regulates CDK2 at the G1/S transition. By maintaining levels of p27, and possibly other CDK inhibitors, it appears to contribute to generating a positive-feedback-driven CDK2 switch to S-phase entry. Our data revealed that Cdh1 depletion impaired this transition and led to prolonged S-phase progression. Together these findings provide new insights into the systems-level role of Cdh1 in its regulation of cell cycle progression and function as a tumor suppressor.
Materials and Methods
siRNAs
Human siRNA validated oligos targeting Cdh1 (sense 5′-AACGAUGUGUCUCCCUACUCC-3′) and Cdc20 (sense 5′-GGUUCAGACCACUCCUAGCAAAC-3′) were purchased from Dharmacon (Lafayette, CO). Luciferase GL2 control siRNA, human cyclin A2, B1, and E1, p27, and CDK2 ON-TARGET plus SMARTpool siRNAs were purchased from Dharmacon. Transfections of siRNA oligos were performed as described previously using Oligofectamine (Invitrogen, Carlsbad, CA) following the manufacturer's instructions (Ma et al., 2012).
Constructs
Human cyclin A2, cyclin E1, Cdh1, and p27 coding sequences were amplified from HeLa cell cDNA by PCR using the following primers: cyclin A2, sense 5′-CGAGAATTCGATGTTGGGCAACTCTGC-3′ (EcoRI), antisense 5′-GCAGGATCCCAGATTTAGTGTCTCTGGTG-3′ (BamHI); cyclin E2, sense 5′-GCACTCGAGATATGCCGAGGGAGCGCAG-3′ (XhoI), antisense 5′-CATACCCGGGATCGCCATTTCCGGCCCG-3′ (SmaI); Cdh1, sense 5′-GCAG AGCTCAGATGGACCAGGACTATGAGCG-3′ (SacI), antisense 5′-CACCCGGGTACCGG ATCCTGGTGAAGAG-3′ (SmaI); p27, sense 5′-CGAGAATTCGATGTCAAACGTGCGAGT G-3′ (RI), antisense 5′-GCAGGATCCCGTTTGACGTCTTCTG-3′ (BamHI). The PCR product was then digested and cloned into a cytomegalovirus-driven mammalian expression vector to produce an N-terminal fusion to an enhanced cyan fluorescent protein (ECFP). The Cdh1 rescue plasmid was constructed by introducing six silent mutations into the binding region of the Cdh1 siRNA, using the following primers: sense, 5′-CGATGACGGCAACGACGTCTCGCCGTAC AGCCTGTCTCCCGTC-3′, antisense 5′-GGGCTGGAGCGCTTGGTGCTAAGGGAATACG-3′. Production of the PCNA-YFP construct was described previously (Hahn et al., 2009).
Cell culture and transfection
HeLa cells and stable HeLa cell lines expressing mCherry-histone-H2B (kindly provided by Claire Walczak, Indiana University, Bloomington, IN) were cultured in complete DMEM (Mediatech, Manassas, VA), 10% (vol/vol) fetal bovine serum (Mediatech), and 1% (vol/vol) penicillin-streptomycin-glutamine (Mediatech) in a humidified 37°C environment with 5% CO2. For siRNA transfections, 2 × 105 asynchronous HeLa cells were plated into six-well plates and blocked with 2.5 mM thymidine (Sigma-Aldrich, S. Louis, MO) for 18 h. After release, cells were transfected with siRNA oligos against luciferase GL2, Cdh1 alone, or Cdh1 along with siRNAs directed against cyclin A2, B2, or E1 using Oligofectamine. Lipofectamine LTX with PLUS reagent (Invitrogen) was used for plasmid transfection. For overexpressing p27 or cyclin E1 in Cdh1-depleted cells, transfection of plasmids (200 ng/well) was performed in six-well plates after 24 h of siRNA transfection. For live-cell imaging, cells were then replated into 96-well imaging plates and 24-well plates. PCNA-YFP biosensor (17 ng/well) was transfected into cells in imaging plates 45 h after siRNA transfection, and 8 h later, live-cell imaging was performed for 17 h. For triple-thymidine block and release, 1.5 × 105 asynchronous HeLa cells were plated into six-well plates and blocked with 2.5 mM thymidine (Sigma-Aldrich) for 18 h. After release, cells were transfected with siRNA oligos and blocked with 2.5 mM thymidine an additional two times for 18 h with 9-h release intervals. Cell samples were collected every 2 h from 18 to 28 h after the third thymidine release and then processed for immunoblot, flow cytometry, and qRT-PCR.
Antibodies
Antibodies specific to Cdh1 (DH01), Aurora A, and Plk1 were purchased from Abcam (Cambridge, MA). Antibodies specific to cyclin A2 (sc-751), cyclin (sc-752), cyclin D (sc-753), cyclin E (sc-247), p27 (sc-528), and Cdc20 (sc-8358) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies specific to α-tubulin (DM1α) were purchased from Sigma-Aldrich. The Skp2 antibody (32-3300), Alexa Fluor 488 anti-mouse antibody (A21202), Alexa Fluor 488 anti-rabbit antibody (A21206), Alexa Fluor 647 anti-mouse antibody (A31571), Alexa Fluor 750 anti-rabbit antibody (A21039), Alex Fluor 680 anti-mouse antibody (A21057), Alexa Fluor 594 rabbit anti-mouse antibody (A11062), Alexa Fluor 488 chicken anti-rat antibody (A21470), Alexa Fluor 594 goat anti-rabbit antibody (A11037), and Alexa Fluor 594 goat anti-chicken (A11039) were purchased from Invitrogen. IRDye 800CW anti-mouse antibody (926-32210) and IRDye 680LT anti-rabbit antibody (926-68021) were purchased from LI-COR Biosciences (Lincoln, NE). Mouse anti-BrdU antibody (347580) was purchased from Becton Dickinson (San Jose, CA). Rat anti-BrdU antibody (OBT0030) was purchased from the Accurate Chemical & Scientific Corporation (Westbury, NY).
Immunoblotting
Total cell lysates were prepared using M-PER mammalian protein extraction reagent (Pierce, Rockford, IL), and protein concentration was determined by bicinchoninic acid assay. Thirty micrograms of protein was separated by 10% or 15% SDS–PAGE gel and transferred to a polyvinylidene fluoride membrane (Immobilon-FL; Millipore, Billerica, MA) using a wet electrophoretic blotting system (Bio-Rad, Hercules, CA). Blots were blocked with Odyssey Blocking Buffer (LI-COR) at room temperature for 1 h and probed with antibody against Cdh1 (0.67 μg/ml) and cyclin A (0.2 μg/ml) diluted in antibody-diluting buffer (ADB; 2% bovine serum albumin [BSA; wt/vol] and 0.1% NaN3 [wt/vol] in Tris-buffered saline and Tween 20 [TBST]). Membranes were then rinsed with TBST three times for 5 min and probed with IRDye 800CW–conjugated anti-mouse secondary antibody (0.2 μg/ml) and IRDye 680LT–conjugated anti-rabbit secondary antibody (0.2 μg/ml) for 1 h at room temperature. After rinsing with TBST, the blots were scanned using the Odyssey infrared imaging system (LI-COR), and the bands were quantified using ImageJ software (National Institutes of Health, Bethesda, MD). For reprobing, the membrane was stripped in stripping buffer (2.5 mM glycine, pH 2.0, 1% SDS) for 30–60 min at room temperature, rinsed twice with TBST for 10 min, and probed for other proteins. Final concentrations of antibodies used were as follows: anti-Aurora A (1 μg/ml), anti-Plk1 (1 μg/ml), anti-cyclin (0.2 μg/ml), anti-cyclin D (0.4 μg/ml), anti-cyclin E (0.2 μg/ml), anti-p27 (0.4 μg/ml), anti-Cdc20 (0.2 μg/ml), anti–α-tubulin (1:5000), anti-Skp2 (0.5 μg/ml), Alexa Fluor 750 anti-rabbit (0.2 μg/ml), and Alexa Fluor 680 anti-mouse (0.2 μg/ml).
Imaging
Live-cell microscopy was performed using a BD pathway 855 High-Content Bioimager system (BD Biosciences, Franklin Lakes, NJ) with a 20× objective (numerical aperture [NA], 0.75) at a humidified 37°C with 5% CO2. Images were taken using Attovision software (BD Biosciences) and analyzed using ImageJ as described previously (Ma et al., 2012). Duration of G1 phase was determined by counting frames between chromosome segregation and the appearance of PCNA-YFP punctum imaged by fluorescence microscopy. Confocal microscopy was performed during DNA fiber analysis on a Nikon A1 scanning confocal microscope (Nikon, Tokyo, Japan) equipped with 561- and 633-nm lasers. Images were taken acquired by 60× oil-immersion objective (NA 1.2) and an Orca-Flash 4.0 sCMOS camera (Hamamatsu, Hamamatsu, Japan) controlled by Nikon Element software. Fixed-cell microscopy for immunofluorescence was performed using a Leica DM5500B (Leica, Wetzlar, Germany) with a 20× objective and a Retiga Exi FAST1384 charge-coupled device camera (Qimaging, Surrey, Canada). Camera and filters were controlled via Image-Pro (Media Cybernetics, Bethesda, MD).
Flow cytometry
Cells frozen in 70% ethanol fixative were thawed, collected by centrifugation at 600 rpm for 5 min, washed with phosphate-buffered saline (PBS), and incubated with 4.2 µg/ml anti–phospho-histone H3 antibody, 2 µg/ml cyclin A2 antibody, 2 µg/ml cyclin B1 antibody, or 2 µg/ml cyclin E1 diluted in ADB (1% BSA [wt/vol] and 0.1% Triton X-100 [vol/vol] in PBS). After 2 h of incubation at room temperature, cells were washed with PBS and stained with 4 µg/ml Alexa Fluor 488–conjugated anti-rabbit and 20 µg/ml Alexa Fluor 647-conjugated anti-mouse secondary antibodies in ADB for an additional 2 h at room temperature. Cells were then rinsed in ADB and resuspended in ADB with 50 μg/ml propidium iodide (PI; Invitrogen) and 100 μg/ml RNase A. For ethynyl-deoxyuridine (EdU) labeling and detection, cells were incubated with 10 μM EdU for 1 h before harvest. Then cells were fixed, permeabilized, and labeled with Alexa Fluor 647 using a Click-iT EdU flow cytometry assay kit (Invitrogen). When analyzed for p27 and cyclin E1 contents, cells were fixed and probed according to recommendations made by the manufacturer (Cell Signaling Technology, Danvers, MA). The staining of other intracellular antigens and PI was performed as described. Quantitation of cyclin A2, cyclin B1, cyclin E1, EdU, and PI content was performed on a FACSCalibur (BD Biosciences), and data were analyzed using FlowJo software (Tree Star, Ashland, OR).
qRT-PCR analysis
Total RNA was isolated and purified from GL2 and Cdh1 siRNA–transfected HeLa cells collected 18–28 h after triple-thymidine block using TRIzol reagent (Invitrogen), according to the manufacturer's instructions. RNA was then treated with DNase 1 (New England BioLabs, Ipswich, MA) and purified with a PureLink RNA mini-kit (Invitrogen). qRT-PCR was performed in triplicate using the One-Step Brilliant II SYBR Green QRT-PCR master mix kit (Agilent Technologies, Santa Clara, CA), sense and antisense primers (50 nM each), and sample RNA from three biological replicates (100 ng). Primers designed for different genes were as follows: cyclin A2, sense, 5′-CATGTCACCGTTCCTCCTTGGAAA-3′, and antisense, 5′-TTCTGGGTCCAGGTAAACTAATGGC-3′; cyclin B1, sense, 5′-TAAGGCGAAGATCAACATGGCAGG-3′, and antisense, 5′-TTT AACAGGCTCAGGTTCTGGCTC-3′; cyclin E1, sense, 5′-TACTTGCTGCTTCGGCCTTGTA TC-3′, and antisense, 5′-TCCAGCAAATCCAAGCTGTCTCTG-3′; PCNA, sense, 5′-AAGAAGG TGTTGGAGGCACTCAAG-3′, and antisense, 5′-TTGGACATACTGGTGAGGTTCACG-3′; and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), sense, 5′-CATGTTCGTCATGGGTGTGAACCA-3′, and antisense, 5′-TAGAGGCAGGG ATGATGTTCTGGA-3′. Accumulation of PCR product was monitored in real time using the Mx3000p system (Stratagene), and the crossing threshold (Ct) was determined using MxPro software. The amplification profile used was 50°C for 30 min and 95°C for 10 min, followed by 40 cycles of 95°C/30 s, 57°C/1 min, and 72°C/1 min. Postamplification dissociation curves were performed to verify specificity of the amplification. Fold changes in gene expression of cyclin A2, cyclin B1, cyclin E1, and PCNA were determined using the ∆∆Ct method with normalization to the expression of GAPDH. All RT-PCR quantifications were performed in triplicate in three independent experiments.
DNA fiber analysis
After 48 h of siRNA transfection, HeLa cells were pulse labeled with 50 μM iododeoxyuridine (IdU) for 10 min, washed, and then pulse labeled with 100 μM chlorodeoxyuridine (CldU) for 20 min. DNA fiber spreading and labeling were performed by following the published protocol (Chastain et al., 2006). Cells were then harvested and diluted in cold PBS to ∼200 cells/μl. A 2-μl sample was streaked onto the slide (Silane-Prep slides; Sigma-Aldrich) by drawing a line across its surface. When the sample was nearly dry, 15 μl of lysis buffer was added to the line of cells. After 10 min of incubation, DNA fibers were spread by tipping the slides at ∼25° to let the buffer migrate to the bottom of the slides. Samples were then air dried for >4 h, fixed in methanol/acetic acid (3:1) for 2 min followed by air drying overnight, and stored at −20°C. For immunostaining, slides were incubated with 2.5 M HCl for 30 min to denature the DNA. The slides were then washed with washing solution and 2× in PBS for 3 min. After blocked in PBS with 5% BSA for 30 min, samples were incubated with mouse and rat anti-BrdU antibodies (1:500) for 1 h. Slides were washed in stringency buffer (10 mM Tris-HCl, pH 7.4, 0.4 M NaCl, 0.02% Tween-20, 0.02% NP40) for 13 min, followed by 2× in PBS for 3 min, and blocked again for 30 min. Samples were labeled with Alexa Fluor 594–conjugated rabbit anti-mouse (2 μg/ml) and Alexa Fluor 488–conjugated chicken anti-rat antibodies (2.67 μg/ml) for 1 h. After treatment with washing solution (5% BSA [wt/vol] in PBS), followed by two rinses with PBS, slides were blocked again for 30 min and then incubated with Alexa Fluor 594–conjugated goat anti-rabbit (2 μg/ml) and Alexa Fluor 488–conjugated goat anti-chicken (2.67 μg/ml) antibodies for 30 min. Slides were then washed with washing solution and twice with PBS and then mounted with ProLong Gold antifade reagent (Invitrogen). DNA fibers were scored from at least eight images for each independent experiment using CASA software (Yaping et al., 2011). The parameters were adjusted to exclude fiber bundles, stretched fibers, and background staining, using following values: minimum length of single-color tracks, 15 pixels; minimum length of segments in two/three-color tracks, 10 pixels; threshold of signal-to-noise ratio, 3; maximum discontinuity, 20%; and maximum track thickness, 10 pixels. In addition, images with >25% fibers detected and >15% discontinuity in one image were excluded from the analysis.
Immunofluorescence
HeLa and U2OS cells were plated into six-well plates containing acid-washed No. 12 circle coverslips (Fisher, Pittsburgh, PA) at 2 × 105 cells/well. After 48 h of siRNA transfection or 2 h of HU treatment, cells grown on the coverslips were fixed with precooled methanol for 20 min at −20°C and rehydrated in PBS for 5 min. Cells were probed with antibody against pS139-H2AX (1:400) diluted in ADB (TBS, 2% BSA, 0.1% Triton X-100, 0.1% sodium azide) for 1.5 h at room temperature. After washing with PBS, cells were stained with Texas red–conjugated anti-rabbit (1: 200) for 1 h at room temperature. Cells were then mounted with Mowiol (Sigma-Aldrich) containing 1 mg/ml 4′,6-diamidino-2-phenylindole (Sigma-Aldrich).
Supplementary Material
Acknowledgments
We thank C. Walczak for providing cell lines, the Indiana Statistical Consulting Center and Y. Dong for assisting with statistical analyses, J. Powers for microscopy assistance, Paul Chastain for guidance with the DNA fiber spreading and analysis, and members of the Pomerening lab for critical review of the manuscript and reviewers for their invaluable assistance in improving it. Live-cell imaging and confocal microscopy were performed in the Indiana Light Microscopy Imaging Center, with grant support from the National Institutes of Health (S10RR025033-01). J.R.P. is a Pew Scholar in the Biomedical Sciences and is supported by the National Institutes of Health (R01GM086526).
Abbreviations used:
- ADB
antibody-diluting buffer
- APC
anaphase-promoting complex
- BSA
bovine serum albumin
- CASA
Computer-Aided Scoring and Analysis
- CDK
cyclin-dependent kinase
- CKI
cyclin-dependent kinase inhibitor
- ECFP
enhanced cyan fluorescent protein
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- ODE
ordinary differential equation
- PCNA
proliferating cell nuclear antigen
- Plk1
Polo-like kinase 1
- qRT-PCR
real-time quantitative reverse transcription PCR
- SCF
SKP1-CUL1-F-box
- YFP
yellow fluorescent protein
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-08-0480) on December 19, 2013.
REFERENCES
- Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer. 2007;6:24. doi: 10.1186/1476-4598-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleem E, Kiyokawa H, Kaldis P. Cdc2-cyclin E complexes regulate the G1/S phase transition. Nat Cell Biol. 2005;7:831–836. doi: 10.1038/ncb1284. [DOI] [PubMed] [Google Scholar]
- Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M. Control of the SCFSkp2-Cks1 ubiquitin ligase by the APC/C-Cdh1 ubiquitin ligase. Nature. 2004;428:190–193. doi: 10.1038/nature02330. [DOI] [PubMed] [Google Scholar]
- Bloom J, Pagano M. Deregulated degradation of the cdk inhibitor p27 and malignant transformation. Semin Cancer Biol. 2003;13:41–47. doi: 10.1016/s1044-579x(02)00098-6. [DOI] [PubMed] [Google Scholar]
- Bornstein G, Bloom J, Sitry-Shevah D, Nakayama K, Pagano M, Hershko A. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J Biol Chem. 2003;278:25752–25757. doi: 10.1074/jbc.M301774200. [DOI] [PubMed] [Google Scholar]
- Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- Castro A, Bernis C, Vigneron S, Labbe J-C, Lorca T. The anaphase-promoting complex: a key factor in the regulation of cell cycle. Oncogene. 2005;24:314–325. doi: 10.1038/sj.onc.1207973. [DOI] [PubMed] [Google Scholar]
- Chastain PD, II, Heffernan TP, Nevis KR, Lin L, Kaufmann WK, Kaufman DG, Cordeiro-Stone M. Checkpoint regulation of replication dynamics in UV-irradiated human cells. Cell Cycle. 2006;5:2160–2167. doi: 10.4161/cc.5.18.3236. [DOI] [PubMed] [Google Scholar]
- Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- Engelbert D, Schnerch D, Baumgarten A, Wasch R. The ubiquitin ligase APCCdh1 is required to maintain genome integrity in primary human cells. Oncogene. 2007;27:907–917. doi: 10.1038/sj.onc.1210703. [DOI] [PubMed] [Google Scholar]
- Gao DM, Inuzuka H, Korenjak M, Tseng A, Wu T, Wan LX, Kirschner M, Dyson N, Wei WY. Cdh1 regulates cell cycle through modulating the claspin/Chk1 and the Rb/E2F1 pathways. Mol Biol Cell. 2009;20:3305–3316. doi: 10.1091/mbc.E09-01-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garci-Higuera I, Manchado E, Dubus P, Canamero M, Mendez J, Moreno S, Malumbres M. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat Cell Biol. 2008;10:802–811. doi: 10.1038/ncb1742. [DOI] [PubMed] [Google Scholar]
- Hahn AT, Jones JT, Meyer T. Quantitative analysis of cell cycle phase durations and PC12 differentiation using fluorescent biosensors. Cell Cycle. 2009;8:1044–1052. doi: 10.4161/cc.8.7.8042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev. 2000;14:2393–2409. doi: 10.1101/gad.813200. [DOI] [PubMed] [Google Scholar]
- Harbour JW, Luo RX, Santi AD, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98:859–869. doi: 10.1016/s0092-8674(00)81519-6. [DOI] [PubMed] [Google Scholar]
- Kamura T, Hara T, Kotoshiba S, Yada M, Ishida N, Imaki H, Hatakeyama S, Nakayama K, Nakayama KI. Degradation of p57Kip2 mediated by SCFSkp2-dependent ubiquitylation. Proc Natl Acad Sci USA. 2003;100:10231–10236. doi: 10.1073/pnas.1831009100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanie T, Onoyama I, Matsumoto A, Yamada M, Nakatsumi H, Tateishi Y, Yamamura S, Tsunematsu R, Matsumoto M, Nakayama KI. Genetic reevaluation of the role of F-box proteins in cyclin D1 degradation. Mol Cell Biol. 2011;32:590–605. doi: 10.1128/MCB.06570-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck JM, Summers MK, Tedesco D, Ekholm-Reed S, Chuang L-C, Jackson PK, Reed SI. Cyclin E overexpression impairs progression through mitosis by inhibiting APCCdh1. J Cell Biol. 2007;178:371–385. doi: 10.1083/jcb.200703202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RW, Peters J-M, Tugendreich S, Rolfe M, Hieter P, Kirschner MW. A 20s complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell. 1995;81:279–288. doi: 10.1016/0092-8674(95)90338-0. [DOI] [PubMed] [Google Scholar]
- Kraft C, Herzog F, Gieffers C, Mechtler K, Hagting A, Pines J, Peters J-M. Mitotic regulation of the human anaphase-promoting complex by phosphorylation. EMBO J. 2003;22:6598–6609. doi: 10.1093/emboj/cdg627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer ER, Gieffers C, Holzl G, Hengstschlager M, Peters JM. Activation of the human anaphase-promoting complex by proteins of the CDC20/Fizzy family. Curr Biol. 1998;8:1207–1210. doi: 10.1016/s0960-9822(07)00510-6. [DOI] [PubMed] [Google Scholar]
- Kramer ER, Scheuringer N, Podtelejnikov AV, Mann M, Peters J-M. Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol Biol Cell. 2000;11:1555–1569. doi: 10.1091/mbc.11.5.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahav-Baratz S, Sudakin V, Ruderman JV, Hershko A. Reversible phosphorylation controls the activity of cyclosome-associated cyclin-ubiquitin ligase. Proc Natl Acad Sci USA. 1995;92:9303–9307. doi: 10.1073/pnas.92.20.9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Yuan X, Wyatt WR, Pomerening JR. Expression of constitutively active CDK1 stabilizes APC-Cdh1 substrates and potentiates premature spindle assembly and checkpoint function in G1 Cells. PLoS One. 2012;7:e33835. doi: 10.1371/journal.pone.0033835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek NP, Sundberg H, McGrew S, Nakayama K, Kyriakidis TR, Roberts JM. A mouse knock-in model exposes sequential proteolytic pathways that regulate p27Kip1 in G1 and S phase. Nature. 2001;413:323–327. doi: 10.1038/35095083. [DOI] [PubMed] [Google Scholar]
- Ohtani K, DeGregori J, Nevins JR. Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci USA. 1995;92:12146–12150. doi: 10.1073/pnas.92.26.12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J-M. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol. 2006;7:644–656. doi: 10.1038/nrm1988. [DOI] [PubMed] [Google Scholar]
- Pomerening JR, Kim SY, Ferrell JE. Systems-level dissection of the cell-cycle oscillator: bypassing positive feedback produces damped oscillations. Cell. 2005;122:565–578. doi: 10.1016/j.cell.2005.06.016. [DOI] [PubMed] [Google Scholar]
- Pomerening JR, Ubersax JA, Ferrell JE., Jr Rapid cycling and precocious termination of G1 phase in cells expressing CDK1AF. Mol Biol Cell. 2008;19:3426–3441. doi: 10.1091/mbc.E08-02-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z, Weiss JN, MacLellan WR. Regulation of the mammalian cell cycle: a model of the G1-to-S transition. Am J Physiol Cell Physiol. 2003;284:C349–C364. doi: 10.1152/ajpcell.00066.2002. [DOI] [PubMed] [Google Scholar]
- Resnitzky D, Gossen M, Bujard H, Reed SI. Acceleration of the G1/S phase transition by expression of cyclins D1 and E with an inducible system. Mol Cell Biol. 1994;14:1669–1679. doi: 10.1128/mcb.14.3.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamaria D, Barriere C, Cerqueira A, Hunt S, Tardy C, Newton K, Caceres JF, Dubus P, Malumbres M, Barbacid M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448:811–815. doi: 10.1038/nature06046. [DOI] [PubMed] [Google Scholar]
- Sigl R, Wandke C, Rauch V, Kirk J, Hunt T, Geley S. Loss of the mammalian APC/C activator FZR1 shortens G1 and lengthens S phase but has little effect on exit from mitosis. J Cell Sci. 2009;122:4208–4217. doi: 10.1242/jcs.054197. [DOI] [PubMed] [Google Scholar]
- Thomson AM, Gillespie PJ, Blow JJ. Replication factory activation can be decoupled from the replication timing program by modulating Cdk levels. J Cell Biol. 2010;188:209–221. doi: 10.1083/jcb.200911037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thron CD. Mathematical analysis of binary activation of a cell cycle kinase which down-regulates its own inhibitor. Biophys Chem. 1999;79:95–106. doi: 10.1016/s0301-4622(99)00041-1. [DOI] [PubMed] [Google Scholar]
- Waesch R, Robbins JA, Cross FR. The emerging role of APC/C-Cdh1 in controlling differentiation, genomic stability and tumor suppression. Oncogene. 2010;29:1–10. doi: 10.1038/onc.2009.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Ayad NG, Wan Y, Zhang G-J, Kirschner MW, Kaelin WG. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 2004;428:194–198. doi: 10.1038/nature02381. [DOI] [PubMed] [Google Scholar]
- Yaping W, Chastain P, Pew-Thian Y, Jie-Zhi C, Kaufman D, Lei G, Dinggang S. Automated DNA fiber tracking and measurement; In: Biomedical Imaging: From Nano to Macro; New York:. Institute of Electrical and Electronics Engineers; 2011. pp. 1349–1352. [Google Scholar]
- Zarkowska T, Mittnacht S. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J Biol Chem. 1997;272:12738–12746. doi: 10.1074/jbc.272.19.12738. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.