

PIP2-binding proteins CAPS and Munc13 are required for Ca2+-triggered vesicle exocytosis. TIRF microscopy localized PIP2, DAG, CAPS, and Munc13. Exocytosis occurred at PIP2-rich sites. CAPS localized to vesicles but required PIP2, whereas Munc13 required stimulus-dependent recruitment to PIP2-rich domains, indicating distinct mechanisms.
Abstract
Phosphoinositides provide compartment-specific signals for membrane trafficking. Plasma membrane phosphatidylinositol 4,5-bisphosphate (PIP2) is required for Ca2+-triggered vesicle exocytosis, but whether vesicles fuse into PIP2-rich membrane domains in live cells and whether PIP2 is metabolized during Ca2+-triggered fusion were unknown. Ca2+-dependent activator protein in secretion 1 (CAPS-1; CADPS/UNC31) and ubMunc13-2 (UNC13B) are PIP2-binding proteins required for Ca2+-triggered vesicle exocytosis in neuroendocrine PC12 cells. These proteins are likely effectors for PIP2, but their localization during exocytosis had not been determined. Using total internal reflection fluorescence microscopy in live cells, we identify PIP2-rich membrane domains at sites of vesicle fusion. CAPS is found to reside on vesicles but depends on plasma membrane PIP2 for its activity. Munc13 is cytoplasmic, but Ca2+-dependent translocation to PIP2-rich plasma membrane domains is required for its activity. The results reveal that vesicle fusion into PIP2-rich membrane domains is facilitated by sequential PIP2-dependent activation of CAPS and PIP2-dependent recruitment of Munc13. PIP2 hydrolysis only occurs under strong Ca2+ influx conditions sufficient to activate phospholipase Cη2 (PLCη2). Such conditions reduce CAPS activity and enhance Munc13 activity, establishing PLCη2 as a Ca2+-dependent modulator of exocytosis. These studies provide a direct view of the spatial distribution of PIP2 linked to vesicle exocytosis via regulation of lipid-dependent protein effectors CAPS and Munc13.
INTRODUCTION
Phosphoinositides provide important compartment-specific signals for membrane trafficking by recruiting and activating specific proteins in cellular membranes (Hurley and Meyer, 2001). In this manner, phosphatidylinositol 4,5-bisphosphate (PIP2) on the plasma membrane plays a key role in regulating actin assembly, endocytosis, exocytosis, phagocytosis, and ion channel function (Martin, 2001; Di Paolo and De Camilli, 2006; Suh and Hille, 2008; Koch and Holt, 2012). Processes near membrane domains of PIP2 may be regulated by local PIP2 synthesis and degradation (Schramp et al., 2012; Zhang et al., 2012). The spatial and temporal regulation of PIP2 was characterized for phagocytosis, where an initial enhanced synthesis of PIP2 in early phagosome formation transitions to a decrease in PIP2, with diacylglycerol (DAG) formation during phagosome closure (Botelho et al., 2000; Flannagan et al., 2012). The dynamics of PIP2 has not been well characterized for other membrane-trafficking events, such as vesicle exocytosis.
Regulated vesicle exocytosis in neuroendocrine cells exhibits a strong requirement for PIP2, but its role is incompletely understood. Dense-core vesicles are transported to the plasma membrane, where they become fusion competent in priming reactions that involve the assembly of soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) protein complexes (Rettig and Neher, 2002; Jahn and Scheller, 2006; Rizo and Rosenmund, 2008). A role for PIP2 in vesicle priming was identified by the ATP requirement for priming involving conversion of PIP to PIP2 (Eberhard et al., 1990; Hay and Martin, 1993; Hay et al., 1995). Levels of PIP2 establish the number of primed vesicles (Olsen et al., 2003; Grishanin et al., 2004; Milosevic et al., 2005) and the rates of sustained secretion (Aikawa and Martin, 2003; Milosevic et al., 2005). PIP2-rich domains in plasma membrane preparations colocalize with a subset of vesicles, which suggests that these domains are preferential sites for exocytosis (Aoyagi et al., 2005; James et al., 2008). Such domains have yet to be identified in live cells, and it is not known whether vesicle fusion occurs into PIP2-rich membrane regions or whether PIP2 is metabolized during Ca2+-triggered exocytosis.
A number of proteins required for vesicle exocytosis are proposed to function via PIP2 binding, including the SNARE protein syntaxin-1 (Aoyagi et al., 2005; James et al., 2008; van den Bogaart et al., 2011) and the Ca2+ sensor synaptotagmin-1 (Bai et al., 2004; Kuo et al., 2011; Koch and Holt, 2012; van den Bogaart et al., 2012). The two major proteins that function in vesicle priming Ca2+-dependent activator protein in secretion 1 (CAPS-1; CADPS) and Munc13-1/2 bind PIP2 (Augustin et al., 1999; Ashery et al., 2000; Grishanin et al., 2004; Jockusch et al., 2007; Liu et al., 2008; Shin et al., 2010). Low-affinity binding by its pleckstrin homology (PH) domain is required for CAPS stimulation of SNARE-dependent liposome fusion (Loyet et al., 1998; Grishanin et al., 2002; James et al., 2008). Higher-affinity, Ca2+-dependent PIP2 binding by the C2B domain of Munc13-2 is required for Ca2+-dependent augmentation of synaptic vesicle exocytosis (Shin et al., 2010). PIP2 may activate or recruit these proteins near sites of vesicle exocytosis in cells, but this has not been determined. CAPS and Munc13 proteins have related C-terminal SNARE protein–binding domains (see later, Figure 2A; Koch et al., 2000; Guan et al., 2008; Pei et al., 2009; Khodthong et al., 2011). Despite sequence relatedness, coexpressed CAPS and Munc13 proteins may not function redundantly, as suggested by the strong impairment of vesicle priming in synapses of CAPS-1/2– or Munc13-1–knockout mice (Augustin et al., 1999; Jockusch et al., 2007). The N-terminal lipid-binding domains of CAPS and Munc13 proteins are distinct (see later, Figure 2A), which may account for this lack of redundancy. However, the lipid-dependent regulation of CAPS and Munc13 proteins at sites of exocytosis remains to be assessed.
FIGURE 2:

CAPS and ubMunc13-2 are corequired for evoked vesicle exocytosis. (A) Schematic of CAPS and Munc13-1/ubMunc13-2 proteins, indicating annotated C2, PH, DUF1041, and MHD1 domains in CAPS, and C2, C1, DUF1041, MHD1, and MHD2 domains in Munc13-1/ubMunc13-2. Dashed lines indicate regions of sequence homology between CAPS and Munc13 proteins. (B) Western blot indicating CAPS and ubMunc13-2 knockdown. β-Actin was used as the loading control. (C) Representative TIRF images of control cells and CAPS- or ubMunc13-2–knockdown cells expressing BDNF-EGFP. Scale bar, 2 μm. The average density of vesicles in the TIRF field is plotted beneath the images. (D) CAPS-knockdown cells stimulated with 56 mM K+ buffer (at time zero) resulted in ∼80% reduction of exocytosis. Recordings were taken at 4 Hz, and events were binned into 15-s increments. Note that no events occur in the absence of stimulation (5.6 mM K+ buffer). (E) ubMunc13-2–knockdown PC12 cells stimulated with 56 mM K+ buffer resulted in ∼90% reduction of exocytosis. Recordings were taken at 4 Hz, and events were binned into 15-s increments. (F) Overexpression of ubMunc13-2 failed to restore evoked exocytosis in CAPS-knockdown cells. Inset, threefold ubMunc13-2 overexpression. (G) Overexpression of CAPS was unable to restore evoked exocytosis in ubMunc13-2–knockdown cells. Inset, overexpression of CAPS-mKate2. Mean values ± SE (n = 15–20 cells for CAPS knockdown and 10–15 cells for ubMunc13-2 knockdown).
In the present work, we use total internal reflection fluorescence microscopy (TIRFM) to image PIP2, DAG, CAPS, and Munc13 at sites of dense-core vesicle exocytosis in live neuroendocrine PC12 cells. We show that PIP2 localizes to domains on the plasma membrane that provide sites for vesicle exocytosis. CAPS resides on vesicles but requires plasma membrane PIP2 for activation. Cytosolic Munc13 exhibits Ca2+-dependent recruitment to PIP2-rich plasma membrane domains, which is required for its activity. PIP2 metabolism to DAG is only detected under stronger Ca2+-influx conditions that activate phospholipase Cη2 (PLCη2), which leads to loss of CAPS function and gain of Munc13 function. These studies provide a direct view of the spatial distribution of PIP2 linked to vesicle exocytosis via regulation of lipid-dependent protein effectors.
RESULTS
PIP2-rich domains localize to sites of vesicle exocytosis
The distribution of PIP2 in membrane preparations from neuroendocrine cells is heterogeneous and punctate (Laux et al., 2000; Aoyagi et al., 2005; Milosevic et al., 2005; James et al., 2008). Subdiffraction domains containing >6 mol% PIP2 colocalize with a subset of dense-core vesicles (James et al., 2008; van den Bogaart et al., 2011). Some sites correspond to syntaxin-1 clusters, which suggests that PIP2 domains constitute preferential sites for vesicle exocytosis (Aoyagi et al., 2005; James et al., 2008; van den Bogaart et al., 2011). To test this directly in live PC12 cells, we imaged PIP2 at sites of vesicle exocytosis by TIRFM. Expression of a fluorescent, PIP2-binding PLCδ1-PH protein resulted in uniform plasma membrane labeling due to high-affinity binding to PIP2, as shown previously (Holz et al., 2000). Therefore we used a PLCδ4-PH fusion protein, which exhibits ∼10-fold-lower affinity for phosphoinositides (Lee et al., 2004). The mKate2-PLCδ4-PH protein partitions onto the plasma membrane into punctate domains (Figure 1A). The specificity of PLCδ4-PH domain binding was confirmed by hydrolyzing PIP2 with a 5-phosphatase (Supplemental Figure S2A). Many of the membrane-proximal vesicles in the evanescent field, identified by brain-derived neurotrophic factor (BDNF)–enhanced green fluorescent protein (EGFP) cargo, colocalized with PIP2-containing membrane domains (Figure 1B). The overlap of BDNF-EGFP pixels (green) with PLCδ4-PH-mKate2 pixels (red) exhibited a Manders coefficient of ∼0.5, which was much greater than random. The results revealed for the first time PIP2-rich domains in the plasma membrane of live neuroendocrine cells.
FIGURE 1:

Evoked vesicle exocytosis occurs at PIP2 domains. (A) PC12 cells expressing PLCδ4-PH-mKate2 exhibit puncta on the plasma membrane viewed by TIRF microscopy. Several cell footprints are shown. (B) In cells coexpressing BDNF-EGFP, many of the dense-core vesicles (green) overlapped with the distribution of PLCδ4-PH-mKate2 puncta (red; right three images, insets, and bottom). Overlap of green pixel distribution with red was assessed by the method of Costes et al. (2004) as a Manders coefficient (M1 = 0.49 ± 0.09 SE, n = 6). A set of rotated images was used to assess random overlap (M1 = 0.06 ± 0.02 SE, n = 6). BDNF-EGFP pixel overlap with PLCδ4-PH-mKate2 pixels was significantly different from random (p < 0.005). The Manders coefficient for PLCδ4-PH-mKate2 pixel overlap with BDNF-EGFP pixels (M2) was 0.10 ± 0.02. Images shown were deconvolved to reduce background. (C) Vesicle exocytosis was stimulated in 56 mM K+ buffer (MS). Images of BDNF-EGFP–containing vesicles and PLCδ4-PH-mKate2 fluorescence were acquired at 4 Hz. Resident (R) vesicles that fused were compared with nonfusing vesicles (N) in the same cells. (D) Similarly, nonresident (NR) vesicles that fused were compared with nonfusing vesicles (N) in the same cells. Nonresident fusion events were defined as absence of a vesicle in the TIRF field for at least 0.5 s before fusion (Supplemental Figure S1). Fluorescence was quantitated across numerous images, as summarized in Supplemental Figure S2, C and D.
Two-channel TIRFM enabled direct imaging of vesicle exocytosis with simultaneous imaging of PIP2 with mKate2-PLCδ4-PH fluorescence. To conduct these studies, we used cells expressing moderate levels of mKate2-PLCδ4-PH to minimize the inhibition of exocytosis. Moderate expression of mKate2-PLCδ4-PH was associated with 46 ± 6% (n = 7) reduction in exocytic events. Vesicle exocytosis was monitored with BDNF-EGFP as vesicle cargo. As shown previously (Lynch et al., 2008), little BDNF-EGFP is released during exocytosis, but there is a marked increase in fluorescence at the time of fusion pore formation due to vesicle deacidification (Supplemental Figure S1). This increase in fluorescence is followed by gradual dimming, which results from fusion pore closure and gradual vesicle reacidification. Using 56 mM K+ depolarization to promote Ca2+ influx optimal for exocytosis, we monitored PIP2 levels by mKate-PLCδ4-PH fluorescence at sites of evoked vesicle exocytosis. PIP2 levels in a region of interest centered on fusing vesicles were higher (Figure 1C, second row) than in a comparable region centered on nonfusing vesicles in the same cells (Figure 1C, fourth row). This was similar for docked vesicles that fused (Figure 1C) and for newly arrived vesicles that fused during stimulation (Figure 1D). The results quantitated across many examples (Supplemental Figure S2, C and D) showed that vesicle exocytosis evoked by moderate stimulation occurs into PIP2-rich membrane domains.
CAPS and ubMunc13-2 are major effectors for PIP2
PIP2 is required for a priming step in the exocytic pathway (Olsen et al., 2003; Grishanin et al., 2004; Milosevic et al., 2005), where CAPS and Munc13 proteins are essential (Ashery et al., 2000; Grishanin et al., 2004; Liu et al., 2008). CAPS and Munc13 may function as PIP2 effector proteins (Grishanin et al., 2004; James et al., 2008; Shin et al., 2010), but the spatial and temporal regulation of these proteins during exocytosis has not been characterized. We confirmed that CAPS-1 is expressed in PC12 cells, and we identified ubMunc13-2 as the major Munc13 protein in PC12 cells (Supplemental Figure S3). The roles of CAPS-1 and ubMunc13-2 in evoked vesicle exocytosis were assessed by reducing the expression of either protein by >90% (Figure 2B). Depletion of either CAPS or ubMunc13-2 did not affect the number of vesicles near the plasma membrane (Figure 2C) but markedly reduced vesicle exocytosis evoked by 56 mM K+ depolarization (Figure 2, D and E). Fusion events from resident or newly arrived vesicles were similarly reduced by CAPS or ubMunc13-2 knockdown (Supplemental Figure S4). Successful rescue experiments with CAPS in CAPS-knockdown cells (see later discussion of Figure 6) or with Munc13-1 in ubMunc13-2–knockdown cells (see Supplemental Figure S7) indicated that off-target effects were not responsible for the inhibition of evoked exocytosis. However, the overexpression of ubMunc13-2 in CAPS-knockdown cells (Figure 2F) or overexpression of CAPS in ubMunc13-2–knockdown cells (Figure 2G) failed to restore evoked vesicle exocytosis. Thus it was apparent that CAPS and ubMunc13-2 function nonredundantly in PC12 cells under moderate stimulation conditions.
FIGURE 6:

CAPS localizes to vesicles and requires PIP2 for activation. (A) Coexpression of BDNF-EGFP and CAPS-mKate2 in PC12 cells revealed that CAPS localizes to dense-core vesicles as viewed by TIRF microscopy. (B) Schematic showing C2, PH, and MHD1 domains in CAPS. Sequences show conservation of CAPS PH domain, with boxed residues indicating sites of mutation (R558D/K560E/K561E; DEE). (C,D) CAPS knockdown by shRNA plasmid markedly attenuates exocytosis evoked by 56 mM K+. Wild-type CAPS rescues exocytosis in the knockdown cells, whereas the CAPS PH-domain mutant (DEE) fails to rescue. (E, F) The CAPS PH-domain mutant DEE localizes to dense-core vesicles based on colocalization with BDNF-EGFP in TIRF microscopy. Line scans between arrowheads indicate colocalization.
Dual role for PIP2 in evoked exocytosis
To assess the role of PIP2 in evoked vesicle exocytosis in live cells, we overexpressed PIP2 hydrolytic enzymes. In the first approach, we expressed a 5-phosphatase-FKBP12 fusion protein that is targeted to a plasma membrane–localized FRB protein upon rapamycin addition (Varnai et al., 2006). Plasma membrane PIP2 was acutely reduced after rapamycin treatment but not in controls, based on the membrane-to-cytosol translocation of a PLCδ1-PH-GFP probe (Figure 3A). This reduction in PIP2 strongly inhibited vesicle exocytosis evoked by 56 mM K+ (termed moderate stimulation [MS]; Figure 3B, MS). In a second approach, we overexpressed PLCη2, a Ca2+-regulated enzyme that hydrolyzes PIP2 (Nakahara et al., 2005). TagRFP-PLCη2 localized to the plasma membrane and generated DAG in response to stimulation as monitored by the cytosol-to-membrane translocation of a protein kinase Cδ (PKCδ)-C1-EGFP fusion protein (Figure 3D). Vesicle exocytosis evoked by moderate stimulation was strongly inhibited by PLCη2 overexpression (Figure 3C, MS). The results indicate that intact PIP2 is required for vesicle exocytosis evoked by moderate stimulation.
FIGURE 3:

PIP2 is required for evoked vesicle exocytosis. (A) Hydrolysis of PIP2 was monitored in epifluorescence microscopy by the membrane-to-cytoplasm translocation of PLCδ1-PH-GFP in cells coexpressing mRFP-FKBP12-5-phosphatase before or after rapamycin (Rapa) treatment. Rapamycin treatment reduced PIP2 levels as inferred from the translocation of PLCδ1-PH-GFP to the cytoplasm, as indicated in line scans. There was no comparable PIP2 reduction in cells expressing mRFP-FKBP12 as controls (unpublished data). (B) Rapamycin-induced recruitment of the FKBP12-5-phosphatase fusion protein inhibited exocytosis evoked by 56 mM K+ (MS) and 95 mM K+ (SS) conditions. Mean ± SE (n = 7–13 cells). (C) Overexpression of PLCη2 inhibited exocytosis evoked by 56 mM K+ (MS) but not that evoked by 95 mM K+ (SS). Mean ± SE (n = 11–13 cells). (D) Representative epifluorescence images showing the localization of tagRFP-PLCη2 at the plasma membrane (middle) and its activation at 56 mM K+ (MS) and 95 mM K+ (SS) to generate DAG, detected by the translocation of a PKCδ1 C1 domain–EGFP fusion protein to the plasma membrane (top), as documented by line scans across the cells (bottom). Right bottom inset, ratio of average pixel fluorescence intensity of C1-EGFP on the plasma membrane to total (mean values ± SD, n = 3–6, *p < 0.0005, ns = nonsignificant).
When we elicited vesicle exocytosis under stronger conditions, the results were quite different. Depolarization at 95 mM K+ promotes a twofold greater Ca2+ rise (to ∼0.8 μM; termed strong stimulation [SS]) than does 56 mM K+ (unpublished data). PIP2 was still required, as indicated by the inhibition of strong stimulation–evoked exocytosis by acute activation of a 5-phosphatase (Figure 3B, SS). However, the requirement for PIP2 appeared to be indirect, based on the finding that PLCη2 overexpression failed to inhibit vesicle exocytosis elicited by strong stimulation (Figure 3C, SS). The results suggested that the requirement for PIP2 was in part because it served as a precursor for DAG generation. To detect plasma membrane DAG, we monitored the membrane translocation of a PKCδ-C1-EGFP protein by TIRFM. There was little DAG generation promoted by moderate stimulation, whereas DAG generation in response to strong stimulation was robust (Figure 4A). The DAG generated under strong stimulation conditions had a punctate appearance consistent with it arising from the metabolism of PIP2 in membrane domains. The results suggested that evoked vesicle exocytosis is dependent on intact PIP2 under moderate stimulation conditions but that exocytosis under strong stimulation conditions is supported in part by DAG generated from PIP2.
FIGURE 4:

Strong Ca2+ influx switches the protein requirements for exocytosis. (A) DAG levels increased after incubation in strong (95 mM K+, SS) but not moderate (56 mM K+, MS) stimulation conditions. DAG was monitored by TIRF microscopy in cells expressing PKCδ-C1-EGFP. Average intensity of C1-EGFP in the TIRF field (bottom; mean ± SE, n = 5–7 cells). (B) Rates and numbers of exocytic events were similar for moderate (MS) and strong stimulation (SS) buffers (added at time zero). No events occur in 5.6 mM K+ buffer. (C) Strong stimulation evoked exocytosis in CAPS-knockdown cells to ∼80% of that in control levels. (D) ubMunc13-2 knockdown inhibited evoked exocytosis in both moderate (MS) and strong (SS) stimulation buffers. (E) The number of cumulative exocytic events observed at 300 s in B–D. Exocytosis in CAPS-knockdown cells was significantly different (*p < 0.05) for MS and SS conditions, whereas CAPS-knockdown cells did not differ from control cells under SS conditions (ns). Mean ± SE, n = 15 cells.
Strong stimulation bypasses the requirement for CAPS but not for ubMunc13-2
These results suggested that the lipid dependence of vesicle exocytosis may differ with stimulus strength. Therefore we determined whether the requirements for the lipid-dependent protein effectors CAPS and ubMunc13-2 differed for moderate and strong stimulation conditions. The overall rates and extent of evoked vesicle exocytosis did not differ for moderate and strong stimulation conditions (Figure 4B). However, we found that exocytosis evoked by strong stimulation did not require CAPS (Figure 4C), which contrasted with the strict requirement for CAPS in exocytosis evoked by moderate stimulation (Figures 4C and 2D). By contrast, evoked vesicle exocytosis at both moderate and strong stimulation required ubMunc13-2 (Figure 4D). These differing requirements, summarized in Figure 4E, indicate that the increased metabolism of PIP2 at elevated Ca2+ evoked by strong stimulation reduced the dependence of exocytosis on CAPS but retained a dependence on ubMunc13-2. The results indicated that both the lipids (PIP2 vs. PIP2/DAG) and priming factors (CAPS/Munc13 vs. Munc13) regulating vesicle exocytosis differ with stimulus strength (MS vs. SS).
PLCη2 is required for the shift in priming factor requirements
That DAG generation occurred under strong but not moderate stimulation conditions (Figure 4A) indicated that a PLC was activated at higher Ca2+ levels. This was consistent with the decreased mKate2-PLCδ4-PH fluorescence observed at sites of exocytosis under strong stimulation conditions (Supplemental Figure S2, B–D). Several PLC isozymes are expressed in PC12 cells (Homma et al., 1989), but we found that the cells also express PLCη2, a strongly Ca2+-activated PLC (Nakahara et al., 2005). To determine its role in evoked vesicle exocytosis, we knocked down PLCη2 by >80% (Figure 5A), but this had little effect on vesicle exocytosis evoked by either moderate or strong stimulation (Figure 5B). However, when PLCη2 and CAPS were knocked down together (Figure 5A), exocytosis evoked by strong stimulation was fully inhibited (Figure 5, C and D). Thus loss of a requirement for CAPS in strong stimulation conditions (Figure 4, C and E) required PIP2 hydrolysis by PLCη2. This suggests that CAPS is not active under strong stimulation conditions because PIP2 is reduced to below a threshold level needed for its activation. Conversely, based on the inhibition by the C1-domain antagonist calphostin C (Betz et al., 1998), ubMunc13-2 required its DAG-binding C1 domain only under strong stimulation conditions, for which DAG was generated (Supplemental Figure S5). Thus the loss of CAPS function and gain of Munc13 function under strong stimulation conditions appeared to result from the partial conversion of PIP2 to DAG.
FIGURE 5:

PLCη2 functions as a Ca2+-dependent modulator of exocytosis. (A) Western blot showing knockdown of PLCη2 mediated by siRNA (left) or shRNA (right). Positions of 100- and 150-kDa standards are indicated. (B) Knockdown of PLCη2 with siRNA did not affect total number of exocytic events, regardless of stimulation conditions. (C) Double knockdown of CAPS and PLCη2 with shRNA plasmids inhibited exocytosis under strong stimulation (SS) condition. This contrasted with the lack of effect of CAPS knockdown (Figure 4C) or PLCη2 knockdown (B) under the same conditions. (D) Cumulative exocytic events at 300 s. Exocytosis evoked by SS conditions differed significantly (***p < 0.0005) for CAPS knockdown and the double knockdown (mean ± SE, n = 5–10 cells).
CAPS resides on vesicles but requires activation by PIP2
Because our results indicated that PIP2 is present at sites of vesicle exocytosis, it was important to determine the localization of CAPS and Munc13 proteins during exocytosis. CAPS exhibited a punctate distribution near the plasma membrane that corresponded to its localization on dense-core vesicles (Figure 6A; Grishanin et al., 2002). It was unclear, however, whether CAPS on vesicles required PIP2 binding for its activity. We found that a PH-domain mutant (R558D/K560E/K561E) of CAPS (Figure 6B), which fails to bind PIP2 and is not activated by PIP2 in vitro (Grishanin et al., 2002; James et al., 2008), was unable to restore vesicle exocytosis in CAPS-depleted cells under moderate stimulation conditions (Figure 6, C and D). This was in spite of the normal localization of the CAPS mutant protein on vesicles (Figure 6, E and F). Thus CAPS activity for exocytosis required binding to PIP2, which may be mediated through trans interactions between CAPS on the vesicle and PIP2 on the plasma membrane.
Stimulation induces Ca2+-dependent Munc13 translocation to sites of exocytosis
The localization of Munc13 proteins during vesicle exocytosis elicited by Ca2+ influx into neuroendocrine cells has not been determined. We localized a functional Munc13-1-GFP protein (Ashery et al., 2000), which has a domain organization similar to that of ubMunc13-2. In TIRFM studies, Munc13-1-GFP exhibited a diffuse cytosolic localization similar to that of GFP (Figure 7A). However, upon stimulation, Munc13-1 was translocated into clusters at the plasma membrane (Figure 7A). This was not observed for GFP, which ruled out clustering as resulting from changes in surface membrane topology. Moreover, clusters of Munc13-1 colocalized in part with vesicles at the plasma membrane (Figure 7B). Thirty percent of membrane-proximal vesicles in the TIRF field colocalized with Munc13-1 puncta after stimulation (Manders coefficient, 0.3 ± 0.02, SE, n = 7 cells).
FIGURE 7:

Stimulation promotes Munc13 translocation to sites of exocytosis. All images are an average of four consecutive images taken 250 ms apart. The contrast and brightness of images were adjusted equally. (A) TIRF microscopy of PC12 cells expressing either EGFP or Munc13-1-EGFP. Cells were stimulated with MS buffer, and images were taken 120 s poststimulation. The white box region is displayed in the zoomed image. Scale bar, 5 μm (four upper images), 3 μm (zoomed image). (B) Representative TIRF images showing the localization of vesicles containing NPY-tagRFP and Munc13-1-EGFP translocated in response to MS buffer. Scale bar, 5 μm. Vesicles colocalized with Munc13-1 with a Manders coefficient of 0.3 ± 0.02 (± SE, n = 7 cells). (C) Time course of the change in fluorescence intensity for cells expressing EGFP or Munc13-EGFP, with zero representing the time of stimulation (MS). Fluorescence from total cell footprint was measured. (D) Change in fluorescence intensity in cells expressing EGFP or Munc13-1-EGFP after 120 s of stimulation (MS). The change for Munc13-1-EGFP and EGFP differed significantly (p < 0.05, mean ± SE, n = 6 cells). (E) Graph displaying the average density of Munc13-1 clusters under both MS and SS conditions. The cluster density poststimulation differed from the density prestimulation (**p < 0.005), but there was no significant difference in cluster densities post-MS and post-SS. (F) Representative images showing that the H567K mutation in the C1 domain did not inhibit Munc13-1 translocation to the plasma membrane. The graph displays the average density of Munc13-1 H567K clusters before and after stimulation, where a significant (**p < 0.005) difference was observed. (G) Representative images showing that the D705N/D711N mutations in the C2B domain (Munc13 DN) prevent Munc13-1 translocation to the plasma membrane. These mutations disrupt Ca2+-dependent PIP2 binding (Shin et al., 2010). The graph displays the average density of Munc13 DN clusters before and after stimulation, which did not differ (mean ± SE, n = 6–8 cells).
The translocation of Munc13-1 to the plasma membrane occurred rapidly after stimulation (Figure 7, C and D) and was the same for moderate and strong stimulation (Figure 7E), which indicated that lower Ca2+ increases were sufficient to induce translocation. Munc13-1 harboring a mutant C1 domain (H567K) that lacks phorbol ester binding (Betz et al., 1998; Rhee et al., 2002; Basu et al., 2007) underwent a similar translocation in response to moderate (unpublished data) as well as strong stimulation (Figure 7F), indicating that the C1 domain does not mediate Ca2+-dependent Munc13-1 translocation. By contrast, Munc13-1 harboring D705N/D711N mutations in its C2B domain, which abolish Ca2+-dependent interactions with PIP2 (Shin et al., 2010), failed to undergo Ca2+-dependent translocation under either moderate (unpublished data) or strong (Figure 7G) stimulation conditions. Consistent with the PIP2-binding properties of its C2B domain, Munc13-1 was found to translocate to PIP2 domains on the plasma membrane, as indicated by the complete suppression of translocation by overexpression of the PIP2-binding PLCδ1 PH domain (Supplemental Figure S6). Finally, we assessed the activity of the Munc13-1 C2B(D705N/D711N) mutant in evoked vesicle exocytosis. Whereas expression of a wild-type Munc13-1 protein reversed the inhibition of evoked exocytosis by ubMunc13-2 knockdown, the Munc13-1 C2B(D705N/D711N) mutant failed to show rescue (Supplemental Figure S7). Thus the Ca2+-dependent translocation of Munc13-1 to PIP2-rich membrane domains mediated by its C2B domain is essential for the activity of Munc13 in vesicle priming.
DISCUSSION
PIP2 was found to be essential for Ca2+-dependent vesicle exocytosis in neuroendocrine cells (Eberhard et al., 1990; Hay and Martin, 1993; Hay et al., 1995). Although subsequent studies clarified the roles of PIP2 in vesicle exocytosis, several questions about PIP2 localization, metabolism, and mechanism remained. The key new findings of the present work are as follows: 1) PIP2 in live cells segregates into high-concentration domains on the plasma membrane; 2) evoked vesicle fusion occurs into PIP2-rich domains in the plasma membrane; 3) two essential vesicle priming proteins, CAPS and Munc13, are regulated by PIP2 but by distinct mechanisms; 4) PIP2 metabolism to DAG does not accompany Ca2+-triggered vesicle exocytosis under Ca2+-influx conditions optimal for eliciting exocytosis; but 5) PIP2 metabolism to DAG is promoted by high Ca2+ concentrations via PLCη2 activation; and 6) PLCη2 activation shifts the dependence of exocytosis on CAPS and Munc13 to dependence on Munc13 alone. Overall PIP2 plays an important role in spatial coordination of vesicle exocytosis, as summarized in Figure 8.
FIGURE 8:
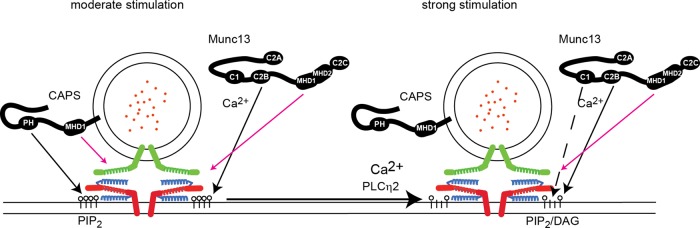
Summary of roles for PIP2, CAPS, Munc13, PLCη2, and DAG in vesicle priming in PC12 cells. Under moderate stimulation conditions (56 mM K+, peak [Ca2+]I = 400 nM), CAPS and ubMunc13-2 are both required for evoked vesicle exocytosis (left). Vesicle-associated CAPS is activated by PIP2, and Munc13 undergoes Ca2+-dependent recruitment to PIP2 domains. At exocytic sites, CAPS and Munc13 promote SNARE complex assembly (syntaxin1, red; SNAP25, blue; VAMP2, green) through Munc homology domain (MHD) interactions. Under strong stimulation conditions (95 mM K+, peak [Ca2+]I = 800 nM), PLCη2 activation leads to partial PIP2 hydrolysis and DAG generation (right). Decreased PIP2 prevents CAPS activation but supports Ca2+-dependent Munc13 recruitment. Mixed domains of PIP2 and DAG enable further C1 domain–dependent Munc13 activation.
Vesicle exocytosis at PIP2-rich membrane domains
Previous studies identified high-concentration domains of PIP2 in isolated cell membranes (Aoyagi et al., 2005; Milosevic et al., 2005; James et al., 2008; van den Bogaart et al., 2011) or in fixed cells (Laux et al., 2000). Here we characterized similar PIP2 domains in the plasma membrane of live cells for the first time. A lower-affinity PIP2-binding PLCδ4-PH probe (Lee et al., 2004) was used to detect membrane domains of PIP2 by TIRFM. A subset of dense-core vesicles colocalized with the PIP2 domains, whereas many of the more numerous PIP2 domains did not colocalize with vesicles. Other PIP2-dependent processes, such as actin assembly, constitutive exocytosis, endocytosis, and ion channel regulation, likely occur at these other PIP2 domains, depending on their protein composition.
The basis for PIP2 domain formation has not been determined. One model for domain formation suggests that it is mediated by charge interactions with proteins containing clusters of basic residues (Laux et al., 2000; McLaughlin and Murray, 2005). PIP2 domains that function in vesicle exocytosis may be organized in part by syntaxin-1, a SNARE protein with a membrane-proximal cluster of basic residues (Aoyagi et al., 2005; James et al., 2008; van den Bogaart et al., 2011). Takahashi and coworkers found that ∼13% of the vesicles in PC12 cell membrane preparations resided at PIP2- or PIP2- and syntaxin-containing sites (Aoyagi et al., 2005). These authors found that depolarization of cells in 60 mM K+ reduced colocalizing vesicles to ∼3% in membrane preparations, which led to the proposal that vesicles fuse preferentially at PIP2-rich domains (Aoyagi et al., 2005). Here we demonstrated this directly in live-cell imaging by TIRFM (Figure 1, C and D, and Supplemental Figure S2) at moderate stimulation conditions (56 mM K+), for which little hydrolysis of PIP2 to DAG occurs (Figure 4A). Arrival of a Ca2+ signal sufficient to optimally trigger exocytosis was not sufficient to promote PIP2 hydrolysis. By contrast, higher levels of Ca2+ promoted by strong stimulation (95 mM K+) evoked exocytosis at sites containing lower levels of PIP2 (Supplemental Figure S2) accompanied by the generation of DAG (Figure 4A). It is apparent that vesicle fusion in cells can occur into membrane domains enriched with PIP2 and/or PIP2 and DAG, depending on stimulation conditions. That vesicle exocytosis occurs at sites enriched in PIP2 and/or DAG is consistent with the proposed functional roles for many lipid-dependent proteins in vesicle exocytosis, including CAPS, Munc13, rabphilin, Mints, phospholipase D, SCAMP2, syntaxin, and synaptotagmin (Koch and Holt, 2012).
CAPS and Munc13 as PIP2 effector proteins
Vesicles transit through priming steps that confer fusion competence for which PIP2 is required (Aikawa and Martin, 2003; Olsen et al., 2003; Milosevic et al., 2005). CAPS and Munc13 proteins, which bind PIP2 (Loyet et al., 1998; Grishanin et al., 2002; James et al., 2008; Shin et al., 2010), were identified as essential for vesicle priming (Walent et al., 1992; Augustin et al., 1999; Grishanin et al., 2004; Liu et al., 2008), but the mechanisms by which these proteins function as PIP2 effectors were unclear. CAPS is cytosolic but also localizes to dense-core vesicles (Grishanin et al., 2002), which is the functional pool of CAPS (G.K., unpublished data). We confirmed the vesicle localization of CAPS and found that its activity, but not vesicle localization, required binding to PIP2 (Figure 6). The interaction of vesicle-bound CAPS with PIP2 may be a trans interaction from the vesicle to the plasma membrane, but we cannot exclude the possible exchange of CAPS from the vesicle to the plasma membrane, given the affinity of CAPS for PIP2 and plasma membrane SNARE proteins (Daily et al., 2010). Nonetheless, it appears that CAPS is delivered to sites of exocytosis on dense-core vesicles.
In vertebrate neurons, Munc13-1 is prelocalized at active zones in proximity to synaptic vesicle release sites through N-terminal-domain interactions with proteins in the cytomatrix of the active zone, such as RIM (Andrews-Zwilling et al., 2006). By contrast, Munc13-1 is cytosolic in neuroendocrine cells (Ashery et al., 2000), and it was not known how Munc13 proteins reach sites of exocytosis. Here we show for the first time that ubMunc13-2 is required for regulated dense-core vesicle exocytosis in PC12 cells along with CAPS-1 (Figure 2). Because of a domain organization similar to ubMunc13-2, we used Munc13-1 for localization studies. Expressed Munc13-1 is cytosolic in PC12 cells, but moderate or strong Ca2+ influx promoted its rapid translocation to PIP2 domains on the plasma membrane that correspond in part to vesicle-docking sites (Figure 7 and Supplemental Figure S6). The stimulation-dependent translocation of Munc13-1 used its Ca2+-dependent, PIP2-binding C2B domain but not its DAG-binding C1 domain (Figure 7). Previous studies showed phorbol ester- or DAG-induced, C1 domain–dependent translocation of Munc13-1 to unspecified membrane sites (Lackner et al., 1999; Ashery et al., 2000). By contrast, our results reveal an essential role for the C2B domain in Munc13 translocation during Ca2+ influx. Moreover, Ca2+-dependent translocation of Munc13 is required for its role in Ca2+-evoked dense-core vesicle exocytosis (Supplemental Figure S7). These results extend the view of Munc13 as a Ca2+-dependent PIP2-binding effector for vesicle exocytosis (Shin et al., 2010). Although the C1 domain of Munc13-1 was not required for Ca2+-dependent membrane recruitment, the activity of Munc13 was enhanced by DAG under strong stimulation conditions, as indicated by its ability to compensate for lack of CAPS activity (Figure 4) and by calphostin C sensitivity of exocytosis evoked by strong stimulation conditions (Supplemental Figure S5). A parsimonious model, strikingly similar to the proposed activation sequence for PKC (Oancea and Meyer, 1998), suggests that the initial Ca2+-dependent recruitment of Munc13 to PIP2 via its C2B domain leads to further activation via its adjacent C1 domain if DAG is available at the same membrane site. Thus the high Ca2+-dependent activation of PLCη2 will generate local membrane domains with reduced PIP2 and increased DAG. Although such membrane domains are inadequate to activate CAPS, a low-affinity PIP2-binding protein (Grishanin et al., 2002; James et al., 2008), they are sufficient to recruit the high-affinity PIP2-binding Munc13 via its C2B domain (Shin et al., 2010) and further activate Munc13 by DAG binding to its C1 domain.
CAPS and Munc13 proteins exhibit sequence similarity in C-terminal regions (Figure 2A) essential for SNARE-protein binding (Koch et al., 2000; Khodthong et al., 2011; Li et al., 2011). A key finding of the present work is that CAPS-1 and ubMunc13-2 function nonredundantly in PC12 cells (Figures 2 and 4), similar to what is inferred from synaptic transmission defects in CAPS- or Munc13-1–knockout mice (Augustin et al., 1999; Jockusch et al., 2007). The findings imply that these proteins function in a complementary manner, and our localization studies (Figures 6 and 7) suggest that these proteins may function sequentially. CAPS, delivered to sites of exocytosis on vesicles, could initiate SNARE-protein complex formation upon activation by plasma membrane PIP2 in resting cells by using its MHD1 domain (Khodthong et al., 2011). After stimulation and its Ca2+- and PIP2-dependent recruitment to sites of exocytosis, ubMunc13-2 could further stabilize SNARE-protein complexes, possibly using its MHD2 domain (Li et al., 2011). Sequential activation is consistent with the reported CAPS requirement for the function of overexpressed Munc13-1 in chromaffin cells (Liu et al., 2010). At high Ca2+ levels (0.8 μM in PC12 cells), the recruitment of Munc13 to PIP2 may be accompanied by DAG-induced conformational changes that enable Munc13 to functionally replace CAPS, possibly using both its MHD2 and MHD1 domains.
The similar time courses for evoked vesicle exocytosis (Figure 4B) and Munc13 recruitment (Figure 7C) indicate that Munc13 arrival at exocytic sites could be rate limiting for vesicle exocytosis in PC12 cells. Detailed comparisons indicate that Munc13 arrival precedes exocytosis at most sites by several seconds (M.Y., unpublished results). The possibility that Ca2+-dependent translocation of Munc13 to sites of exocytosis rate-limits evoked vesicle exocytosis would explain the absence of a ready releasable vesicle pool in resting cells and the long latencies between stimulation and secretion observed for PC12 cells (Kasai et al., 2012; Martin, 2003).
Role of PLCη2 as a Ca2+-dependent modulator of Ca2+-dependent exocytosis
A key finding in the present work is that dense-core vesicle exocytosis uses two alternative mechanisms with different protein and lipid requirements, depending on stimulus strength (Figure 8). Under moderate stimulation conditions, exocytosis depends on PIP2 and uses both CAPS-1 and ubMunc13-2 (Figures 2 and 3). Under strong stimulation conditions, exocytosis depends on PIP2 and DAG and uses ubMunc13-2 (Figures 3 and 4). The latter is similar to the effect of high-frequency stimulation, which overcomes impaired synaptic transmission in CAPS-1/2–knockout mice (Jockusch et al., 2007). Strong but not moderate stimulation activates PLCη2 to generate DAG (Figure 4A), and the present work indicates that PLCη2 activation alters the protein requirements for vesicle exocytosis. Strong stimulation bypasses the requirement for CAPS in wild-type cells but does not do so in PLCη2-depleted cells (Figure 5). PLCη2 was characterized as a neural/neuroendocrine-specific PLC, but its role was unknown (Nakahara et al., 2005; Kanemaru et al., 2010). The present work shows that PLCη2 acts as a Ca2+-dependent switch that shifts exocytosis from a CAPS/Munc13-dependent mode to a Munc13-dependent mode. PLCη2 may be the unidentified enzyme required for Munc13-dependent synaptic augmentation (Rosenmund et al., 2002).
In summary (Figure 8), the present work uses TIRF imaging to show that PIP2 is enriched at sites of vesicle exocytosis in live cells and participates directly in the activation and recruitment of lipid-dependent priming factors, CAPS and Munc13. PLCη2 is found to be a Ca2+-regulated switch activated by strong Ca2+ influx. PLCη2 activation reduces PIP2 in membrane domains to below a threshold for CAPS activation but is sufficient for the recruitment of Munc13, which is further activated by DAG.
MATERIALS AND METHODS
DNA constructs
The plasmid encoding BDNF-EGFP was provided by V. Lessmann (Johannes Gutenberg Universität, Mainz, Germany). The plasmid CAPS-mKate2 was generated by subcloning rat CAPS from a CAPS-mychis pcDNA3.1 plasmid into pmKate2-N1 from Evrogen (Moscow, Russia) using XhoI and KpnI. The ubMunc13-2 expression plasmid was generated by cloning ubMunc13-2 from PC12 cDNA into pcDNA3.1 using XhoI/KpnI. PKCδC1-EGFP plasmid was provided by S. Grinstein (Hospital for Sick Children, Toronto, Canada). Plasmids encoding PLCδ4-PH-EGFP, cyan fluorescent protein (CFP)-FRB, monomer red fluorescent protein (mRFP)-FKBP12-only, and mRFP-FKBP12-5-ptase were provided by T. Balla (National Institute of Health, Bethesda, MD). EGFP-mousePLCη2 (EGFP-PLCη2) plasmid was provided by K. Fukami (Tokyo University of Pharmacy and Life Science, Tokyo, Japan). TagRFP-PLCη2 and mKate2-PLCη2 were generated by inserting BspEI/BamHI and BglII/SalI fragments of EGFP-PLCη2 into the BspEI and BamHI sites and BglII and SalI sites of TagRFP-C and mKate2-C vector (Evrogen), respectively. PLCδ4-PH-mKate2 plasmid was generated by subcloning rat PLCδ4-PH from a PLCδ4-PH-EGFP plasmid into KpnI and SmaI sites of pmKate2-N. Munc13-1-EGFP plasmid was provided by J. Rettig (Universität des Saarlandes, Saarbrücken, Germany), and site-directed mutagenesis (Stratagene, Santa Clara, CA) was used to generate point mutations in Munc13-1-EGFP. The C1-domain mutation H576K was introduced using the forward primer 5-TCCTGCACCACGCCGAAAAACTTCGAGGTGTGG-3 and the reverse primer 5-CCACACCTCGAAGTTTTTCGGCGTGGTGCAGGA-3. The C2 mutations D705N/D711N were made consecutively with the D705N introduced first using the forward primer 5-GGCTTGCAGGCCAAGAACAAGACAGGATCCA-3 and the reverse primer 5-TGGATCCTGTCTTGTTCTTGGCCTGCAAGCC-3. The D711N mutation was introduced next with the forward primer 5-AGAACAAGACAGGATCCAGTAACCCTTATGTCAC-3 and the reverse primer 5-GTGACATAAGGGTTACTGGATCCTGTCTTGTTCT-3.
The short hairpin RNA (shRNA) plasmid for CAPS was generated by ligating oligonucleotides encoding shRNAs into pSHAG-1 vector (generously provided by G. Hannon, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY) with BamHI and BseRI. Rat CAPS mRNA sequence corresponding to nucleotides 3839–3866 (ACAGUGACGAGGAAGAUGAAGAAGACGA) was targeted. The oligonucleotide sequences were 5′-TCGTCTTCTTCATCTTCCTCGTCACTGTGAAGCTTGATAGTGATGAGGAGGATGAGGAAGACGACTATTTTTT-3′ (sense) and 5′-GATCAAAAAATAGTCTTCCTCATCCTCCTCATCACTATCAAGCTTCAGCACAGTGACGAGGAAGATGAAGAAGACGAC-3′ (antisense). The specificity of the knockdown construct was verified by a BLAST search of the public databases. Knockdown of ubMunc13-2 was performed with endoribonuclease-prepared small RNAs (esiRNAs) as described (Kittler et al., 2005). PC12 cell cDNA was generated from isolated mRNA, and a 400–base pair esiRNA target sequence was selected using the Web server DEQOR (Henschel et al., 2004). PCR was used to amplify this region using the forward primer 5′-GCTAATACGACTCACTATAGGGAGAGTCAGGATGAAGGTGCAAGAA-3′ and the reverse primer 5′-GCTAATACGACTCACTATAGGGAG ACTTCATTGTGGAGCCACTTCA-3′. A T7 promoter sequence was appended to the 5′ end of both primers, and the 400–base pair PCR product was used as a template for an in vitro transcription reaction (Applied Biosystems, Foster City, CA). Transcribed RNA was annealed to generate double-stranded RNA and digested for 72 h at 37°C with DICER (Finnzymes, Vantaa, Finland). The esiRNA was purified over a Q-Sepharose column, precipitated in isopropyl alcohol, washed with ethanol, dried, and resuspended in transfection buffer. Concentrations of esiRNA were determined by measuring the OD260 and confirmed on 4% agarose gel. As a control, esiRNA targeting mCherry was generated. Knockdown of ubMunc13-2 was alternatively conducted by transfection with an shRNA knockdown construct (target sequence CACAACTCACTGAGGATAGACCTGTCTAC; OriGene, Rockville, MD) with a sham vector as control. Knockdown of PLCη2 was conducted with pSM2-PLCη2 (or empty) shRNA plasmids (Open Biosystems, Huntsville, AL) or siRNA.
Antibodies and reagents
Anti-mouse PLCη2 polyclonal antibody was kindly provided by K. Fukami. The anti–glyceraldehyde 3-phosphate dehydrogenase monoclonal antibody was purchased from Ambion (Austin, TX) and used at a 1:1000 dilution. The anti-actin monoclonal antibody was purchased from Sigma-Aldrich (St. Louis, MO) and used at a 1:8000 dilution. The anti–Munc13-1 polyclonal antibody was purchased from Synaptic Systems (Göttingen, Germany) and used at a 1:1000 dilution. The anti–Munc13-2 monoclonal antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and used at a 1:250 dilution. The anti–Munc13-4 polyclonal antibody was provided by H. Horiuchi (Tohoku University, Sendai, Japan) and used at 1:1000 dilution. The anti-CAPS polyclonal antibody was generated against full-length CAPS protein, purified by protein A–agarose chromatography, and used at 1:1000 dilution. Fluo-4 AM was purchased from Molecular Probes (Eugene, OR). Calphostin C was purchased from LKT Laboratories (St. Paul, MN). Phorbol 12-myristate 13-acetate and rapamycin were purchased from Sigma-Aldrich.
Cell culture and transfection
PC12 cells were cultured in DMEM (Sigma-Aldrich) supplemented with 5% horse serum and 5% calf serum at 37°C in a 10% CO2 atmosphere at constant humidity. PC12 cells (grown to ∼80% confluency in a 10-cm dish) were suspended in 0.5 ml of cytomix buffer (25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 120 mM KCl, 10 mM KH2PO4, 0.15 mM CaCl2, 5 mM MgCl2, 2 mM ethylene glycol tetraacetic acid, pH 7.6) and were transfected with 10–50 μg of plasmid DNA. Transfections for plasmid DNA or siRNA were performed by electroporation using an ECM 830 (BTX, Holliston, MA) set at 8 ms, one pulse at 230 or 90 V, respectively. Alternatively, transfections were conducted with 10 μg of CAPS shRNA plasmid or empty vector using an Electroporator II (Invitrogen, Carlsbad, CA) set at 71 μF and 330 V. Cells were plated on poly-l-lysine and collagen-coated 35-mm glass-bottom dishes (MatTek, Ashland, MA) or six-well dishes for 72–96 h with subsequent TIRF analysis or Western blotting.
Cell imaging
Live-cell imaging was conducted on a Nikon TIRF microscope evanescent wave imaging system with a TE2000-U inverted microscope (Nikon, Kawasaki, Japan) and an Apo TIRF 100×/numerical aperture 1.45 (Nikon) objective lens. EGFP fluorescence was excited with a 488-nm laser line and mKate2 with a 514-nm laser. Images were acquired (usually at 4 Hz) with a CoolSNAP-ES Digital Monochrome charge-coupled device (CCD) camera (Photometrics, Tucson, AZ) or an Evolve Digital Monochrome electron-multiplying CCD camera (Photometrics) controlled by MetaMorph software (Universal Imaging, Downingtown, PA). Data analysis was done using ImageJ (National Institutes of Health, Bethesda, MD) or MetaMorph.
Imaging was conducted in a basal buffer (15 mM HEPES, pH 7.4, 145 mM NaCl, 5.6 mM KCl, 2.2 mM CaCl2, 0.5 mM MgCl2, 5.6 mM glucose, 0.5 mM ascorbic acid, 0.1% bovine serum albumin) or with stimulation buffers for moderate (basal buffer with 95 mM NaCl, 56 mM KCl) or strong (basal buffer with 56 mM NaCl, 95 mM KCl) stimulation at room temperature. Calcium imaging studies were conducted by loading cells with Fluo-4 by incubation with 2 mM of Fluo-4AM and 0.02% Pluronic F-127 (Molecular Probes) at room temperature for 30 min in the dark, followed by incubation in basal buffer at 37°C for 20 min for subsequent imaging at 488 nm. The K+ buffers, 56 and 95 mM, increased intracellular Ca2+ from ∼50 to ∼400 and ∼800 nM, respectively, within 5 s. Rapamycin-induced PIP2 hydrolysis was conducted as described (Varnai et al., 2006). Briefly, PC12 cells were transfected with plasmids encoding CFP-FRB, mRFP-FKBP12-5-ptase (or mRFP-FKBP-only), and BDNF-EGFP or PLCδ1-PH-EGFP for 48 h. Medium was replaced with basal buffer, rapamycin to 100 nM was added, and cells were stimulated with 56 or 95 mM K+ buffer. To detect PIP2 and vesicle exocytosis, cells were transfected with plasmids encoding mKate2-PLCδ4-PH and BDNF-EGFP for 48 h and imaged by TIRF microscopy. Consecutive images from recordings at 4 Hz were averaged over 2 s and deconvolved for analysis. Images were analyzed in Fiji with autothresholding and the JACoP plug-in using the method of Costes et al. (2004) to calculate the Manders coefficients. The Costes randomization p values were close to 1.0. To directly assess random overlap, rotated images were used to calculate a Manders coefficient for random. DAG generation on the plasma membrane was detected in cells expressing C1-EGFP. Munc13-1-EGFP translocation to the plasma membrane was quantitated by averaging four consecutive images before stimulation and four images taken 120 s after stimulation unless otherwise indicated.
Assays for evoked exocytosis were conducted with PC12 cells expressing BDNF-EGFP as previously described (Lynch et al., 2008). There is little BDNF-EGFP release during dense-core vesicle exocytosis. Fusion pore formation during exocytosis is accompanied by increased fluorescence from vesicle deacidification. The fluorescence subsequently decreases gradually as the result of fusion pore closure and vesicle reacidification. These fluorescence changes, shown for representative vesicles in Supplemental Figure S1, are the hallmark features of vesicle fusion and are readily distinguished from vesicles that approach and leave the plasma membrane. Images were acquired at 250-ms intervals, and fusion events were manually counted. An alternative assay for secretion was used for the study of Supplemental Figure S7. PC12 cells stably expressing NPY-EGFP were transfected with ubMunc13-2 shRNA construct or sham vector with or without coexpression of rat Munc13-1-ECFP and seeded into 96-well plates. At 4 d posttransfection, cells were stimulated with 56 mM K+ buffer, and secretion of NPY-EGFP was monitored 10 min poststimulation. Percentage secretion was calculated as 100 × GFPsupernatant/(GFPsupernatant + GFPcells).
For fixed-cell imaging, PC12 cells were transfected with plasmids encoding C1-EGFP and tagRFP or tagRFP-PLCη2. Cells were plated on poly-l-lysine and collagen-coated coverslips. After 48 h of incubation, cells were washed with basal buffer and stimulated with 56 or 95 mM K+ buffer for 2 min. Cells were immediately fixed with 3.7% formaldehyde in phosphate-buffered saline. Coverslips were mounted on slides with Mowiol 4-88 Reagent (EMD Biosciences, San Diego, CA) and imaged on a Nikon C1 laser scanning confocal microscope with a 60× oil immersion objective with numerical aperture 1.4.
Supplementary Material
Acknowledgments
This work was supported by U.S. Public Health Service Grants DK040428 and DK025861 to T.F.J.M. We acknowledge the generous contributions of plasmids by T. Balla, S. Grinstein, K. Fukami, and J. Rettig.
Abbreviations used:
- BDNF
brain-derived neurotrophic factor
- CAPS
Ca2+-dependent activator protein in secretion
- DAG
diacylglycerol
- PH
pleckstrin homology
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PKC
protein kinase C
- PLC
phospholipase C
- TIRFM
total internal reflection fluorescence microscopy
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-11-0829) on December 19, 2013.
REFERENCES
- Aikawa Y, Martin TF. ARF6 regulates a plasma membrane pool of phosphatidylinositol(4,5)bisphosphate required for regulated exocytosis. J Cell Biol. 2003;162:647–659. doi: 10.1083/jcb.200212142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews-Zwilling YS, Kawabe H, Reim K, Varoqueaux F, Brose N. Binding to Rab3A-interacting molecule RIM regulates the presynaptic recruitment of Munc13-1 and ubMunc13-2. J Biol Chem. 2006;281:19720–19731. doi: 10.1074/jbc.M601421200. [DOI] [PubMed] [Google Scholar]
- Aoyagi K, Sugaya T, Umeda M, Yamamoto S, Terakawa S, Takahashi M. The activation of exocytotic sites by the formation of phosphatidylinositol 4,5-bisphosphate microdomains at syntaxin clusters. J Biol Chem. 2005;280:17346–17352. doi: 10.1074/jbc.M413307200. [DOI] [PubMed] [Google Scholar]
- Ashery U, Varoqueaux F, Voets T, Betz A, Thakur P, Koch H, Neher E, Brose N, Rettig J. Munc13-1 acts as a priming factor for large dense-core vesicles in bovine chromaffin cells. EMBO J. 2000;19:3586–3596. doi: 10.1093/emboj/19.14.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin I, Rosenmund C, Sudhof TC, Brose N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature. 1999;400:457–461. doi: 10.1038/22768. [DOI] [PubMed] [Google Scholar]
- Bai J, Tucker WC, Chapman ER. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat Struct Mol Biol. 2004;11:36–44. doi: 10.1038/nsmb709. [DOI] [PubMed] [Google Scholar]
- Basu J, Betz A, Brose N, Rosenmund C. Munc13-1 C1 domain activation lowers the energy barrier for synaptic vesicle fusion. J Neurosci. 2007;27:1200–1210. doi: 10.1523/JNEUROSCI.4908-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Sudhof TC, Rettig J, Brose N. Munc13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron. 1998;21:123–136. doi: 10.1016/s0896-6273(00)80520-6. [DOI] [PubMed] [Google Scholar]
- Botelho RJ, Teruel M, Dierckman R, Anderson R, Wells A, York JD, Meyer T, Grinstein S. Localized biphasic changes in phosphatidylinositol-4,5-bisphosphate at sites of phagocytosis. J Cell Biol. 2000;151:1353–1368. doi: 10.1083/jcb.151.7.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daily NJ, Boswell KL, James DJ, Martin TF. Novel interactions of CAPS (Ca2+-dependent activator protein for secretion) with the three neuronal SNARE proteins required for vesicle fusion. J Biol Chem. 2010;285:35320–35329. doi: 10.1074/jbc.M110.145169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- Eberhard DA, Cooper CL, Low MG, Holz RW. Evidence that the inositol phospholipids are necessary for exocytosis. Loss of inositol phospholipids and inhibition of secretion in permeabilized cells caused by a bacterial phospholipase C and removal of ATP. Biochem J. 1990;268:15–25. doi: 10.1042/bj2680015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannagan RS, Jaumouille V, Grinstein S. The cell biology of phagocytosis. Annu Rev Pathol. 2012;7:61–98. doi: 10.1146/annurev-pathol-011811-132445. [DOI] [PubMed] [Google Scholar]
- Grishanin RN, Klenchin VA, Loyet KM, Kowalchyk JA, Ann K, Martin TF. Membrane association domains in Ca2+-dependent activator protein for secretion mediate plasma membrane and dense-core vesicle binding required for Ca2+-dependent exocytosis. J Biol Chem. 2002;277:22025–22034. doi: 10.1074/jbc.M201614200. [DOI] [PubMed] [Google Scholar]
- Grishanin RN, Kowalchyk JA, Klenchin VA, Ann K, Earles CA, Chapman ER, Gerona RR, Martin TF. CAPS acts at a prefusion step in dense-core vesicle exocytosis as a PIP2 binding protein. Neuron. 2004;43:551–562. doi: 10.1016/j.neuron.2004.07.028. [DOI] [PubMed] [Google Scholar]
- Guan R, Dai H, Rizo J. Binding of the Munc13-1 MUN domain to membrane-anchored SNARE complexes. Biochemistry. 2008;47:1474–1481. doi: 10.1021/bi702345m. [DOI] [PubMed] [Google Scholar]
- Hay JC, Fisette PL, Jenkins GH, Fukami K, Takenawa T, Anderson RA, Martin TF. ATP-dependent inositide phosphorylation required for Ca(2+)-activated secretion. Nature. 1995;374:173–177. doi: 10.1038/374173a0. [DOI] [PubMed] [Google Scholar]
- Hay JC, Martin TF. Phosphatidylinositol transfer protein required for ATP-dependent priming of Ca(2+)-activated secretion. Nature. 1993;366:572–575. doi: 10.1038/366572a0. [DOI] [PubMed] [Google Scholar]
- Henschel A, Buchholz F, Habermann B. DEQOR: a web-based tool for the design and quality control of siRNAs. Nucleic Acids Res. 2004;32:W113–W120. doi: 10.1093/nar/gkh408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz RW, Hlubek MD, Sorensen SD, Fisher SK, Balla T, Ozaki S, Prestwich GD, Stuenkel EL, Bittner MA. A pleckstrin homology domain specific for phosphatidylinositol 4, 5-bisphosphate (PtdIns-4,5-P2) and fused to green fluorescent protein identifies plasma membrane PtdIns-4,5-P2 as being important in exocytosis. J Biol Chem. 2000;275:17878–17885. doi: 10.1074/jbc.M000925200. [DOI] [PubMed] [Google Scholar]
- Homma Y, Takenawa T, Emori Y, Sorimachi H, Suzuki K. Tissue- and cell type-specific expression of mRNAs for four types of inositol phospholipid-specific phospholipase C. Biochem Biophys Res Commun. 1989;164:406–412. doi: 10.1016/0006-291x(89)91734-8. [DOI] [PubMed] [Google Scholar]
- Hurley JH, Meyer T. Subcellular targeting by membrane lipids. Curr Opin Cell Biol. 2001;13:146–152. doi: 10.1016/s0955-0674(00)00191-5. [DOI] [PubMed] [Google Scholar]
- Jahn R, Scheller RH. SNAREs—engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- James DJ, Khodthong C, Kowalchyk JA, Martin TF. Phosphatidylinositol 4,5-bisphosphate regulates SNARE-dependent membrane fusion. J Cell Biol. 2008;182:355–366. doi: 10.1083/jcb.200801056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jockusch WJ, Speidel D, Sigler A, Sorensen JB, Varoqueaux F, Rhee JS, Brose N. CAPS-1 and CAPS-2 are essential synaptic vesicle priming proteins. Cell. 2007;131:796–808. doi: 10.1016/j.cell.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Kanemaru K, Nakahara M, Nakamura Y, Hashiguchi Y, Kouchi Z, Yamaguchi H, Oshima N, Kiyonari H, Fukami K. Phospholipase C-eta2 is highly expressed in the habenula and retina. Gene Expr Patterns. 2010;10:119–126. doi: 10.1016/j.gep.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Kasai H, Takahashi N, Tokumaru H. Distinct initial SNARE configurations underlying the diversity of exocytosis. Physiol Rev. 2012;92:1915–1964. doi: 10.1152/physrev.00007.2012. [DOI] [PubMed] [Google Scholar]
- Khodthong C, Kabachinski G, James DJ, Martin TF. Munc13 homology domain-1 in CAPS/UNC31 mediates SNARE binding required for priming vesicle exocytosis. Cell Metab. 2011;14:254–263. doi: 10.1016/j.cmet.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler R, Heninger AK, Franke K, Habermann B, Buchholz F. Production of endoribonuclease-prepared short interfering RNAs for gene silencing in mammalian cells. Nat Methods. 2005;2:779–784. doi: 10.1038/nmeth1005-779. [DOI] [PubMed] [Google Scholar]
- Koch H, Hofmann K, Brose N. Definition of Munc13-homology-domains and characterization of a novel ubiquitously expressed Munc13 isoform. Biochem J. 2000;349:247–253. doi: 10.1042/0264-6021:3490247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch M, Holt M. Coupling exo- and endocytosis: an essential role for PIP(2) at the synapse. Biochim Biophys Acta. 2012;1821:1114–1132. doi: 10.1016/j.bbalip.2012.02.008. [DOI] [PubMed] [Google Scholar]
- Kuo W, Herrick DZ, Cafiso DS. Phosphatidylinositol 4,5-bisphosphate alters synaptotagmin 1 membrane docking and drives opposing bilayers closer together. Biochemistry. 2011;50:2633–2641. doi: 10.1021/bi200049c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner MR, Nurrish SJ, Kaplan JM. Facilitation of synaptic transmission by EGL-30 Gqalpha and EGL-8 PLCbeta: DAG binding to UNC-13 is required to stimulate acetylcholine release. Neuron. 1999;24:335–346. doi: 10.1016/s0896-6273(00)80848-x. [DOI] [PubMed] [Google Scholar]
- Laux T, Fukami K, Thelen M, Golub T, Frey D, Caroni P. GAP43, MARCKS, and CAP23 modulate PI(4,5)P(2) at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. J Cell Biol. 2000;149:1455–1472. doi: 10.1083/jcb.149.7.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SB, Varnai P, Balla A, Jalink K, Rhee SG, Balla T. The pleckstrin homology domain of phosphoinositide-specific phospholipase Cdelta4 is not a critical determinant of the membrane localization of the enzyme. J Biol Chem. 2004;279:24362–24371. doi: 10.1074/jbc.M312772200. [DOI] [PubMed] [Google Scholar]
- Li W, Ma C, Guan R, Xu Y, Tomchick DR, Rizo J. The crystal structure of a Munc13 C-terminal module exhibits a remarkable similarity to vesicle tethering factors. Structure. 2011;19:1443–1455. doi: 10.1016/j.str.2011.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, et al. Two distinct secretory vesicle-priming steps in adrenal chromaffin cells. J Cell Biol. 2010;190:1067–1077. doi: 10.1083/jcb.201001164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Schirra C, Stevens DR, Matti U, Speidel D, Hof D, Bruns D, Brose N, Rettig J. CAPS facilitates filling of the rapidly releasable pool of large dense-core vesicles. J Neurosci. 2008;28:5594–5601. doi: 10.1523/JNEUROSCI.5672-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loyet KM, Kowalchyk JA, Chaudhary A, Chen J, Prestwich GD, Martin TF. Specific binding of phosphatidylinositol 4,5-bisphosphate to calcium-dependent activator protein for secretion (CAPS), a potential phosphoinositide effector protein for regulated exocytosis. J Biol Chem. 1998;273:8337–8343. doi: 10.1074/jbc.273.14.8337. [DOI] [PubMed] [Google Scholar]
- Lynch KL, Gerona RR, Kielar DM, Martens S, McMahon HT, Martin TF. Synaptotagmin-1 utilizes membrane bending and SNARE binding to drive fusion pore expansion. Mol Biol Cell. 2008;19:5093–5103. doi: 10.1091/mbc.E08-03-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TF. PI(4,5)P(2) regulation of surface membrane traffic. Curr Opin Cell Biol. 2001;13:493–499. doi: 10.1016/s0955-0674(00)00241-6. [DOI] [PubMed] [Google Scholar]
- Martin TF. Tuning exocytosis for speed: fast and slow modes. Biochim Biophys Acta. 2003;1641:157–165. doi: 10.1016/s0167-4889(03)00093-4. [DOI] [PubMed] [Google Scholar]
- McLaughlin S, Murray D. Plasma membrane phosphoinositide organization by protein electrostatics. Nature. 2005;438:605–611. doi: 10.1038/nature04398. [DOI] [PubMed] [Google Scholar]
- Milosevic I, Sorensen JB, Lang T, Krauss M, Nagy G, Haucke V, Jahn R, Neher E. Plasmalemmal phosphatidylinositol-4,5-bisphosphate level regulates the releasable vesicle pool size in chromaffin cells. J Neurosci. 2005;25:2557–2565. doi: 10.1523/JNEUROSCI.3761-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara M, Shimozawa M, Nakamura Y, Irino Y, Morita M, Kudo Y, Fukami K. A novel phospholipase C, PLC(eta)2, is a neuron-specific isozyme. J Biol Chem. 2005;280:29128–29134. doi: 10.1074/jbc.M503817200. [DOI] [PubMed] [Google Scholar]
- Oancea E, Meyer T. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell. 1998;95:307–318. doi: 10.1016/s0092-8674(00)81763-8. [DOI] [PubMed] [Google Scholar]
- Olsen HL, et al. Phosphatidylinositol 4-kinase serves as a metabolic sensor and regulates priming of secretory granules in pancreatic beta cells. Proc Natl Acad Sci USA. 2003;100:5187–5192. doi: 10.1073/pnas.0931282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei J, Ma C, Rizo J, Grishin NV. Remote homology between Munc13 MUN domain and vesicle tethering complexes. J Mol Biol. 2009;391:509–517. doi: 10.1016/j.jmb.2009.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig J, Neher E. Emerging roles of presynaptic proteins in Ca++-triggered exocytosis. Science. 2002;298:781–785. doi: 10.1126/science.1075375. [DOI] [PubMed] [Google Scholar]
- Rhee JS, et al. Beta phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell. 2002;108:121–133. doi: 10.1016/s0092-8674(01)00635-3. [DOI] [PubMed] [Google Scholar]
- Rizo J, Rosenmund C. Synaptic vesicle fusion. Nat Struct Mol Biol. 2008;15:665–674. doi: 10.1038/nsmb.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Sigler A, Augustin I, Reim K, Brose N, Rhee JS. Differential control of vesicle priming and short-term plasticity by Munc13 isoforms. Neuron. 2002;33:411–424. doi: 10.1016/s0896-6273(02)00568-8. [DOI] [PubMed] [Google Scholar]
- Schramp M, Hedman A, Li W, Tan X, Anderson R. PIP kinases from the cell membrane to the nucleus. Subcell Biochem. 2012;58:25–59. doi: 10.1007/978-94-007-3012-0_2. [DOI] [PubMed] [Google Scholar]
- Shin OH, et al. Munc13 C2B domain is an activity-dependent Ca2+ regulator of synaptic exocytosis. Nat Struct Mol Biol. 2010;17:280–288. doi: 10.1038/nsmb.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Hille B. PIP2 is a necessary cofactor for ion channel function: how and why. Annu Rev Biophys. 2008;37:175–195. doi: 10.1146/annurev.biophys.37.032807.125859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaart G, Meyenberg K, Diederichsen U, Jahn R. Phosphatidylinositol 4,5-bisphosphate increases Ca2+ affinity of synaptotagmin-1 by 40-fold. J Biol Chem. 2012;287:16447–16453. doi: 10.1074/jbc.M112.343418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaart G, et al. Membrane protein sequestering by ionic protein-lipid interactions. Nature. 2011;479:552–555. doi: 10.1038/nature10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnai P, Thyagarajan B, Rohacs T, Balla T. Rapidly inducible changes in phosphatidylinositol 4,5-bisphosphate levels influence multiple regulatory functions of the lipid in intact living cells. J Cell Biol. 2006;175:377–382. doi: 10.1083/jcb.200607116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walent JH, Porter BW, Martin TF. A novel 145 kd brain cytosolic protein reconstitutes Ca(2+)-regulated secretion in permeable neuroendocrine cells. Cell. 1992;70:765–775. doi: 10.1016/0092-8674(92)90310-9. [DOI] [PubMed] [Google Scholar]
- Zhang L, Mao YS, Janmey PA, Yin HL. Phosphatidylinositol 4, 5 bisphosphate and the actin cytoskeleton. Subcell Biochem. 2012;59:177–215. doi: 10.1007/978-94-007-3015-1_6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.