

Abstract
Regulatory regions of the human genome can be modified through epigenetic processes during prenatal life to make an individual more likely to suffer chronic diseases when they reach adulthood. The modification of chromatin and DNA contributes to a larger well-documented process known as “programming” whereby stressors in the womb give rise to adult onset diseases, including cancer. It is now well known that death from ischemic heart disease is related to birth weight; the lower the birth weight, the higher the risk of death from cardiovascular disease as well as type 2 diabetes and osteoporosis. Recent epidemiological data link rapid growth in the womb to metabolic disease and obesity and also to breast and lung cancers. There is increasing evidence that “marked” regions of DNA can become “unmarked” under the influence of dietary nutrients. This gives hope for reversing propensities for cancers and other diseases that were acquired in the womb. For several cancers, the size and shape of the placenta are associated with a person’s cardiovascular and cancer risks as are maternal body mass index and height. The features of placental growth and nutrient transport properties that lead to adult disease have been little studied. In conclusion, several cancers have their origins in the womb, including lung and breast cancer. More research is needed to determine the epigenetic processes that underlie the programming of these diseases.
I. INTRODUCTION TO PROGRAMMING
Over the past 20 years, evidence for a link between environmental conditions in the womb and disease risk in offspring has been growing (Gluckman et al., 2008). The hypothesis that the intrauterine environment affects disease risk in adulthood now enjoys widespread support. While the associations between fetal growth and later adult disease may partly represent the pleiotropic effects of genes transmitted from mother to child, maternally mediated modulation of gene expression in offspring, through the environment that a mother’s body provides, appears to be more important than a purely heritable genetic risk. The association between an environmental stress in the womb and disease outcome in later life is called “programming” (Barker, 1998; Thornburg and Louey, 2005). Many stressors that lead to fetal programming have been identified including nutritional factors like over- and undernutrition, high corticosteroid exposure, and fetal hypoxia. In animal models and human studies alike, malnutrition in its various forms affects a host of development processes that manifest as disease in adulthood.
The programming story began when David Barker’s team in United Kingdom showed that the risk for mortality from ischemic heart disease was inversely related to the birth weight of residents of Hertfordshire, United Kingdom (Barker et al., 1989). This relationship showed a graded effect across the entire birth weight spectrum. The low birth weight–high adult disease risk relationship is now known to be very strong for many chronic conditions, including hypertension, coronary artery disease, type 2 diabetes, and osteoporosis (Gluckman et al., 2008). In animal models, any insult that reduces the flow of nutrients from mother to fetus leads to programmed offspring that suffer cardiovascular and metabolic disturbances for life (McMillen and Robinson, 2005).
While the link between nutrient flow and later disease is now documented in dozens of human and animal studies (Gluckman et al., 2008), the mechanisms by which environmental stressors in the womb alter the developing embryo and fetus remain a mystery (Fig. 3.1). Figure 3.1 shows, in diagrammatic form, the major steps and modifiers to which the genome is sensitive and that lead to programming. On the one hand, developmental plasticity allows an organism to adapt to environmental conditions to improve its odds of survival, but on the other hand, it allows for the modification of the epigenome in ways that increase the risk for chronic adult onset diseases. While it is now certain that epigenetic mechanisms are important in mediating the enduring effects of fetal malnutrition (Burdge et al., 2007), the extent to which epigenetic processes underlie even common chronic diseases in humans is unknown. Also, the extent to which epigenetic modifications can be reversed after birth is not known.
Figure 3.1.

Flow diagram illustrating the process of programming. Developmental plasticity allows a number of gene expression options during development. Stressors like malnutrition, excess cortisol, or hypoxia may lead to changes in gene expression in the embryo that predispose the offspring to disease in later life. The effect on the fetus will depend on its gender, its stage of gestation, the nutrient environment, and its genetic background. Female offspring may give birth to offspring that are programmed, repeating the cycle in the next generation.
In addition to epigenetic mechanisms, another accommodation to malnutrition in the fetus is the “trading off” of anatomic structures that then predispose to later disease. Perhaps the best example is the reduction in nephron number that accompanies a nutritional stress. Because nephron number in each kidney is set before birth in many animals and in people, an inadequate nephron endowment at birth cannot be reversed in later life. The variation in nephron number in the apparently normal human kidney is large, ranging from some 300,000 to greater than 1.8 million (Zandi-Nejad et al., 2006). Brenner hypothesized that a reduction in nephron number predicts the risk for adult onset systemic hypertension; animal studies generally support this hypothesis (Zandi-Nejad et al., 2006).
Additional compromised structural changes in the fetus are associated with future disease states and include reductions in liver size (Barker 2002; Gentili et al., 2009), skeletal muscle endowment (Baker et al., 2010), elastin in blood vessels (Martyn and Greenwald, 1997), and numbers of working cardio-myocytes in the heart (Jonker et al., 2010). However, many aspects of programming do not appear to be related to changes in organ structure. For example, changes in appetite, brain function, and tissue metabolism that accompany over-and undernutrition during fetal life appear to involve intricate but permanent changes in hormonal or cellular processes. For these, the mechanisms by which genes and environment interact must yet be determined.
It is fortunate that the relationship between fetal weight and disease outcome in adults was robust enough to be detected in Barker’s early studies because it was profoundly important in the discovery of the programming process. It is now known, however, that many stressors lead to programming without affecting fetal weight at birth. Thus, in any given birth weight category in a population, there would be a range of growth trajectories which different fetuses travel to gain that final weight, which raises the possibility that even cohorts with similar birth weights could have distinct disease predispositions. As the field becomes more sophisticated, it will undoubtedly become possible to unravel the varied pathways of fetal growth that leads to different disease outcomes. That process of discovery will necessarily include determining the effects of variations in the genome that predispose to programming effects. There is increasing evidence that genetic background modifies the effects of birth weight on later disease. Three examples will illustrate different degrees to which the genome and birth weight interact:
-
Eriksson et al. (2002( found that Pro12Pro and the Pro12Ala polymorphisms of the PPAR-γ2 gene affected insulin resistance in 152 elderly people depending upon their body size at birth. The Pro12Pro polymorphism of the PPAR-γ2 gene was associated with increased insulin resistance (P<0.002) and elevated insulin concentrations (P<0.003) only in individuals who had low birth weight. Individuals with the Pro12Ala polymorphism did not show the relationship.
Thus, there is an urgent need for studies of polymorphisms and their relationship to prenatal nutritional factors that are known to be associated with enduring risks for disease.
In another study, bone mineral density in the spine was found to be higher among people of genotype BB who were in the lowest third of the birth weight distribution compared in those with the Bb or bb genotype (P<0.01) after adjustment for age, sex, and current weight. However, people with the genotype BB and who were also in the highest birth weight category had the lowest bone mineral density compared with people with the Bb or bb genotype (P=0.04).
In a cohort study offemale twins (4000 subjects), significant intrapair correlation was found between birth weight and bone mass, even between monozygous twins (Dennison et al., 2001). This suggests that the intrauterine environment dominates the relationship between birth weight and bone mass compared to genomic inheritance.
II. PROGRAMMING AND CANCER
There is increasing evidence that the high birth weight baby is also programmed for vulnerability to adult onset diseases including cancer. For studies reported thus far, cancers have not been linked to low birth weight conditions. Macrosomic babies born to diabetic mothers have increased risks for obesity and type 2 diabetes and the metabolic syndrome in adulthood. Only in the past few years has evidence arisen linking adult onset cancers with intrauterine environment. The possibility that the origins of metastatic disease and metabolic disease are linked through common mechanisms that regulate prenatal growth has not been investigated thus far but would appear to be a fertile area of study.
In a recent Finnish study, adult onset lung cancer was linked to newborns with a large ponderal index (weight/length3) if the mother’s height was below the median. These data suggest that risks carried by birth size are modified by maternal phenotype (Eriksson et al., 2010). These studies also showed that the surface area of the chorionic plate of the delivered placenta was also related to the disease (Barker et al., 2010).
The relationship between birth weight and breast cancer has been studied more extensively than for other cancers. In several studies, birth weight has been shown to be positively associated with rates of breast cancer. There has been growing interest in the potential association between maternal dietary habits and adult breast cancer risk in offspring. In a study from the Finnish Birth Cohort, Barker et al. (2008a) showed that the width and roundness of a woman’s hips predicted breast cancer in her daughters. A similar relationship was found for ovarian cancer (Barker et al., 2008b). The hazard ratio for breast cancer in daughters was 3.7 (95% CI: 2.1–6.6) if the distance between the iliac crests was greater than 30 cm and if they were born at or after 40 weeks gestation. The shape of the female body pelvis is determined by the growth factors and hormones to which the pelvis is exposed during puberty. High pubertal levels of estrogen cause the pelvis to widen and become rounded.
If peak plasma estrogen levels are related to a girl’s nutrition during and preceding puberty, a high calorie diet during that period development could lead to pelvic changes and play an important role in her cancer risk and the risk of her offspring. Women with wide, round hips may impart a higher estrogen exposure to their embryos in the womb. One potential explanation for the relationship between pelvic size/shape and breast cancer in daughters is the exposure of early fetal breast stem cells to maternal estrogen. This hypothesis has been suggested (Barker et al., 2008a). While estrogen is known to be carcinogenic, a direct toxic effect of the hormone on the breast cell is not the only potential explanation for its actions. It is also possible that the high estrogen levels of the mother lead to epigenetic changes in affected breast tissues and that these changes lead to vulnerability for the cancer in later life. The role of epigenetic regulation of estrogen receptors is being investigated (Leader et al., 2006)
Several studies have been based on the hypothesis that a highly estrogenic environment will result in epigenetic modifications and increased breast cancer risk (Hilakivi-Clarke and de Assis 2006). Human studies have mostly targeted indirect measures of the fetal estrogen environment, including birth size, gestational age, and birth weight (as reviewed by Ruder et al., 2008). Thus, the field has not yet provided mechanistic answers to question regarding the role played by estrogens in the developing breast. A few studies have attempted to characterize maternal dietary intake, as it relates directly to adult breast cancer risk, they provide is evidence for increased risk in offspring with maternal consumption of fat and phytoestrogens such as genistein (Ruder et al., 2008).
There is evidence that other steroid hormones are influential in programming offspring through epigenetic mechanisms. For example, Meaney’s laboratory (McGowan et al., 2008) has shown that the programmed changes in brain function in newborn rat pups that occur in response to maternal licking are mediated by glucocorticoid actions. Increased levels of pup licking/grooming during the first week of life leads to increased expression of the glucocorticoid receptor (GR) in the hippocampus, augmented glucocorticoid feedback sensitivity, and suppressed hypothalamic pituitary stress responses compared to offspring reared by mothers who did not groom their offspring (Francis et al., 1999).
The epigenetic pathways in this sequence of events are known. 5-Hydroxytryptamine receptors in the rat pup hippocampus are activated by maternal licking and grooming. This leads to increased cAMP and phosphorylation of the transcription factor, NGFI-A, and the recruitment of histone acetyl-tranferase/creb-binding protein to the glucocorticoid exon 17 promoter. Acetylation of histone tails at the binding site facilitates its demethylation. In pups whose mothers do not groom, this gene regulation process is reduced, leading to differential epigenetic programming of the promoter region of the GR gene between pups. The HDAC inhibitor, TSA, increases histone acetylation and facilitates demethylation and epigenetic activation of the gene in the offspring of the low grooming mothers. On the other hand, the administration of methionine to adult offspring of attentive mothers leads to increased S-adenosylmethionine (SAM) inhibition of demethylation, increased DNA methylation, and reduced activity of the GR exon 17 promoter. Thus, the surprising part of the story is that the epigenetic state is reversible in the adult under specific dietary conditions.
The influence of the GR is highly significant for many aspects of programming because the GR is important to a number of developmental processes and it is so often subject to epigenetic regulation. For example, modification of GR expression has been documented in lung, liver, adrenal gland, and kidney in the offspring of animals that were malnourished during pregnancy (Brennan et al., 2005; Gnanalingham et al., 2005; Whorwood et al., 2001).
III. DYNAMIC CHANGES IN THE EPIGENOME DURING MAMMALIAN DEVELOPMENT
A longstanding conceptual problem in development biology is that only a small fraction of the genome is transcribed into RNA in a given cell type, and the specific fraction that is transcribed is different for different cell types. The epigenome explains how cell-specific gene expression occurs. The main components of the epigenome are DNA methylation and histone tail modifications, most notably lysine acetylation and methylation (Vaissiere et al., 2008). Lysine acetylation marks actively expressed regions of the genome, whereas histone methylation can mark transcribed or repressed regions depending upon the specific lysine residue that is modified.
While the genome is stable throughout life, the epigenome is dynamic (Turker, 1999). Establishment of an individual’s epigenome begins after fertilization when gamete-specific DNA methylation patterns are removed at different rates (Fulka et al., 2008). Paternal genome DNA methylation in humans and rodents is actively removed shortly after fertilization, before the first cell division, via an active process that directly removes methyl groups from cytosine bases or the methylated cytosine bases in toto. The specific mechanism has not yet been elucidated. In contrast, DNA methylation of the maternal genome is removed via a passive process that is believed to occur over several cell divisions because the maintenance DNA methyltransferase contributed by the oocyte (DNMT1o) cannot move into the nucleus of the developing embryo. The exception to this rule is at the eight-cell stage when DNMT1o moves into the nucleus, apparently to protect maternally transmitted imprinted loci (Howell et al., 2001).
The first cellular differentiation step occurs at about the time of blasto-cyst implantation when the trophoectoderm and inner cell mass (ICM) cells emerge (Howell et al., 2001). At that time widespread genomic DNA methylation returns, with higher levels in the ICM cells. A simplistic view of this process is that it is required to convert gamete specific methylation patterns to those that are initially compatible with totipotency and eventually consistent with differentiated cell types (Gopalakrishnan et al., 2008; Turker, 1999). Importantly, formation of somatic cell DNA methylation patterns continues during embryonic development, infancy, and childhood, as cellular growth and differentiation proceed (Gopalakrishnan et al., 2008). DNA methylation changes continue, albeit at a much slower pace, until death (Issa, 1999, 2000), though adult DNA methylation patterns are quite stable relative to the dynamic changes that occur during development.
The parental gametic genomes also exhibit different levels of histone modifications, in large part because paternal gametes use protamines instead of histones to package DNA. Shortly after fertilization, the protamines are replaced by maternally contributed histones, which are then available for modification. Early modifications during the first few cell divisions include methylation of histone 3 lysine 9 and histone 3 lysine 27; both are repressive marks. An additional modification that occurs is histone 3 lysine 4 methylation, which is an activating mark (Albert and Peters, 2009; Corry et al., 2009). Current thought is that the bivalent modification of both lysine 27 and lysine 4 methylation at a variety of loci allows for rapid activation or repression, respectively, during differentiation by removing a specific modification (Bernstein et al., 2006). The tight relation between DNA methylation and histone modifications (Vaissiere et al., 2008), and the presence of embryonic specific patterns of histone modifications (Balch et al., 2007; Ohm et al., 2007), likely leads to additional locus-specific changes in histone modifications, as fetal development continues into childhood development.
IV. ENVIRONMENTAL PERTURBATION OF THE EPIGENOME AND THE DEVELOPMENTAL ORIGINS OF DISEASE
As a general rule, it’s easier to move an object in motion than at rest. This rule could help provide a basis for the nascent field of environmental epigenetics, which simply means that environmental exposures can alter the epigenome, particularly during the dynamic phases of embryonic development (Baccarelli and Bollati, 2009, 2010). In other words, dynamic changes programmed to occur in the epigenome during development and then stabilize as an optimal epigenome in the adult could lead to a suboptimal epigenome if environmental exposures “moved” the developing epigenome. Alternatively, though not exclusively, the altered epigenome could at times represent an adaptive response to environmental exposure that conditions the fetus to give rise to an adult better adjusted to the conditions that it faced during development (Tang and Ho, 2007). According to one theory the fetus “assumes” that any unusual environmental condition it faces in utero will continue after birth, and therefore adapts itself for this condition (Gluckman and Hanson, 2004).
The adaptive response concept is consistent with the programming hypothesis (Barker, 1990), as demonstrated by the established relation between nutritional deprivation during fetal development and increased incidence of obesity, diabetes, and heart disease in the adult (Roseboom et al., 2001). One explanation for this phenomenon, termed the thrifty phenotype, is that the epigenome of a nutritionally deprived fetus is reset to more efficiently utilize food resources to improve its chances for survival in a challenged environment (Ross and Milner, 2007). Experimental evidence consistent with an epigenetic basis for this hypothesis is beginning to emerge. For example, blood DNA in persons conceived in the Netherlands during a war-induced famine in winter 1944–1945 exhibited decreased levels of DNA methylation at the imprinted IFG2 locus (Heijmans et al., 2009). This locus is normally expressed only from the maternally inherited allele and it plays an important role in growth and development. According to the adaptive hypothesis, increased expression throughout life would lead to a normal weight in the adult under conditions in which food resources were scarce, but would instead lead to obesity if food was readily available. Interestingly, famine exposure late in pregnancy had no effect on IGF2 methylation, suggesting that the relevant epigenome had stabilized by that time (Heijmans et al., 2009). Other genes with increased or decreased DNA methylation associated with nutritional deprivation in utero have also been reported, in some cases with gender specific changes (Dobosy et al., 2008; Tobi et al., 2009), suggesting that nutritional deprivation in utero leads to widespread modification of the epigenome.
The programming hypothesis, as put forward by Barker, assumes that an adaptive epigenetic response could subsequently have maladaptive consequences if environmental conditions change. However, the possibility also exists that pathological epigenetic changes could occur from toxic environmental exposures early in life (Baccarelli and Bollati, 2009, 2010). For example, the estrogenic drug, diethylstilbestrol (DES), was widely prescribed until 1971 based on the erroneous belief that it could prevent miscarriage (Langston, 2008); instead it turned into a “biological time bomb” (Herbst et al., 1971) causing a variety of reproductive problems in the daughters of exposed women (Rubin, 2007). The observed pathologies in daughters of exposed women may have been caused by epigenetic mechanisms, as borne out by results from rodent models (Bromer et al., 2009; Li et al., 1997; McLachlan, 2006; Miller et al., 1998; Ruden et al., 2005).
A story similar to that for DES may be playing out in the modern environment with the now ubiquitous endocrine disrupting chemical bisphenol-A, which has been shown to decrease DNA methylation of Hoxa10 gene in the uteri of female mice exposed in utero (Bromer et al., 2010). This effect is opposite to increased methylation induced by DES on this gene (Bromer et al., 2009). BPA exposure in utero also alters coat color in the agouti mouse model (Dolinoy et al., 2007b), which is a sensitive detector of changes in genomic DNA methylation (Dolinoy et al., 2007a). A variety of other environmental chemicals are believed to have epigenetic effects, and in some cases may have aberrant effects that last not only over the lifetime of an individual, but also over subsequent generations (Baccarelli and Bollati, 2009, 2010). Information describing the pathways by which these chemicals perturb the developing epigenome is clearly needed.
An additional, and again not exclusive, explanation for altered epigenomes in the environmentally exposed, developing fetus is based on the common observation that DNA methylation patterns are variable, even, for example, in identical twins (Fraga et al., 2005; Petronis et al., 2003). Thus, the epigenome of each cell is somewhat unique. A concept termed “epigenetic gambling” suggests that this variation allows preferential amplification of cells whose epigenomes can best survive in particular environments (Martin, 2009). The epigenetic gambling concept could help explain both adaptive changes in the epigenome, as envisioned for the programming hypothesis, and pathological changes that could occur from chemical exposures because in both cases selective processes (e.g., a toxic environmental exposure) coupled with variable epigenomes could lead to individuals with markedly different epigenomes as adults.
V. MECHANISMS INVOLVED IN DIETARY MODULATION OF EPIGENETIC MODIFICATION
While animal and human studies have linked dietary factors to epigenetic regulation, it has been challenging to determine the exact mechanisms that nutrients play in gene regulation. A number of biologically active constituents of food have been discovered including vitamins B6 and B12, methionine and folate (Chmurzynska, 2010). These compounds are potential methyl donors and deficiencies in these nutrients can affect DNA methylation status in mammals.
One carbon metabolism (Fig. 3.2) represents the best understood mechanism of diet regulated DNA methylation. In this scheme, methionine is generated by methylation of homocysteine via the folate- and B12-dependent methionine synthase reaction and the betaine-homocysteine methyltransferase reaction. Transfer of adenosine to methionine by methionine adenosyltransferase, gives the product SAM, which is the principal donor for a host of intracellular methyl transfer reactions. As a consequence of methyl group transfer, SAM is converted to S-adenosylhomocysteine (SAH), which binds with high affinity to methyltransferases and induces potent product inhibition (De Cabo et al., 1995). The ratio of SAM:SAH is, therefore, a crucial determinant of the methylation capacity. Perturbations in this system may be caused by dietary imbalances affecting the supply of methyl donors such as folic acid (FA) or by genetic polymorphisms affecting regulatory enzymes. Folate is not synthesized and is thus provided only by the diet. It has the potential to affect the SAM:SAH ratio and thereby an individual’s methylation status.
Figure 3.2.
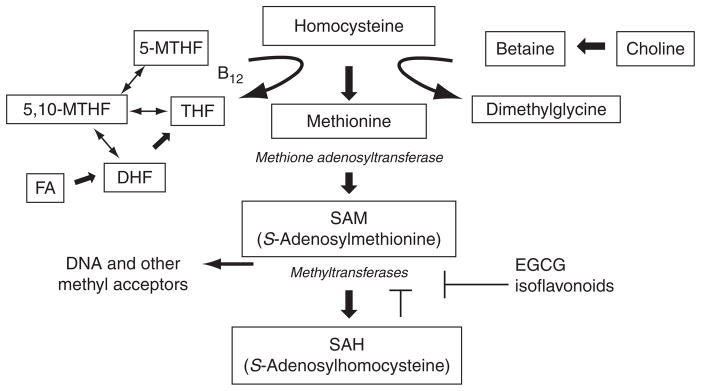
A summary of one-carbon metabolism and its role in DNA methylation (adapted from Johnson and Belshaw, 2008).
Epidemiological data suggest that low folate status is associated with a higher risk of colorectal cancer (Giovannucci, 2002) while data for other cancers remain unconclusive. As illustrated in Fig. 3.2, low folate status is likely to result in elevated levels of homocysteine and SAH and lead to DNA hypomethylation. Colonic adenocarcinoma cell lines and human adenocarcinoma cells (Stempak et al., 2005) demonstrate demethylation and remethylation of p53 when folate levels in the medium are altered (Wasson et al., 2006).
The application of animal studies to human disease is not always straightforward. Rats maintained on diets deficient in folate showed no methylation changes in the colon either genome wide or at specific genes (Choi et al., 2003; Duthie et al., 2000; Kim, 2005). In contrast, similar feeding studies deficient in methyl donors including folate displayed high variation in gene-specific DNA hypo- and hypermethylation in the liver (Pogribny et al., 1995, 2006). These differences in effect are probably attributed to modulation of expression of the DNA methylation machinery including DNMT enzymes and methyl CpG-binding proteins (Ghoshal et al., 2006). The SAM:SAH ratio may become different in colon versus liver in response to a methyl donor deficiency (Kim, 2005). A different effect was observed in mice in response to a folate deficient diet. Folate and choline deficiency in the ApcMin/+ mouse resulted in DNA hypomethylation in the small intestine and was correlated with tumor multiplicity (Sibani et al., 2002), though dietary factors may also play a role because a genetic deficiency in DNA methylation leads to decreased tumor formation in the ApcMin/+ mouse (Yamada et al., 2005). These variations in the results between animal models have added to the complexity and challenge in translating animal data to the human population.
Several folate deficiency studies have shown a correlation between DNA methylation status and cancers in colon (Pufulete et al., 2003, 2005), cervix (Fowler et al., 1998) and lung (Piyathilake et al., 2000). The variability in response to folate deficiency prompted the investigation of polymorphisms in the one carbon metabolism and association of the methylenetetrahydrofolate reducatse gene (MTHFR) responsible for the reduction of 5,10-methyleneterathydrofolate (5,10-MTHF) to 5-methyltertahydrofolate (5-MTHF) (Fig. 3.2). Two main polymorphisms in MTHFR have been identified; the carriers respond differently to folate deficiency with regard to polyp risk in the colon but not to other cancers (Giovannucci, 2002; Martinez et al., 2006).
Other substances including selenium (Davis and Uthus, 2002) and phytochemicals, such as epigallocatechin-3 gallate (Fang et al., 2003), alter DNA methylation status; both act through the inhibition of DNA methyltransferase (DNMT). Phytoestrogens, including the isoflavonoid genestein, may be beneficial prostate cancer prevention through its epigenetic actions (reviewed in Molinié and Georgel, 2009). The list of dietary compounds that have purported epigenetic regulatory actions is growing rapidly; most phytochemicals seem to regulate DNA methylation but others are known to affect histone acetylation.
While the mechanisms underlying global dietary regulation of DNA methylation remain unclear, even less is known about the regulation of histone acetylation by nutrients. However, this latter topic has been studied more intensely over the last few years, perhaps due to the now recognized interacting roles that methylation and acetylation play in regulating gene expression. Histone acetylation leads to chromatin remodeling and a derepression of transcription. Histone deacetylase (HDAC) inhibitors have become a recent focus for cancer prevention and therapy by virtue of their ability to “reactivate” the expression of epigenetically silenced genes, including those involved in differentiation, cell cycle regulation, apoptosis, angiogenesis, invasion, and metastasis (Dashwood et al., 2006). Several natural HDAC inhibitors have been shown to affect the growth and survival of tumor cells in vitro and in vivo. Among them three dietary chemopreventive agents, butyrate, diallyl disulfide, and sulforaphane, have HDAC inhibitory activity (Myzak and Dashwood, 2006). These compounds have been shown to protect against prostate cancer. Other dietary agents such as biotin, lipoic acid, garlic organosulfur compounds, and metabolites of vitamin E have structural features compatible with HDAC inhibition.
It is becoming clear that many dietary compounds and regiments acting as methyl donors or as inhibitors of enzymatic activity can be used to regulate epigenetic modifications. It is also clear that these same compounds have chemopreventive activities. The correlation between the two activities are becoming stronger although more studies will be needed to include the genetic effect of genetic polymorphisms on these events and how they may influence human disease, starting at conception.
VI. MATERNAL DIET AND RISK OF CANCER IN OFFSPRING: HUMAN STUDIES
Determining the association between maternal diet and the risk of cancer in offspring in humans presents a number of challenges, not least of which is logistic; connecting dietary habits during pregnancy to disease some years if not decades later is very difficult. However, once it was known that nutrient intake by a mother could alter fetal development and augment disease risk for life, the need for such research became apparent. Early studies focused on gestational dietary exposure to procarcinogens and their effects on the development of childhood tumors. Popular hypotheses were based upon prenatal exposure to carcinogens and the role of chemopreventive agents in DNA repair. These studies followed a classic model of carcinogen exposure and biologic response. More recent work investigating maternal diet as it relates to adult onset cancers such as breast cancer is shifting to an epigenetic model of cancer risk (Dalvai and Bystricky, 2010; Morrow et al., 2009).
There are a number of associations between compounds in the diet and later cancers. These are mentioned here because they are potential programming agents in development. Human studies of maternal nutrient intake and risk of acute lymphoblastic leukemia (ALL) have focused primarily on dietary compounds and combinations of foods suggested to impact DNA damage and DNA repair. Two of seven case control studies published since 1994 used short surveys focused primarily on meat intake (Peters et al., 1994; Sarasua and Savitz, 1994) and two looked only at vitamin supplement use (Shu et al., 1988; Thompson et al., 2001).
The remaining studies evaluated comprehensive nutrient intake during pregnancy using a food frequency questionnaire. Two of these investigations reported a reduction in risk for leukemias with greater fruit and vegetable intake (Jensen et al., 2004; Petridou et al., 2005). Ross et al. (1996) evaluated fruit and vegetables as sources of DNA topoisomerase II (DNAt2) inhibitors and reported an increase in the risk of acute myelogenous leukemia (AML) in infancy, but not ALL, with increasing fresh fruit and vegetable intake. A larger study by this group reported a reduction in risk of overall leukemia with an index of fresh fruits and vegetables and other dietary sources of DNAt2 inhibitors (Spector et al., 2005). Protective effects were also reported for supplemental use of a vitamin A and D fortified cod liver oil or folate (Shu et al., 1988; Thompson et al., 2001). Epigenetic events have not been the focus of this research, though the findings are consistent with the hypothesis that the associations with fruit and vegetable consumption and folate supplementation are related to the role of folate as a methyl donor or sulforphane, found in cruciferous vegetables, as an histone deacetylase inhibitor. Jensen et al. (2004) reported an unexpected reduction in risk of ALL with high maternal consumption of protein sources (Spector et al., 2005). While the authors suggest this may reflect higher levels of the antioxidant tripeptide glutathione, more recent work suggests that maternal low-protein diets may impact DNA methylation of a number of genes.
In comparison to studies on lymphoblastic leukemia, there has been a wider array of studies investigating the association between maternal dietary intake and pediatric brain tumors. Much of this work began by identifying the associations between consumption of dietary compounds that are known as neural carcinogens in animals, such as N-nitroso compounds and the occurrence of brain tumors. Cured meats commonly contain high amounts of N-nitroso compounds added during the curing process; hot dogs in particular have been implicated as a risk factor in brain tumor development. A meta-analysis published in 2004 reported on the findings of seven studies of maternal consumption of cured meats and risk of brain tumors in offspring (Huncharek and Kupelnick, 2004). This group reported a pooled 68% increase in relative risk of having a child with a brain tumor with increasing cured meat consumption. The carcino-genesis of N-nitroso compounds may be dramatically altered by intake of other bioactive dietary compounds such as the antioxidants in fruits and vegetables. A recent international case-control study found a reduction for risk of brain tumors in the children of mothers who consumed large amounts of yellow-orange vegetables, fresh fish, and grains (Pogoda et al., 2009).
High maternal fruit and vegetable consumption during pregnancy was evaluated in a case-control study of retinoblastoma (Orjuela et al., 2005). The investigators found a reduction in the risk of sporadic retinoblastoma in offspring of mothers with higher fruit and vegetable consumption and consequently, higher folate intake. These investigators argued for an adequate folate status during pregnancy to protect against increased uracil misincorporation or hypomethylation.
VII. THE PLACENTA AS A PROGRAMMING FACTOR
The placenta is the organ through which the embryo and fetus must acquire its nutrients. The size and shape of an individual’s placenta are associated with increased risks for high blood pressure, heart failure and lung cancer (Barker et al., 2010). These relationships persist even in cases where birth weight is not associated with risk. The mechanisms by which placental growth powerfully affects disease risk are not known. However, a common feature of the placenta-disease risk relationship is its dependence upon maternal phenotype, typically her body mass index and/or height. The effects of these phenotypic traits can be further dependent upon the sex of the offspring. In addition, it is clear that the independent phenotypic traits of the placenta itself have predictive value. This suggests that placental thickness and dimensions are related to independent biological processes that affect fetal development.
The molecules that regulate epigenetic actions in the fetus are transferred from the mother by receptor mediated transport proteins. Essential amino acids like methionine are actively transported by several independent transporter systems (Desforges and Sibley, 2010). The components of folate metabolism and transport have been recently investigated (Solanky et al., 2010). Figure 3.3 shows the potential positions of transporters and key metabolic enzymes for one carbon metabolism in the human placenta. These studies demonstrate that the folate transport system and the homocysteine metabolism system are established early in gestation. The transport of methionine and folate are dependent upon a continuous maternal supply. More research is required to understand how molecules that are important in fetal epigenetic processes are acquired by the placenta.
Figure 3.3.

Proposed model for folate transport across human placental syncytiotrophoblast. Folate receptor alpha (FRa), localized to the microvillous membrane (MVM) surface binds 5-MTHF (folate). Colocalization of FRa and PCFT to MVM allows internalization of both transporters into an endosomal structure. Following acidification of the endosome by vacuolar proton ATPase, a favorable H+ gradient exists allowing the H+-coupled movement of folate by PCFT into the cytoplasm. FRa and PCFT are then recycled back to the MVM surface. RFC at the MVM surface provides an alternative folate uptake mechanism which would be favored at physiological pH. Efflux across the BM does not involve FRa or PCFT. Instead, folate is transported across BM via an exchange mechanism by RFC. Other transport mechanisms localized to BM may also play a role in transporting folate across the basal plasma membrane. From Solankey et al. (2004) with permission.
VIII. CONCLUSIONS
Programming is the process by which stressors in the womb lead to disease in the offspring. The programming effect is known to be a powerful process that may account for a large portion of chronic disease in populations world-wide. It also explains, in part, why diseases come and go over the ages. The inverse relationship between term birth weight and cardiovascular disease, obesity, type 2 diabetes, and osteoporosis is due to the developmental plasticity which allows alternate expression patterns in the embryo and fetus. These patterns of adaptation may have “predictive” survival value. Depending on the stage of gestation, the changes in gene expression may stabilize at slightly aberrant levels that lead to permanent structural changes in the developing organs. The under endowment of nephrons in the kidney is an example. One mechanism by which plasticity might bring enduring changes is the epigenetic modification of gene function. Thus, vulnerabilities for cancer may arise in the womb. Understanding the mechanisms that underlie epigenetic changes in the womb may offer preventative and therapeutic strategies in the future.
Acknowledgments
This work was supported by the National Institutes of Health and the M. Lowell Edwards Endowment.
References
- Albert M, Peters AH. Genetic and epigenetic control of early mouse development. Curr Opin Genet Dev. 2009;19:113–121. doi: 10.1016/j.gde.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Antoniades L, MacGregor AJ, Andrew T, Spector TD. Association of birth weight with osteoporosis and osteoarthritis in adult twins. Rheumatology. 2003;42:791–796. doi: 10.1093/rheumatology/keg227. [DOI] [PubMed] [Google Scholar]
- Baccarelli A, Bollati V. Epigenetics and environmental chemicals. Curr Opin Pediatr. 2009;21:243–251. doi: 10.1097/mop.0b013e32832925cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker J, Workman M, Bedrick E, Frey MA, Hurtado M, Pearson O. Brains versus brawn: An empirical test of Barker’s brain sparing model. Am J Hum Biol. 2010;22:206–215. doi: 10.1002/ajhb.20979. [DOI] [PubMed] [Google Scholar]
- Balch C, Nephew KP, Huang TH, Bapat SA. Epigenetic “bivalently marked” process of cancer stem cell-driven tumorigenesis. Bioessays. 2007;29:842–845. doi: 10.1002/bies.20619. [DOI] [PubMed] [Google Scholar]
- Barker DJ. The fetal and infant origins of adult disease. BMJ. 1990;301:1111. doi: 10.1136/bmj.301.6761.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJP. Mothers, Babies and Health in Later Life. Churchill Livingstone; Edinburgh: 1998. [Google Scholar]
- Barker DJ. Fetal programming of coronary heart disease. Trends Endocrinol Metab. 2002;13:364–368. doi: 10.1016/s1043-2760(02)00689-6. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Winter PD, Osmond C. Weight in infancy and death from ischaemic heart disease. Lancet. 1989;ii:577–580. doi: 10.1016/s0140-6736(89)90710-1. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Osmond C, Thornburg KL, Kajantie E, Forsen TJ, Eriksson JG. A possible link between the pubertal growth of girls and breast cancer in their daughters. Am J Hum Biol. 2008a;20:127–131. doi: 10.1002/ajhb.20688. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Osmond C, Thornburg KL, Kajantie E, Eriksson JG. A possible link between the pubertal growth of girls and ovarian cancer in their daughters. Am J Hum Biol. 2008b;20:659–662. doi: 10.1002/ajhb.20789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ, Thornburg KL, Osmond C, Kajantie E, Eriksson JG. The prenatal origins of lung cancer. II. The placenta. Am J Hum Biol. 2010 doi: 10.1002/ajhb.21041. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Bollati V, Baccarelli A. Environmental epigenetics. Heredity. 2010 doi: 10.1038/hdy.2010.2. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan KA, Gopalakrishnan GS, Kurlak L, Rhind SM, Kyle CE, Brooks AN, Rae MT, Olson DM, Stephenson T, Symonds ME. Impact of maternal undernutrition and fetal number on glucocorticoid, growth hormone and insulin-like growth factor receptor mRNA abundance in the ovine fetal kidney. Reproduction. 2005;129:151–159. doi: 10.1530/rep.1.00229. [DOI] [PubMed] [Google Scholar]
- Bromer JG, Wu J, Zhou Y, Taylor HS. Hypermethylation of homeobox A10 by in utero diethylstilbestrol exposure: An epigenetic mechanism for altered developmental programming. Endocrinology. 2009;150:3376–3382. doi: 10.1210/en.2009-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromer JG, Zhou Y, Taylor MB, Doherty L, Taylor HS. Bisphenol-A exposure in utero leads to epigenetic alterations in the developmental programming of uterine estrogen response. FASEB J. 2010 doi: 10.1096/fj.09-140533. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdge GC, Hanson MA, Slater-Jefferies JL, Lillycrop KA. Epigenetic regulation of transcription: A mechanism for inducing variations in phenotype (fetal programming) by differences in nutrition during early life? Br J Nutr. 2007;97:1036–1046. doi: 10.1017/S0007114507682920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmurzynska A. Fetal programming: Link between early nutrition, DNA methylation, and complex diseases. Nutr Rev. 2010;68:87–98. doi: 10.1111/j.1753-4887.2009.00265.x. [DOI] [PubMed] [Google Scholar]
- Choi SW, Friso S, Dolnikowski GG, Bagley PJ, Edmondson AN, Smith DE, Mason JB. Biochemical and molecular aberrations in the rat colon due to folate depletion are age-specific. J Nutr. 2003;133:1206–1212. doi: 10.1093/jn/133.4.1206. [DOI] [PubMed] [Google Scholar]
- Corry GN, Tanasijevic B, Barry ER, Krueger W, Rasmussen TP. Epigenetic regulatory mechanisms during preimplantation development. Birth Defects Res C Embryo Today. 2009;87:297–313. doi: 10.1002/bdrc.20165. [DOI] [PubMed] [Google Scholar]
- Dashwood RH, Myzak MC, Ho E. Dietary HDAC inhibitors: Time to rethink weak ligands in cancer chemoprevention? Carcinogenesis. 2006;27:344–349. doi: 10.1093/carcin/bgi253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CD, Uthus EO. Dietary selenite and azadeoxycytidine treatments affect dimethylhydrazine-induced aberrant crypt formation in rat colon and DNA methylation in HT-29 cells. J Nutr. 2002;132:292–297. doi: 10.1093/jn/132.2.292. [DOI] [PubMed] [Google Scholar]
- De Cabo SF, Santos J, Fernandez-Piqueras J. Molecular and cytological evidence of S-adenosyl-L-homocysteine as an innocuous undermethylating agent in vivo. Cytogenet Cell Genet. 1995;71:187–192. doi: 10.1159/000134104. [DOI] [PubMed] [Google Scholar]
- Dennison EM, Arden NK, Keen RW, et al. Birthweight, vitamin D receptor genotype and the programming of osteoporosis. Paediatr Perinat Epidemiol. 2001;15:211–219. doi: 10.1046/j.1365-3016.2001.00350.x. [DOI] [PubMed] [Google Scholar]
- Desforges M, Sibley CP. Placental nutrient supply and fetal growth. Int J Dev Biol. 2010;54:377–390. doi: 10.1387/ijdb.082765md. [DOI] [PubMed] [Google Scholar]
- Dobosy JR, Fu VX, Desotelle JA, Srinivasan R, Kenowski ML, Almassi N, Weindruch R, Savren J, Jarrardm DF. A methyl-deficient diet modifies histone methylation and alters lgf2 and H19 repression in the prostate. Prostate. 2008;68:1187–1195. doi: 10.1002/pros.20782. [DOI] [PubMed] [Google Scholar]
- Dolinoy DC, Das R, Weidman JR, Jirtle RL. Metastable epialleles, imprinting, and the fetal origins of adult diseases. Pediatr Res. 2007a;61:30R–37R. doi: 10.1203/pdr.0b013e31804575f7. [DOI] [PubMed] [Google Scholar]
- Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci USA. 2007b;104:13056–13061. doi: 10.1073/pnas.0703739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duthie SJ, Grant G, Narayanan S. Increased uracil misincorporation in lymphocytes from folate-deficient rats. Br J Cancer. 2000;83:1532–1537. doi: 10.1054/bjoc.2000.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson JG, Lindi V, Uusitupa M, Forsén TJ, Laakso M, Osmond C, Barker DJ. The effects of the Pro12Ala polymorphism of the peroxisome proliferator-activated receptor-gamma2 gene on insulin sensitivity and insulin metabolism interact with size at birth. Diabetes. 2002;51:2321–2324. doi: 10.2337/diabetes.51.7.2321. [DOI] [PubMed] [Google Scholar]
- Eriksson JG, Thornburg KL, Osmond C, Kajantie E, Barker DJ. The prenatal origins of lung cancer. I. The fetus. Am J Hum Biol. 2010 doi: 10.1002/ajhb.21040. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, Welsh W, Yang CS. Tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003;63:7563–7570. [PubMed] [Google Scholar]
- Fowler BM, Giuliano AR, Piyathilake C, Nour M, Hatch K. Hypomethylation in cervical tissue: Is there a correlation with folate status? Cancer Epidemiol Biomarkers Prev. 1998;7:901–906. [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis DD, Caldji C, Champagne F, Plotsky PM, Meaney MJ. The role of corticotropin-releasing factor—Norepinephrine systems in mediating the effects of early experience on the development of behavioral and endocrine responses to stress. Biol Psychiatry. 1999;46:1153–1166. doi: 10.1016/s0006-3223(99)00237-1. [DOI] [PubMed] [Google Scholar]
- Fulka H, St John JC, Fulka J, Hozak P. Chromatin in early mammalian embryos: Achieving the pluripotent state. Differentiation. 2008;76:3–14. doi: 10.1111/j.1432-0436.2007.00247.x. [DOI] [PubMed] [Google Scholar]
- Gentili S, Morrison JL, McMillen IC. Intrauterine growth restriction and differential patterns of hepatic growth and expression of IGF1, PCK2, and HSDL1 mRNA in the sheep fetus in late gestation. Biol Reprod. 2009;80:1121–1127. doi: 10.1095/biolreprod.108.073569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoshal K, Li X, Datta J, Bai S, Pogribny I, Pogribny M, Huang Y, Young D, Jacob ST. A folate- and methyl-deficient diet alters the expression of DNA methyl-transferases and methyl CpG binding proteins involved in epigenetic gene silencing in livers of F344 rats. J Nutr. 2006;136:1522–1527. doi: 10.1093/jn/136.6.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannucci E. Epidemiologic studies of folate and colorectal neoplasia: A review. J Nutr. 2002;132:2350S–2355S. doi: 10.1093/jn/132.8.2350S. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA. Developmental origins of disease paradigm: A mechanistic and evolutionary perspective. Pediatr Res. 2004;56:311–317. doi: 10.1203/01.PDR.0000135998.08025.FB. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359:61–73. doi: 10.1056/NEJMra0708473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnanalingham MG, Mostyn A, Dandrea J, Yakubu DP, Symonds ME, Stephenson T. Ontogeny and nutritional programming of uncoupling protein-2 and glucocorticoid receptor mRNA in the ovinelung. J Physiol. 2005;565:159–169. doi: 10.1113/jphysiol.2005.083246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan S, Van Emburgh BO, Robertson KD. DNA methylation in development and human disease. Mutat Res. 2008;647:30–38. doi: 10.1016/j.mrfmmm.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijmans BT, Tobi EW, Lumey LH, Slagboom PE. The epigenome: Archive of the prenatal environment. Epigenetics. 2009;4:526–531. doi: 10.4161/epi.4.8.10265. [DOI] [PubMed] [Google Scholar]
- Herbst AL, Ulfelder H, Poskanzer DC. Adenocarcinoma of the vagina. Association of maternal stilbestrol therapy with tumor appearance in young women. N Engl J Med. 1971;284:878–881. doi: 10.1056/NEJM197104222841604. [DOI] [PubMed] [Google Scholar]
- Hilakivi-Clarke L, de Assis S. Fetal origins of breast cancer. Trends Endocrinol Metab. 2006;17:340–348. doi: 10.1016/j.tem.2006.09.002. [DOI] [PubMed] [Google Scholar]
- Howell CY, Bestor TH, Ding F, Latham KE, Mertineit C, Trasler JM, Chaillet JR. Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell. 2001;104:829–838. doi: 10.1016/s0092-8674(01)00280-x. [DOI] [PubMed] [Google Scholar]
- Huncharek M, Kupelnick B. A meta-analysis of maternal cured meat consumption during pregnancy and the risk of childhood brain tumors. Neuroepidemiology. 2004;23:78–84. doi: 10.1159/000073979. [DOI] [PubMed] [Google Scholar]
- Issa JP. Aging, DNA methylation and cancer. Crit Rev Oncol Hematol. 1999;32:31–43. doi: 10.1016/s1040-8428(99)00019-0. [DOI] [PubMed] [Google Scholar]
- Issa JP. CpG-island methylation in aging and cancer. Curr Top Microbiol Immunol. 2000;249:101–118. doi: 10.1007/978-3-642-59696-4_7. [DOI] [PubMed] [Google Scholar]
- Jensen CD, Block G, Buffler P, Ma X, Selvin S, Month S. Maternal dietary risk factors in childhood acute lymphoblastic leukemia (United States) Cancer Causes Control. 2004;15:559–570. doi: 10.1023/B:CACO.0000036161.98734.17. [DOI] [PubMed] [Google Scholar]
- Johnson IT, Belshaw NJ. Environment, diet and CpG island methylation: Epigenetic signals in gastrointestinal neoplasia. Food Chem Toxicol. 2008;46:1346–1359. doi: 10.1016/j.fct.2007.09.101. [DOI] [PubMed] [Google Scholar]
- Jonker SS, Giraud MK, Giraud GD, Chattergoon NN, Louey S, Davis LE, Faber JJ, Thornburg KL. Cardiomyocyte enlargement, proliferation and maturation during chronic fetal anaemia in sheep. Exp Physiol. 2010;95:131–139. doi: 10.1113/expphysiol.2009.049379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YI. Nutritional epigenetics: Impact of folate deficiency on DNA methylation and colon cancer susceptibility. J Nutr. 2005;13:2703–2709. doi: 10.1093/jn/135.11.2703. [DOI] [PubMed] [Google Scholar]
- Langston N. The Retreat from precaution: Regulating diethylstilbestrol (DES), endocrine disruptors, and environmental health. Environ Hist. 2008;13:1–26. [Google Scholar]
- Li S, Washburn KA, Moore R, Uno T, Teng C, Newbold RR, McLachlan JA, Negishi M. Developmental exposure to diethylstilbestrol elicits demethylation of estrogen-responsive lactoferrin gene in mouse uterus. Cancer Res. 1997;57:4356–4359. [PubMed] [Google Scholar]
- Martin GM. Epigenetic gambling and epigenetic drift as an antagonistic pleiotropic mechanism of aging. Aging Cell. 2009;8:761–764. doi: 10.1111/j.1474-9726.2009.00515.x. [DOI] [PubMed] [Google Scholar]
- Martinez ME, Thompson P, Jacobs ET, Giovannucci E, Jiang R, Klimecki W, Alberts DS. Dietary factors and biomarkers involved in the methylenetetrahydrofolate reductase genotype-colorectal adenoma pathway. Gastroenterology. 2006;131:1706–1716. doi: 10.1053/j.gastro.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Martyn CN, Greenwald SE. Impaired synthesis of elastin in walls of aorta and large conduit arteries during early development as an initiating event in pathogenesis of systemic hypertension. Lancet. 1997;350:953–955. doi: 10.1016/s0140-6736(96)10508-0. [DOI] [PubMed] [Google Scholar]
- McGowan PO, Meaney MJ, Szyf M. Diet and the epigenetic (re)programming of phenotypic differences in behavior. Brain Res. 2008;1237:12–24. doi: 10.1016/j.brainres.2008.07.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan JA. Commentary: Prenatal exposure to diethylstilbestrol (DES): A continuing story. Int J Epidemiol. 2006;35:868–870. doi: 10.1093/ije/dyl140. [DOI] [PubMed] [Google Scholar]
- McMillen IC, Robinson JS. Developmental origins of the metabolic syndrome: Prediction, plasticity, and programming. Physiol Rev. 2005;85:571–633. doi: 10.1152/physrev.00053.2003. [DOI] [PubMed] [Google Scholar]
- Miller C, Degenhardt K, Sassoon DA. Fetal exposure to DES results in de-regulation of Wnt7a during uterine morphogenesis. Nat Genet. 1998;20:228–230. doi: 10.1038/3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinié B, Georgel P. Genetic and epigenetic regulations of prostate cancer by genistein. Drug News Perspect. 2009;22:247–254. doi: 10.1358/dnp.2009.22.5.1378633. [DOI] [PubMed] [Google Scholar]
- Myzak M, Dashwood RH. Histone deacetylases as targets for dietary cancer preventive agents: Lessons learned with butyrate, diallyl disulfide, and sulforaphane. Curr Drug Targets. 2006;7:443–452. doi: 10.2174/138945006776359467. [DOI] [PubMed] [Google Scholar]
- Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007;39:237–242. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orjuela MA, Titievsky L, Liu X, Ramirez-Ortiz M, Ponce-Castaneda V, Lecona E, Molina E, Beaverson K, Abramson DH, Mueller NE. Fruit and vegetable intake during pregnancy and risk for development of sporadic retinoblastoma. Cancer Epidemiol Biomarkers Prev. 2005;14:1433–1440. doi: 10.1158/1055-9965.EPI-04-0427. [DOI] [PubMed] [Google Scholar]
- Peters JM, Preston-Martin S, London SJ, Bowman JD, Buckley JD, Thomas DC. Processed meats and risk of childhood leukemia (California, USA) Cancer Causes Control. 1994;5:195–202. doi: 10.1007/BF01830266. [DOI] [PubMed] [Google Scholar]
- Petridou E, Ntouvelis E, Dessypris N, Terzidis A, Trichopoulos D. Maternal diet and acute lymphoblastic leukemia in young children. Cancer Epidemiol Biomarkers Prev. 2005;14:1935–1939. doi: 10.1158/1055-9965.EPI-05-0090. [DOI] [PubMed] [Google Scholar]
- Petronis A, Gottesman II, Kan P, Kennedy JL, Basile VS, Paterson AD, Popendikyte V. Monozygotic twins exhibit numerous epigenetic differences: Clues to twin discordance? Schizophr Bull. 2003;29:169–178. doi: 10.1093/oxfordjournals.schbul.a006988. [DOI] [PubMed] [Google Scholar]
- Piyathilake CJ, Johanning GL, Macaluso M, Whiteside M, Oelschlager DK, Heimburger DC, Grizzle WE. Localized folate and vitamin B-12 deficiency in squamous cell lung cancer is associated with global DNA hypomethylation. Nutr Cancer. 2000;37:99–107. doi: 10.1207/S15327914NC3701_13. [DOI] [PubMed] [Google Scholar]
- Pogoda JM, Preston-Martin S, Howe G, Lubin F, Mueller BA, Holly EA, Filippini G, Peris-Bonet R, McCredie MR, Cordier S, Choi W. An international case-control study of maternal diet during pregnancy and childhood brain tumor risk: A histology-specific an alysis by food group. Ann Epidemiol. 2009;19:148–160. doi: 10.1016/j.annepidem.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogribny IP, Basnakian AG, Miller BJ, Lopatina NG, Poirier LA, James SJ. Breaks in genomic DNA and within the p53 gene are associated with hypomethylation in livers of folate/methyl-deficient rats. Cancer Res. 1995;55:1894–1901. [PubMed] [Google Scholar]
- Pogribny IP, Ross SA, Wise C, Pogribna M, Jones EA, Tryndyak VP, James SJ, Dragan YP, Poirier LA. Irreversible global DNA hypomethylation as a key step in hepatocarcinogenesis induced by dietary methyl deficiency. Mutat Res. 2006;593:80–87. doi: 10.1016/j.mrfmmm.2005.06.028. [DOI] [PubMed] [Google Scholar]
- Pufulete M, Al-Ghnaniem R, Leather AJ, Appleby P, Gout S, Terry C, Emery PW, Sanders TA. Folate status, genomic DNA hypomethylation, and risk of colorectal adenoma and cancer: A case control study. Gastroenterology. 2003;124:1240–1248. doi: 10.1016/s0016-5085(03)00279-8. [DOI] [PubMed] [Google Scholar]
- Pufulete M, Al-Ghnaniem R, Rennie JA, Appleby P, Harris N, Gout S, Emery PW, Sanders TA. Influence of folate status on genomic DNA methylation in colonic mucosa of subjects without colorectal adenoma or cancer. Br J Cancer. 2005;92:838–842. doi: 10.1038/sj.bjc.6602439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseboom TJ, van der Meulen JH, Ravelli AC, Osmond C, Barker DJ, Bleker OP. Effects of prenatal exposure to the Dutch famine on adult disease in later life: An overview. Twin Res. 2001;4:293–298. doi: 10.1375/1369052012605. [DOI] [PubMed] [Google Scholar]
- Ross SA, Milner JA. Epigenetic modulation and cancer: Effect of metabolic syndrome? Am J Clin Nutr. 2007;86:872–877. doi: 10.1093/ajcn/86.3.872S. [DOI] [PubMed] [Google Scholar]
- Ross JA, Potter JD, Reaman GH, Pendergrass TW, Robison LL. Maternal exposure to potential inhibitors of DNA topoisomerase II and infant leukemia (United States): A report from the Children’s Cancer Group. Cancer Causes Control. 1996;7:581–590. doi: 10.1007/BF00051700. [DOI] [PubMed] [Google Scholar]
- Rubin MM. Antenatal exposure to DES: Lessons learned…future concerns. Obstet Gynecol Surv. 2007;62:548–555. doi: 10.1097/01.ogx.0000271138.31234.d7. [DOI] [PubMed] [Google Scholar]
- Ruden DM, Xiao L, Garfinkel MD, Lu X. Hsp90 and environmental impacts on epigenetic states: A model for the trans-generational effects of diethylstibesterol on uterine development and cancer. Hum Mol Genet. 2005;14:R149–R155. doi: 10.1093/hmg/ddi103. [DOI] [PubMed] [Google Scholar]
- Ruder EH, Dorgan JF, Kranz S, Kris-Etherton PM, Hartman TJ. Examining breast cancer growth and lifestyle risk factors: Early life, childhood, and adolescence. Clin Breast Cancer. 2008;8:334–342. doi: 10.3816/CBC.2008.n.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarasua S, Savitz DA. Cured and broiled meat consumption in relation to childhood cancer: Denver, Colorado (United States) Cancer Causes Control. 1994;5:141–148. doi: 10.1007/BF01830260. [DOI] [PubMed] [Google Scholar]
- Shu XO, Gao YT, Brinton LA, Linet MS, Tu JT, Zheng W, Fraumeni JF., Jr A population-based case-control study of childhood leukemia in Shanghai. Cancer. 1988;62:635–644. doi: 10.1002/1097-0142(19880801)62:3<635::aid-cncr2820620332>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Sibani S, Melnyk S, Pogribny IP, Wang W, Hiou-Tim F, Deng L, Trasler J, James SJ, Rozen R. Studies of methionine cycle intermediates (SAM, SAH), DNA methylation and the impact of folate deficiency on tumor numbers in Min mice. Carcinogenesis. 2002;23:61–65. doi: 10.1093/carcin/23.1.61. [DOI] [PubMed] [Google Scholar]
- Solanky N, Requena Jimenez A, D’Souza SW, Sibley CP, Glazier JD. Expression of folate transporters in human placenta and implications for homocysteine metabolism. Placenta. 2010;31:134–143. doi: 10.1016/j.placenta.2009.11.017. [DOI] [PubMed] [Google Scholar]
- Spector LG, Xie Y, Robison LL, Heerema NA, Hilden JM, Lange B, Felix CA, Davies SM, Slavin J, Potter JD, Blair CK, Reaman GH, et al. Maternal diet and infant leukemia: The DNA topoisomerase II inhibitor hypothesis: A report from the children’s oncology group. Cancer Epidemiol Biomarkers Prev. 2005;14:651–655. doi: 10.1158/1055-9965.EPI-04-0602. [DOI] [PubMed] [Google Scholar]
- Stempak JM, Sohn KJ, Chiang EP, Shane B, Kim YI. Cell and stage of transformation-specific effects of folate deficiency on methionine cycle intermediates and DNA methylation in an in vitro model. Carcinogenesis. 2005;26:981–990. doi: 10.1093/carcin/bgi037. [DOI] [PubMed] [Google Scholar]
- Tang WY, Ho SM. Epigenetic reprogramming and imprinting in origins of disease. Rev Endocr Metab Disord. 2007;8:173–182. doi: 10.1007/s11154-007-9042-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JR, Gerald PF, Willoughby ML, Armstrong BK. Maternal folate supplementation in pregnancy and protection against acute lymphoblastic leukaemia in childhood: A case-control study. Lancet. 2001;358:1935–1940. doi: 10.1016/S0140-6736(01)06959-8. [DOI] [PubMed] [Google Scholar]
- Thornburg KL, Louey S. Fetal roots of cardiac disease. Heart. 2005;91:867–868. doi: 10.1136/hrt.2004.047407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, Slagboom PE, Heijmans BT. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. 2009;18:4046–4053. doi: 10.1093/hmg/ddp353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turker MS. The establishment and maintenance of DNA methylation patterns in mouse somatic cells. Semin Cancer Biol. 1999;9:329–337. doi: 10.1006/scbi.1999.0133. [DOI] [PubMed] [Google Scholar]
- Vaissiere T, Sawan C, Herceg Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res. 2008;659:40–48. doi: 10.1016/j.mrrev.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Wasson GR, McGlynn AP, McNulty H, O’Reilly SL, McKelvey-Martin VJ, McKerr G, Strain JJ, Scott J, Downes CS. Global DNA and p53 region-specific hypomethylation in human colonic cells is induced by folate depletion and reversed by folate supplementation. J Nutr. 2006;136:2748–2753. doi: 10.1093/jn/136.11.2748. [DOI] [PubMed] [Google Scholar]
- Whorwood CB, Firth KM, Budge H, Symonds ME. Maternal undernutrition during early to midgestationprograms tissue-specific alterations in the expression of the glucocorticoid receptor, 11 betahydroxysteroiddehydrogenase isoforms, and type 1 angiotensin ii receptor in neonatal sheep. Endocrinology. 2001;142:2854–2864. doi: 10.1210/endo.142.7.8264. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Jackson-Grusby L, Linhart H, Meissner A, Eden A, Lin H, Jaenisch R. Opposing effects of DNA hypomethylation on intestinal and liver carcinogenesis. Proc Natl Acad Sci USA. 2005;102:13580–13585. doi: 10.1073/pnas.0506612102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandi-Nejad K, Luyckx VA, Brenner BM. Adult hypertension and kidney disease: The role of fetal programming. Hypertension. 2006;47:502–508. doi: 10.1161/01.HYP.0000198544.09909.1a. [DOI] [PubMed] [Google Scholar]