

Abstract
The LIM domain-containing TRIP6 (Thyroid Hormone Receptor-interacting Protein 6) is a focal adhesion molecule known to regulate lysophosphatidic acid (LPA)-induced cell migration through interaction with the LPA2 receptor. LPA stimulation targets TRIP6 to the focal adhesion complexes and promotes c-Src-dependent phosphorylation of TRIP6 at Tyr-55, which creates a docking site for the Crk Src homology 2 domain, thereby promoting LPA-induced morphological changes and cell migration. Here we further demonstrate that a switch from c-Src-mediated phosphorylation to PTPL1/Fas-associated phosphatase-1-dependent dephosphorylation serves as an inhibitory feedback control mechanism of TRIP6 function in LPA-induced cell migration. PTPL1 dephosphorylates phosphotyrosine 55 of TRIP6 in vitro and inhibits LPA-induced tyrosine phosphorylation of TRIP6 in cells. This negative regulation requires a direct protein-protein interaction between these two molecules and the phosphatase activity of PTPL1. In contrast to c-Src, PTPL1 prevents TRIP6 turnover at the sites of adhesions. As a result, LPA-induced association of TRIP6 with Crk and the function of TRIP6 to promote LPA-induced morphological changes and cell migration are inhibited by PTPL1. Together, these results reveal a novel mechanism by which PTPL1 phosphatase plays a counteracting role in regulating TRIP6 function in LPA-induced cell migration.
The LIM domain-containing TRIP6, also known as ZRP-1 (Zyxin-related Protein 1), is a zyxin family member that has been implicated in cell motility and transcriptional control (1). Originally discovered as an interacting protein of the nuclear thyroid hormone receptor in a yeast two-hybrid screen (2), TRIP6 was later identified as a focal adhesion molecule with the capability to shuttle between cell surface and nucleus (3). TRIP6 is structurally similar to zyxin, LPP (Lipoma Preferred Partner) and Ajuba (1). They possess a proline-rich region and nuclear export signals at their amino termini and three LIM domains (named by the initials of Lin-11, Isl-1, and Mec-3) at their carboxyl termini. Through the LIM domain-mediated protein-protein interactions, TRIP6 forms complexes with several molecules involved in actin rearrangement, cell adhesion, and migration, at least including p130cas (4), CasL/HEF1 (4), endoglin (5), supervillin (6), and the LPA2 receptor (7). In addition, the most carboxyl-terminal LIM3 and PDZ-binding domain of TRIP6 have been demonstrated to mediate the interaction with the second PDZ domain of human PTPL1/Fas-associated phosphatase 1 (3) and its mouse homologue, PTP-BL (8). However, the functional significance for this interaction remains to be elucidated.
Lysophosphatidic acid (LPA),3 a growth factor-like phospholipid, mediates diverse biological responses such as cell migration, cell proliferation, and cell survival through the activation of G protein-coupled LPA receptors (9). Among the five membrane-bound LPA receptors (10 –14), the LPA1, LPA2, and LPA3 receptors of the EDG (Endothelial Differentiation Gene) family are structurally similar to each other except for the carboxyl-terminal tails, suggesting that this region may specifically regulate the unique protein-protein interactions and functions of each receptor. Previously we have demonstrated that the carboxyl-terminal tail of the LPA2 receptor, but not LPA1 or LPA3 receptor, interacts with the LIM domains of TRIP6 (7). This association promotes LPA-dependent recruitment of TRIP6 to the focal adhesion sites where it forms complexes with p130cas, focal adhesion kinase, paxillin, and c-Src. The function of TRIP6 in cell motility is regulated by c-Src-mediated phosphorylation at Tyr-55 (15). This phosphorylation is required for TRIP6 coupling to the Crk Src homology 2 domain and ERK (extracellular signal-regulated kinase) activation, thereby enhancing LPA-induced morphological changes and chemotaxis.
Cell migration is a dynamic process that requires a tight coordination of various signaling molecules involved in cell adhesion and migration. Several tyrosine kinases and phosphatases have been shown to regulate these signaling events through reversible tyrosine phosphorylation and dephosphorylation of their substrates (16). In particular, the focal adhesion kinase-Src-mediated pathways play a fundamental role in regulating adhesion turnover and disassembly during cell migration (17). To understand how tyrosine phosphorylation and dephosphorylation of TRIP6 modulate its function in cell motility, we explored whether PTPL1 phosphatase is a candidate responsible for dephosphorylation of TRIP6. Human PTPL1, also known as FAP-1 (Fas-associated Phosphatase 1), PTP1E, and PTPN13 and its mouse homologue, PTP-BL, are ~270-kDa cytosolic tyrosine phosphatases that contain an amino-terminal FERM domain, five PDZ motifs, and a carboxyl-terminal tyrosine phosphatase catalytic domain (18). The FERM domain functions as membrane-cytoskeleton linkers and may play a role in cytokinesis (19). Several regulators of actin cytoskeleton have been shown to interact with the PDZ domains of PTPL1, at least including TRIP6, PARG (PTPL1-associated RhoGAP), APC (adenomatous polyposis coli), ephrinB, PRK2 (protein kinase C-related kinase-2), CRIP2 (cysteine-rich intestinal protein 2), and RIL (Reversion-induced LIM), implicating a role for PTPL1 in actin dynamics (18). A number of molecules have been identified as PTPL1 substrates, at least including ephrinB, RIL, IκB, and c-Src (20–23); however, the functional consequences of their dephosphorylation are still poorly understood.
In this report, we demonstrate that PTPL1 dephosphorylates c-Src-phosphorylated TRIP6 in vitro and inhibits LPA-induced tyrosine phosphorylation of TRIP6 in cells through a direct protein-protein interaction. This dephosphorylation reduces TRIP6 turnover from the sites of adhesions and inhibits TRIP6 coupling to Crk. As a result, the function of TRIP6 in promoting LPA-induced morphological changes and cell migration is attenuated by PTPL1. Furthermore, LPA-induced cell migration is enhanced by suppression of endogenous PTPL1 expression but is reduced by overexpression of PTPL1, suggesting that PTPL1 acts as a negative regulator of cell motility.
EXPERIMENTAL PROCEDURES
Plasmid Construction
A TRIP6 cDNA fragment encoding amino acids 1–396 was amplified by polymerase chain reaction (PCR) and inserted into pCMV-Tag2A (Stratagene) to generate the FLAG-TRIP6-Δ-(397–476) expression vector. The pEYFP-TRIP6 was constructed by replacing the green fluorescent protein (GFP) cDNA sequences of pEGFP-TRIP6 with a cDNA fragment encoding yellow fluorescent protein (YFP). A cDNA fragment encoding GFP or a cyan fluorescent protein (CFP) was inserted into pCMV5-HA-PTPL1 (24) such that HA-PTPL1 was tagged in-frame with GFP or CFP at its amino terminus, respectively. To express a GFP-HA-PTPL1-ΔCD mutant lacking the catalytic domain of PTPL1, a SacI cDNA fragment encoding amino acids 1–2277 of PTPL1 was removed from pCMV-HA-PTPL1 and inserted into pEGFP-C2. To express the recombinant PTPL1 catalytic domain (designated PTPL1-CD), a cDNA fragment encoding amino acids 2087–2485 of PTPL1 was amplified by PCR from pCMV5-HA-PTPL1 and inserted into pGEX-6P3 (Amersham Biosciences). To inhibit the expression of PTP-BL, a mouse homologue of PTPL1, the pSUPER vector was used to direct the expression of a small interfering RNA (siRNA) of PTP-BL (designated pSUPER-si(m)PTPL1) that specifically targets the 21-nt sequences of PTP-BL, 5′-GAGTGAGCATTGCTGACCCTG-3′. A control siRNA (designated siScramble) expression vector was constructed by inserting the 19-nt sequences, 5′-GCGCGCTTTG-TAGGATTCG-3′ that do not target any known cellular RNA, into pSUPER vector. The entire sequences of each cDNA clone were verified by automatic DNA sequencing.
In Vitro Phosphorylation and Dephosphorylation Assays
Escherichia coli BL21(DE3)(LysS) was transformed with pGEX-6P3-TRIP6 or pGEX-6P3-PTPL1-CD. GST-TRIP6 and GST-PTPL1-CD were purified as described previously (7). GST-PTPL1-CD was further digested with PreScission protease (Amersham Biosciences) to cleave GST. 1 μg of GST-TRIP6 was first phosphorylated by recombinant p60c-Src (Upstate Biotechnology) as described previously (15). Following reaction, GST-TRIP6-immobilized glutathione beads were washed four times to remove c-Src. These beads were then incubated with 0.1 or 0.5 μg of PTPL1-CD for 10 or 30 min in the phosphatase buffer (25 mM imidazole-HCl, pH 7.2, 1 mg/ml BSA, and 1 mM dithiothreitol). After SDS-PAGE, the immunoblot was probed with an horseradish peroxidase-conjugated anti-phosphotyrosine antibody (PY20H; Santa Cruz Biotechnology) to detect phosphorylated TRIP6. The blot was then stripped and reprobed with an anti-TRIP6 antibody (Bethyl Laboratories).
Co-immunoprecipitation, Immunoblotting, and Tyrosine Phosphorylation of TRIP6
Transfected HEK 293T cells were starved overnight followed by incubation with 2 μM LPA for 15 min. Co-immunoprecipitation was performed as described previously (7). Immunoblotting was performed using the antibodies specific to human TRIP6 (Bethyl Laboratories), mouse TRIP6 (BD Biosciences), vinculin (Sigma), PTPL1, FLAG epitope, GFP, HA epitope, or β-actin (Santa Cruz Biotechnology).
To detect tyrosine phosphorylation of the endogenous TRIP6, FLAG-TRIP6, or FLAG-TRIP6-Δ-(397–476), transfected SYF + c-Src or HEK 293T cells were starved in 0.1% fatty acid-free BSA-containing Dulbecco’s modified Eagle’s medium for 8 h (SYR + c-Src cells) or overnight (HEK 293T cells) followed by incubation with 2 μM LPA for 15 min. The endogenous TRIP6 and transfected FLAG-TRIP6 were immunoprecipitated with an anti-TRIP6 mouse antibody (BD Biosciences) and anti-FLAG M2 monoclonal antibody-conjugated agarose beads from the lysates, respectively. After SDS-PAGE, tyrosine-phosphorylated TRIP6 was detected by immunoblotting using a horseradish peroxidase-conjugated anti-phosphotyrosine antibody (PY20H; Santa Cruz Biotechnology). Total tyrosine phosphoproteins in SYF + c-Src MEFs (mouse embryonic fibroblasts) were immunoprecipitated with anti-phosphotyrosine antibody-conjugated agarose beads, and the immunoblot was probed with a horseradish peroxidase-conjugated anti-phosphotyrosine PY20H antibody (Santa Cruz Biotechnology). Tyrosine phosphorylation of endogenous c-Src-Y416 in the whole cell lysates of SYF + c-Src MEFs was detected by immunoblotting using an anti-phospho-c-Src-Y416 antibody (Cell Signaling).
Time-lapse Imaging of Live Cells
SYF + c-Src MEFs expressing YFP or CFP fusion proteins of TRIP6 or PTPL1 were plated on glass-bottomed 35-mm tissue culture dishes (MatTek Corp.). Cells were washed twice with phenol red-free Dulbecco’s modified Eagle’s medium/F-12 containing 1% fatty acid-free BSA and 10 mM HEPES, pH 7.4, and then incubated with 10 μM LPA. The expression of YFP and CFP fusion proteins was visualized by Olympus IX70 inverted fluorescence microscope using a × 100 objective. The YFP images were acquired every 20 s for 30 min with a Photometrics 1400 charge-coupled device camera under the control of IPLab software (Scanlytics, Inc.).
Transwell Cell Migration Assays
Transfected SKOV-3 cells were subjected to a transwell cell migration assay as described previously (15).
TRIP6 Turnover
SYF + c-Src MEFs expressing YFP-PTPL1 or control YFP were starved for 8 h, followed by incubation with 2 μM LPA for 15, 30, and 45 min. Cells were harvested on ice in the Triton-X-containing buffer (1% Triton-X, 10% glycerol, 150 mM NaCl, 50 mM HEPES, pH 7.4, 1 mM EDTA, and 1 mM EGTA) supplemented with a mixture of protease inhibitors and phosphatase inhibitors for 30 min. The detergent-resistant cytoskeletal fractions were collected by centrifugation at 14,000 × g for 15 min. The pellets were dissolved in SDS lysis buffer, boiled, and sonicated to disrupt the cytoskeleton. Immunoblotting was performed to detect TRIP6, β-actin, and vinculin using antibodies specific to these proteins.
RESULTS AND DISCUSSION
PTPL1 Inhibits LPA-induced Tyrosine Phosphorylation of TRIP6 through Direct Interaction with TRIP6
Previously it has been shown that the carboxyl-terminal LIM3 and PDZ-binding domain of TRIP6 interacts with the second PDZ domain of human PTPL1 and its mouse homologue PTP-BL in the yeast two-hybrid systems (3, 8). However, the functional significance for this interaction has not yet been elucidated. In search of the candidate phosphatase of TRIP6, we tested the ability of PTPL1 to dephosphorylate phosphotyrosine 55 of TRIP6 in vitro. We first phosphorylated GST-TRIP6 with recombinant c-Src kinase. Following the reaction, c-Src was removed by extensive washing and GST-TRIP6 was incubated with the catalytic domain of PTPL1 for 15 or 30 min. The result showed that the c-Src-phosphorylated TRIP6 was dephosphorylated in the presence of PTPL1 in a dose- and time-dependent manner (Fig. 1A). To investigate whether PTPL1 regulates TRIP6 phosphorylation directly or acts as an adaptor by recruiting other phosphatases to TRIP6, control GFP, GFP-PTPL1, or a GFP-PTPL1-ΔCD mutant that is capable of binding to TRIP6 but lacks the catalytic domain of tyrosine phosphatase was overexpressed in Src−/−, Yes−/−, Fyn−/− triple knock-out MEFs in which c-Src had been reconstituted (designated SYF + c-Src MEFs). Tyrosine phosphorylation of TRIP6 was determined after LPA stimulation for 15 min. As shown in Fig. 1B, LPA-induced tyrosine phosphorylation of endogenous TRIP6 was significantly reduced by overexpression of PTPL1, but not PTPL1-ΔCD. This result indicates that the phosphatase activity of PTPL1 is required for the reduction of TRIP6 phosphorylation.
FIGURE 1. PTPL1 mediates dephosphorylation of phosphotyrosine 55 of TRIP6 in vitro and inhibits LPA-induced tyrosine phosphorylation of TRIP6 in cells.

A, PTPL1 dephosphorylates c-Src-phosphorylated TRIP6 in a dose- and time-dependent manner in vitro. 1 μg of purified recombinant GST-TRIP6 was phosphorylated by c-Src in vitro. After extensive washing to remove c-Src, phosphorylated GST-TRIP6 was incubated with 0.1 or 0.5 μg of the catalytic domain of PTPL1 (PTPL1-CD) for 30 min (left panel) or with 0.5 μg of PTPL1-CD for 10 or 30 min (right panel). After SDS-PAGE, the immunoblot was probed with an anti-phosphotyrosine antibody (PY20H) to detect phosphorylated TRIP6. The immunoblot was then stripped and reprobed with a TRIP6-specific antibody. B, overexpression of PTPL1, but not a PTPL1-ΔCD mutant that lacks the catalytic domain of phosphatase, eliminates LPA-induced tyrosine phosphorylation of TRIP6 in SYF + c-Src cells. SYF + c-Src cells were transiently transfected with pEGFP, pEGFP-PTPL1, or pEGFP-PTPL1-ΔCD. Cells were starved in 0.1% fatty acid-free BSA-containing medium for 8 h followed by incubation with 2 μM LPA for 15 min. The endogenous TRIP6 was immunoprecipitated with a TRIP6-specific monoclonal antibody or a control mouse IgG. After SDS-PAGE, the immunoblot was probed with an anti-phosphotyrosine antibody to detect tyrosine-phosphorylated TRIP6 and then was stripped and reprobed with a rabbit anti-TRIP6 antibody. The right panel shows the expression of endogenous TRIP6, endogenous PTP-BL, GFP-PTPL1, GFP-PTPL1-ΔCD, and GFP in the whole cell lysates. C, inhibition of PTPL1 expression enhances LPA-induced tyrosine phosphorylation of TRIP6 in SYF + c-Src cells. SYF + c-Src cells were transiently transfected with a control pSUPER-siScramble vector expressing a control siRNA or pSUPER-si(m)PTPL1 vector expressing an siRNA of PTP-BL, a mouse homologue of PTPL1. LPA-induced tyrosine phosphorylation of TRIP6 was determined as described in panel B. The bottom two panels show the expression of endogenous PTP-BL and TRIP6 in the whole cell lysates, respectively. Data shown in each figure are representative of three to four independent experiments.
To understand whether PTPL1 negatively regulates TRIP6 phosphorylation at physiological conditions, we inhibited the expression of PTP-BL by a specific siRNA (designated si(m)PTPL1) and examined LPA-induced tyrosine phosphorylation of TRIP6 in SYF + c-Src MEFs. Under this condition, LPA-induced tyrosine phosphorylation of TRIP6 was enhanced, suggesting a physiological relevance for this regulation (Fig. 1C).
Previously it has been reported that PTP-BL dephosphorylates phosphotyrosine 416 of c-Src, thereby preventing auto-phosphorylation of c-Src and inactivating its kinase activity (20, 23). Although PTPL1 may mediate TRIP6 dephosphorylation through a direct interaction with TRIP6, it is also possible that PTPL1 indirectly inhibits tyrosine phosphorylation of TRIP6 by attenuating c-Src kinase activity. Perhaps because the expression and kinase activity of the reconstituted c-Src are high in SYF + c-Src MEFs, we did not observe a significant alteration of c-Src-Y416 phosphorylation or the total levels of tyrosine phosphoproteins by the inhibition of PTP-BL expression with its specific siRNA (supplemental Fig. S1).
So next we compared tyrosine phosphorylation of wild-type TRIP6 and a TRIP6-Δ-(397– 476) deletion mutant that lacks LIM3 and PDZ-binding domains required for PTPL1 interaction but retains the ability to bind to the LPA2 receptor. The LPA-promoted association of PTPL1 with TRIP6, but not TRIP6-Δ-(397–476) mutant, was first confirmed in HEK 293T cells, which express substantial amounts of endogenous PTPL1 (Fig. 2A). Moreover, c-Src-mediated tyrosine phosphorylation of TRIP6, but not TRIP6-Δ-(397–476) mutant, was attenuated by overexpression of PTPL1 (Fig. 2B, lanes 3, 4 compared with lanes 1, 2, and lanes 7, 8 compared with lanes 5, 6). Together, these results suggest that PTPL1 mediates TRIP6 dephosphorylation through a direct protein-protein interaction.
FIGURE 2. Deletion of the carboxyl-terminal LIM3 and PDZ-binding domains of TRIP6 disrupts its interaction with PTPL1 and abolishes PTPL1-mediated dephosphorylation of TRIP6.
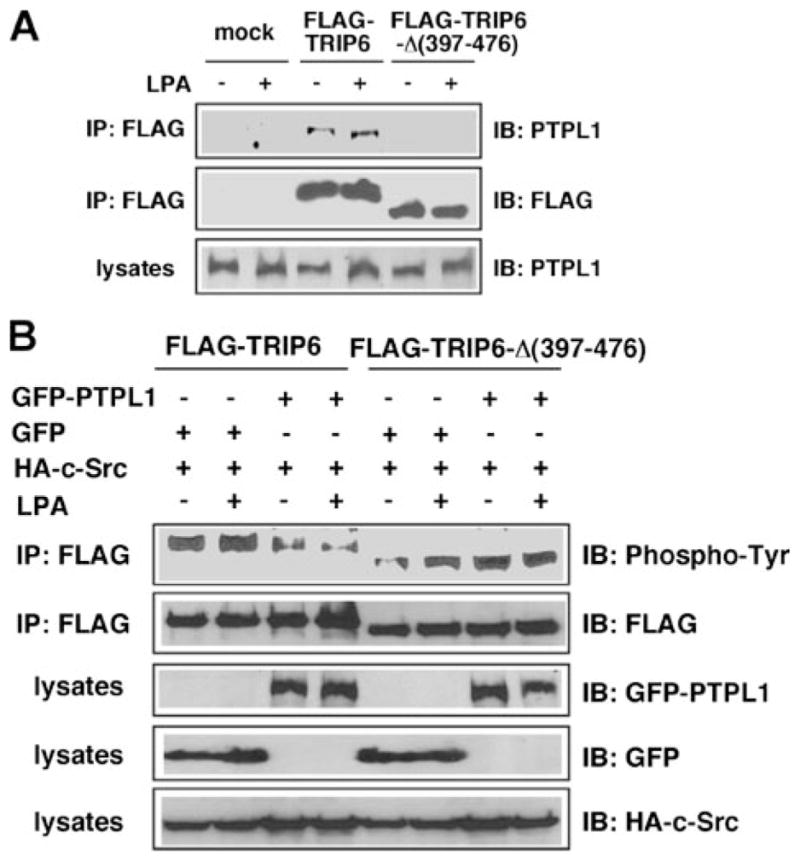
A, a TRIP6-Δ-(397–476) mutant that lacks the LIM3 and PDZ-binding domains cannot bind to PTPL1. HEK 293T cells transiently expressing FLAG-TRIP6 or FLAG-TRIP6-Δ-(397–476) were starved overnight and then stimulated with LPA for 15 min. TRIP6 or TRIP6-Δ-(397–476) was immunoprecipitated with an anti-FLAG M2 monoclonal antibody. After SDS-PAGE, the immunoblot was probed with an anti-PTPL1 antibody. The blot was then stripped and reprobed with an anti-FLAG antibody to detect the immunoprecipitated TRIP6 or TRIP6-Δ-(397–476). The bottom panel shows the expression of endogenous PTPL1 in the whole cell lysates. B, PTPL1 overexpression does not affect tyrosine phosphorylation of TRIP6-Δ-(397–476). FLAG-TRIP6 or FLAG-TRIP6-Δ-(397–476) was expressed in HEK 293T with HA-c-Src, GFP, or GFP-PTPL1 as indicated. After starvation overnight, cells were stimulated with LPA for 15 min. The levels of tyrosine-phosphorylated TRIP6 or TRIP6-Δ-(397–476) were determined as described in panel A. The bottom three panels show the expression of GFP-PTPL1, GFP, and HA-c-Src in the whole cell lysates, respectively. The result shown is representative of three independent experiments.
PTPL1 Reduces TRIP6 Turnover at the Sites of Adhesions and Inhibits LPA-induced Morphological Changes in SYF + c-Src Cells
Previously we have shown that in serum-free conditions TRIP6 is diffusely present in the cytosol. Upon stimulation with LPA for 15–20 min, TRIP6 is targeted to focal adhesions and co-localized with actin cytoskeleton (7). c-Src-mediated phosphorylation of TRIP6 at Tyr-55 does not affect these signaling events but is important for TRIP6 turnover at the sites of adhesions (15). Accordingly, PTPL1-dependent dephosphorylation may regulate TRIP6 turnover at focal adhesions. To examine the turnover of cytoskeleton-associated TRIP6 following LPA stimulation, we set up an experiment to determine LPA-induced accumulation of TRIP6 in the detergent-resistant cytoskeletal fractions. Therefore, SYF + c-Src cells expressing YFP-PTPL1 or control YFP were starved for 8 h, followed by stimulation with LPA for 15, 30, or 45 min, and the Triton-X-insoluble fractions were collected. Our results showed that LPA stimulation rapidly induced the accumulation of TRIP6 in the insoluble cytoskeletal fractions (Fig. 3A). It reached the highest level after 15 min of treatment. Subsequently, TRIP6 was dissociated from cytoskeleton; however, the turnover of cytoskeleton-associated TRIP6 was significantly delayed in cells over-expressing PTPL1 (Fig. 3B). Compared with TRIP6, significant amounts of vinculin were already present in the insoluble cytoskeletal fractions. In the control SYF + c-Src cells, LPA stimulation slightly increased the accumulation of vinculin in the detergent-resistant fractions but the turnover of vinculin was much slower than TRIP6 (Fig. 3A). In the cells overexpressing PTPL1, LPA stimulation did not further increase the accumulation or induce the turnover of vinculin in detergent-resistant cytoskeletal fractions. These results suggest that LPA induces a rapid assembly and disassembly of focal adhesion molecules in SYF + c-Src cells; however, this dynamic process is retarded by PTPL1 overexpression.
FIGURE 3. PTPL1 reduces the turnover of cytoskeleton-associated TRIP6 and inhibits LPA-induced morphological changes.

A and B, LPA induces a rapid accumulation and turnover of TRIP6 in the detergent-resistant cytoskeletal fractions, whereas PTPL1 slows this dynamic process. A, SYF + c-Src fibroblasts transiently expressing YFP or YFP-PTPL1 were starved for 8 h, followed by stimulation with LPA for 15, 30, or 45 min, and then harvested in 1% Triton X-100-containing buffer on ice for 30 min. The insoluble cytoskeletal fractions were collected by centrifugation at 14,000 × g and dissolved in SDS lysis buffer. After boiling and sonication to disrupt cytoskeleton, equal amounts of protein were subjected to immunoblotting using the antibodies specific to TRIP6, β-actin, and vinculin, respectively. The right panel shows the expression of YFP-PTPL1 in the whole cell lysates. B, the expression levels of TRIP6 in the insoluble cytoskeletal fractions were quantified using NIH IMAGE J software program and were compared with that in the absence of LPA. The result shown is a representative from three independent experiments. C, the ability of TRIP6 to promote LPA-induced morphological changes is inhibited by PTPL1. SYF + c-Src cells transiently expressing YFP or YFP-TRIP6 with CFP or CFP-PTPL1 were washed twice with phenol red-free Dulbecco’s modified Eagle’s medium/F-12 containing 1% fatty acid-free BSA and then stimulated with 10 μM LPA for 20 min. The YFP images of live cells were acquired every 20 s for 20 min by inverted fluorescence microscope using a × 100 objective under the control of IPLab software. Results shown are the images captured at various times and are representative of five independent experiments.
LPA stimulation rapidly induces cytoskeletal rearrangement with cellular morphological changes through the coordinate activation of Rho GTPases (25). Previously we have demonstrated that overexpression of TRIP6, but not TRIP6-Y55F mutant, promoted LPA-induced morphological changes in SYF + c-Src MEFs. However, overexpression of TRIP6 in Src-null SYF MEFs not only does not promote LPA-induced morphological changes but even retards this dynamic process (15). One explanation is that TRIP6 forms stable complexes with other signaling molecules at the sites of adhesions and the accumulated mature adhesions perturb LPA-induced adhesion disassembly. Analogously, PTPL1-dependent dephosphorylation of TRIP6 may negatively regulate LPA-induced morphological changes. To explore this possibility, time-lapse imaging of live cells was performed in SYF + c-Src cells expressing YFP or YFP-TRIP6 with either CFP or CFP-PTPL1. Cells were washed twice with 1% fatty acid-free BSA-containing medium, and the expression of these proteins was verified by fluorescence microscopy. Subsequently, LPA was added to the cells and the YFP images of live cells were acquired by time-lapse fluorescence microscopy. Our results showed that the cells expressing control YFP and CFP started to retract their cell bodies soon after LPA stimulation for 5 min and almost completely rounded up after 15 min of treatment (Fig. 3C, panel one, and supplemental video S1, YFP+CFP.mov). However, this process became much slower when CFP-PTPL1 was co-expressed (Fig. 3C, panel two, and supplemental video S2, CFP+PTPL1.mov). Overexpression of YFP-TRIP6 accelerated the rate of LPA-induced morphological changes, and cells started to contract soon after LPA treatment for 1 min (Fig. 3C, panel three, and supplemental video S3, YFP-TRIP6+CFP.mov). In contrast, in cells co-expressing YFP-TRIP6 and CFP-PTPL1, LPA-induced morphological changes were almost completely blocked (Fig. 3C, panel four, and supplemental video S4, YFP-TRIP6+CFP-PTPL1.mov). It is apparent that the number of focal adhesions and the accumulation of YFP-TRIP6 at the sites of adhesions were increased when CFP-PTP-L1 was co-expressed. These results suggest that the relative expression of phosphorylated and dephosphorylated TRIP6 is a critical determinant of TRIP6 function in LPA-induced morphological changes.
PTPL1 Reduces LPA-induced Association of TRIP6 with Crk and Attenuates TRIP6 Function in LPA-induced Cell Migration
Previously we have demonstrated that the pY55QAP motif of human TRIP6 or pY55QPP motif of mouse TRIP6 creates a docking site for the Crk Src homology 2 domain once Tyr-55 is phosphorylated by c-Src (15). Because PTPL1 mediates dephosphorylation of phosphotyrosine 55 of TRIP6, it would be expected to negatively regulate this association. Indeed, our result showed that LPA-induced interaction of TRIP6 with CrkI was significantly reduced by PTPL1 (Fig. 4A). This result implies a role for PTPL1 in regulating TRIP6 function in LPA-induced cell migration.
FIGURE 4. PTPL1 negatively regulates TRIP6 function in Crk coupling and LPA-induced cell migration.
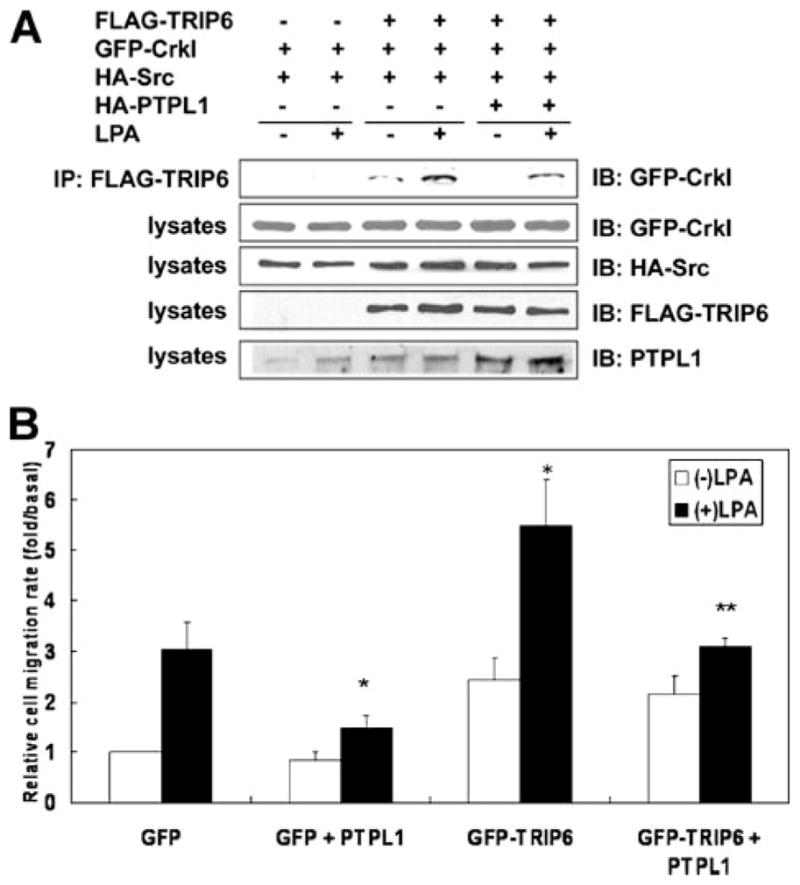
A, PTPL1 attenuates LPA-induced association of TRIP6 with CrkI. HEK 293T cells expressing FLAG-TRIP6, GFP-CrkI, HA-c-Src without or with HA-PTPL1 were starved overnight and then stimulated with LPA for 15 min. TRIP6 was immunoprecipitated with anti-FLAG M2 monoclonal antibody-conjugated agarose beads. After SDS-PAGE, the immunoblot was probed with an anti-GFP antibody to detect co-immunoprecipitated CrkI. The bottom four panels show the expression of GFP-CrkI, HA-c-Src, FLAG-TRIP6, and total PTPL1 in the whole cell lysates, respectively. The result shown is a representative from three independent experiments. B, PTPL1 attenuates the ability of TRIP6 to promote LPA-induced cell migration. pEGFP or pEGFP-TRIP6 was transiently transfected with pCMV5 or pCMV5-HA-PTPL1 in SKOV-3 cells. Cells were washed with 1% fatty acid-free BSA-containing medium and then subjected to a transwell cell migration assay. LPA was added to the bottom chamber of the transwell, and cells were allowed to migrate for 6 h. Cells that migrated to the fibronectin-coated bottom filter were fixed, and the GFP-positive cells were counted under a fluorescence microscope. The migration rate was determined as the relative rate of migrated cells compared with the migrated GFP-expressing cells in the absence of LPA and was normalized by transfection efficiency. The results shown are the mean ± S.E. of three independent experiments. *, p < 0.01 versus LPA-stimulated GFP-expressing cells. **, p < 0.05 versus LPA-stimulated GFP-TRIP6-expressing cells (Student’s t-test).
So next we expressed PTPL1 with TRIP6 in SKOV-3 ovarian cancer cells in which c-Src is highly expressed and examined LPA-induced cell migration. Our result showed that overexpression of PTPL1 reduced LPA-induced transwell cell migration (Fig. 4B). In the absence of ligand stimulation, the relative cell migration rate was slightly higher in cells overexpressing TRIP6. However, the ability of TRIP6 to promote LPA-induced cell migration was significantly attenuated by PTPL1 overexpression.
Because PTPL1 associates with a number of actin-associated molecules, it may play a general role in modulating cell motility through the binding and/or dephosphorylation of other substrates involved in cell adhesion and migration. To test this possibility, we first examined the effect of PTPL1 siRNA on cell motility. Indeed, suppression of endogenous PTPL1 expression significantly enhanced LPA-induced cell migration in SKOV-3 cells, suggesting a physiological relevance of PTPL1 in this regulation (Fig. 5A). To determine whether PTPL1 plays a general role in cell motility or specifically regulates LPA-induced cell migration through the modulation of TRIP6 function, next we expressed PTPL1 with the TRIP6-Δ-(397–476) mutant that lacks the PTPL1-binding domain but retains the ability to bind to the LPA2 receptor. As shown in Fig. 5B, TRIP6-Δ-(397–476) significantly enhanced LPA-induced cell migration. Although PTPL1 attenuated TRIP6-Δ-(397–476) function to promote LPA-induced cell migration, this effect was not statistically significant (p = 0.08, Student’s t-test). Thus, PTPL1-mediated dephosphorylation of TRIP6 plays a particular role in modulating its function in LPA-induced cell migration. Nonetheless, our results do not exclude the possibility that PTPL1 may play a broader role in cell migration induced by other growth factors through the binding and dephosphorylation of additional substrates involved in cell adhesion and migration.
FIGURE 5. PTPL1 negatively regulates LPA-induced cell migration.

A, suppression of PTPL1 expression enhances LPA-induced cell migration. pEGFP was co-transfected with pSUPER-siScramble or pSUPER-siPTPL1 into SKOV-3 cells. LPA-induced transwell cell migration was performed as described in Fig. 4B. The immunoblot shows the expression of GFP and endogenous PTPL1 in the whole cell lysates. The result shown is the mean ± S.E. of three independent experiments. *, p < 0.05 versus LPA-stimulated GFP-expressing cells. B, PTPL1 does not significantly affect the ability of TRIP6-Δ-(397–476) to promote LPA-induced cell migration. pEGFP or pEGFP-TRIP6-Δ-(397–476) was co-transafected with pCMV5 or pCMV5-HA-PTPL1 into SKOV-3 cells. LPA-induced transwell cell migration was performed as described above. The results shown are the mean ± S.E. of three independent experiments. *, p < 0.05 versus LPA-stimulated GFP-expressing cells. **, p = 0.08 versus LPA-stimulated GFP-TRIP6-Δ-(397–476)-expressing cells (Student’s t-test).
In summary, TRIP6 function in LPA-induced adhesion turnover and cell migration is coordinately regulated through c-Src-and PTPL1-dependent reversible tyrosine phosphorylation and dephosphorylation. Another example for this regulation is ephrinB. It has been reported that Src family kinases mediate ephrinB phosphorylation upon the engagement of ephrinB with Eph receptor and regulate ephrinB-stimulated angiogenic sprouting in primary endothelial cells. With delayed kinetics, ephrinB recruits PTP-BL and is dephosphorylated, which switches the Src-mediated signaling to PDZ-dependent signaling (23).
Thus far, several tyrosine phosphatases have been shown to modulate cell motility, at least including PTP-PEST, PTP-α, PTP-1B, SHP-2, and low molecular weight PTP (26–30). Over-expression or inhibition of these tyrosine phosphatases may perturb adhesion turnover and impair cell motility. For example, overexpression of PTP-PEST, a phosphatase for tyrosine-phosphorylated p130cas, leads to the inactivation of Rac1 and defective cell migration (31, 32). Intriguingly, the PTP-PEST−/− MEFs also show impaired cell spreading and migration (27). Thus, appropriate regulation of the tyrosine phosphatase activity of PTP-PEST is important for efficient cell migration. Likewise, overexpression of PTPL1 slows TRIP6 turnover at the sites of adhesions and negatively regulates TRIP6 function to promote LPA-induced cell migration. Because PTPL1 associates with a number of actin-associated molecules, it will be an important task to determine whether PTPL1 plays a wider role in modulating cell motility through dephosphorylation of other substrates involved in cell adhesion and migration.
Supplementary Material
Acknowledgments
We thank Dr. Dario Alessi for providing the HA-PTPL1 expression vector and Albert Tousson at UAB for technical assistance in time-lapse fluorescence microscopy.
Footnotes
This work was supported in part by National Institutes of Health Grant CA100848 (to F.-T. L.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1 and supplemental videos S1–S4.
The abbreviations used are: LPA, lysophosphatidic acid; GFP, green fluorescent protein; YFP, yellow fluorescent protein; CFP, cyan fluorescent protein; siRNA, small interfering RNA; GST, glutathione S-transferase; BSA, bovine serum albumin; HEK, human embryonic kidney; HA, hemagglutinin; MEF, mouse embryonic fibroblast.
References
- 1.Wang Y, Gilmore TD. Biochim Biophys Acta. 2003;1593:115–120. doi: 10.1016/s0167-4889(02)00349-x. [DOI] [PubMed] [Google Scholar]
- 2.Lee JW, Choi HS, Gyuris J, Brent R, Moore DD. Mol Endocrinol. 1995;9:243–254. doi: 10.1210/mend.9.2.7776974. [DOI] [PubMed] [Google Scholar]
- 3.Murthy KK, Clark K, Fortin Y, Shen SH, Banville D. J Biol Chem. 1999;274:20679–20687. doi: 10.1074/jbc.274.29.20679. [DOI] [PubMed] [Google Scholar]
- 4.Yi J, Kloeker S, Jensen CC, Bockholt S, Honda H, Hirai H, Beckerle MC. J Biol Chem. 2002;277:9580–9589. doi: 10.1074/jbc.M106922200. [DOI] [PubMed] [Google Scholar]
- 5.Sanz-Rodriguez F, Guerrero-Esteo M, Botella LM, Banville D, Vary CP, Bernabeu C. J Biol Chem. 2004;279:32858–32868. doi: 10.1074/jbc.M400843200. [DOI] [PubMed] [Google Scholar]
- 6.Takizawa N, Smith TC, Nebl T, Crowley JL, Palmieri SJ, Lifshitz LM, Ehrhardt AG, Hoffman LM, Beckerle MC, Luna EJ. J Cell Biol. 2006;174:447–458. doi: 10.1083/jcb.200512051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu J, Lai YJ, Lin WC, Lin FT. J Biol Chem. 2004;279:10459–10468. doi: 10.1074/jbc.M311891200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cuppen E, van Ham M, Wansink DG, de Leeuw A, Wieringa B, Hendriks W. Eur J Cell Biol. 2000;79:283–293. doi: 10.1078/S0171-9335(04)70031-X. [DOI] [PubMed] [Google Scholar]
- 9.Mills GB, Moolenaar WH. Nat Rev Cancer. 2003;3:582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 10.An S, Bleu T, Huang W, Hallmark OG, Coughlin SR, Goetzl EJ. FEBS Lett. 1997;417:279–282. doi: 10.1016/s0014-5793(97)01301-x. [DOI] [PubMed] [Google Scholar]
- 11.Bandoh K, Aoki J, Hosono H, Kobayashi S, Kobayashi T, Murakami-Murofushi K, Tsujimoto M, Arai H, Inoue K. J Biol Chem. 1999;274:27776–27785. doi: 10.1074/jbc.274.39.27776. [DOI] [PubMed] [Google Scholar]
- 12.Lee CW, Rivera R, Gardell S, Dubin AE, Chun J. J Biol Chem. 2006;281:23589–23597. doi: 10.1074/jbc.M603670200. [DOI] [PubMed] [Google Scholar]
- 13.Noguchi K, Ishii S, Shimizu T. J Biol Chem. 2003;278:25600–25606. doi: 10.1074/jbc.M302648200. [DOI] [PubMed] [Google Scholar]
- 14.Hecht JH, Weiner JA, Post SR, Chun J. J Cell Biol. 1996;135:1071–1083. doi: 10.1083/jcb.135.4.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lai YJ, Chen CS, Lin WC, Lin FT. Mol Cell Biol. 2005;25:5859–5868. doi: 10.1128/MCB.25.14.5859-5868.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Webb DJ, Parsons JT, Horwitz AF. Nat Cell Biol. 2002;4:E97–100. doi: 10.1038/ncb0402-e97. [DOI] [PubMed] [Google Scholar]
- 17.Panetti TS. Front Biosci. 2002;7:d143–150. doi: 10.2741/A771. [DOI] [PubMed] [Google Scholar]
- 18.Erdmann KS. Eur J Biochem. 2003;270:4789–4798. doi: 10.1046/j.1432-1033.2003.03895.x. [DOI] [PubMed] [Google Scholar]
- 19.Herrmann L, Dittmar T, Erdmann KS. Mol Biol Cell. 2003;14:230–240. doi: 10.1091/mbc.E02-04-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roskoski R., Jr Biochem Biophys Res Commun. 2005;331:1–14. doi: 10.1016/j.bbrc.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 21.Nakai Y, Irie S, Sato TA. Eur J Biochem. 2000;267:7170–7175. doi: 10.1046/j.1432-1327.2000.01818.x. [DOI] [PubMed] [Google Scholar]
- 22.Cuppen E, Gerrits H, Pepers B, Wieringa B, Hendriks W. Mol Biol Cell. 1998;9:671–683. doi: 10.1091/mbc.9.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palmer A, Zimmer M, Erdmann KS, Eulenburg V, Porthin A, Heumann R, Deutsch U, Klein R. Mol Cell. 2002;9:725–737. doi: 10.1016/s1097-2765(02)00488-4. [DOI] [PubMed] [Google Scholar]
- 24.Kimber WA, Deak M, Prescott AR, Alessi DR. Biochem J. 2003;376:525–535. doi: 10.1042/BJ20031154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Panetti TS, Magnusson MK, Peyruchaud O, Zhang Q, Cooke ME, Sakai T, Mosher DF. Prostaglandins. 2001;64:93–106. doi: 10.1016/s0090-6980(01)00102-2. [DOI] [PubMed] [Google Scholar]
- 26.Yu DH, Qu CK, Henegariu O, Lu X, Feng GS. J Biol Chem. 1998;273:21125–21131. doi: 10.1074/jbc.273.33.21125. [DOI] [PubMed] [Google Scholar]
- 27.Angers-Loustau A, Cote JF, Charest A, Dowbenko D, Spencer S, Lasky LA, Tremblay ML. J Cell Biol. 1999;144:1019–1031. doi: 10.1083/jcb.144.5.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manes S, Mira E, Gomez-Mouton C, Zhao ZJ, Lacalle RA, Martinez AC. Mol Cell Biol. 1999;19:3125–3135. doi: 10.1128/mcb.19.4.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rigacci S, Rovida E, Sbarba PD, Berti A. J Biol Chem. 2002;277:41631–41636. doi: 10.1074/jbc.M201709200. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Z, Lin SY, Neel BG, Haimovich B. J Biol Chem. 2006;281:1746–1754. doi: 10.1074/jbc.M509590200. [DOI] [PubMed] [Google Scholar]
- 31.Garton AJ, Tonks NK. J Biol Chem. 1999;274:3811–3818. doi: 10.1074/jbc.274.6.3811. [DOI] [PubMed] [Google Scholar]
- 32.Sastry SK, Lyons PD, Schaller MD, Burridge K. J Cell Sci. 2002;115:4305–4316. doi: 10.1242/jcs.00105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.