

Abstract
Developmental and cancer models show Wnt/β-catenin-dependent signaling mediates diverse phenotypic outcomes in the pancreas that are dictated by context, duration and strength of activation. While generally assumed to be pro-tumorigenic, it is unclear to what extent dysregulation of Wnt/β-catenin signaling impacts tumor progression in pancreatic adenocarcinoma (PDAC). In the present study, Wnt/β-catenin activity was characterized across a spectrum of PDAC cell lines and primary tumors. Reporter and gene expression based assays revealed wide heterogeneity in Wnt/β-catenin transcriptional activity across PDAC cell lines and patient tumors, as well as variable responsiveness to exogenous Wnt ligand stimulation. An experimentally-generated, pancreas-specific gene expression signature of Wnt/β-catenin transcriptional activation was used to stratify pathway activation across a cohort of resected, early stage PDAC tumors (N=41). In this cohort, higher Wnt/β-catenin activation was found to significantly correlate with lymphvascular invasion and worse disease specific survival (median survival time 20.3 versus 43.9 months, log rank P=0.03). Supporting the importance of Wnt ligand in mediating autocrine Wnt signaling, Wnt/β-catenin activity was significantly inhibited in PDAC cell lines by WLS gene silencing and the small molecule inhibitor IWP-2, both of which functionally block Wnt ligand processing and secretion. Transcriptional profiling revealed elevated expression of WNT7B occurred in PDAC cell lines with high levels of cell autonomous Wnt/β-catenin activity. Gene knockdown studies in AsPC-1 and HPAF-2 cell lines confirmed WNT7B mediated cell autonomous Wnt/β-catenin activation, as well as an anchorage-independent growth phenotype. Our findings indicate WNT7B can serve as a primary determinant of differential Wnt/β-catenin activation in PDAC. Disrupting the interaction between Wnt ligands and their receptors may be a particularly suitable approach for therapeutic modulation of Wnt/β-catenin signaling in PDAC and other cancer contexts where Wnt activation is mediated by ligand expression rather than mutations in canonical pathway members.
Keywords: pancreatic cancer, WNT7B, Wnt/β-catenin signaling
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is a highly lethal cancer noteworthy for its rapid clinical progression and resistance to therapy (1). While there has been recent significant progress in our understanding of the cellular and molecular basis of PDAC (2, 3), this knowledge has not yet translated into significant improvements in clinical outcomes. PDAC incidence has been slowly rising, while 5-year overall survival is 6% (4). A primary complicating factor is the remarkable genetic and epigenetic heterogeneity of pancreatic cancer (5–7). High throughput sequencing of the human PDAC exome reveals individual tumors average 63 genetic mutations, including a small number of high frequency mutations and a much larger number of heterogeneous lower frequency mutations (6). Interestingly, these highly variable alterations also define a core set of twelve signaling pathways and cellular processes in most PDAC tumors, including Wnt signaling (6).
The Wnt signaling pathway is highly conserved throughout vertebrate species and plays critical roles in development, tissue self-renewal and human diseases, including cancer (8). Wnts are secreted glycoproteins able to act as short-range ligands to activate receptor-mediated signaling via Frizzled (Fzd) receptors and LRP5/6 co-receptors or other novel receptor complexes. Numerous additional extracellular and intracellular proteins further transduce or regulate Wnt signaling (8–10). Multiple Wnt and Fzd isoforms exist and are variably expressed spatially and temporally during development and in adult organs. This complex pattern of expression contributes to a broad range of signaling outcomes that are highly context-dependent (9–11). Wnt signaling occurs through either Wnt/β-catenin-dependent (“canonical”) or β-catenin-independent (“non-canonical”) pathways. Wnt/β-catenin signaling involves an intracellular signaling cascade that stabilizes β-catenin (encoded by CTNNB1), a highly versatile protein involved in both cell adhesion and nuclear transcription. The net result is accumulation of β-catenin in the cytosol and its translocation to the nucleus where it complexes with the TCF/LEF family of transcription factors to activate target gene expression (8, 9).
Our understanding of Wnt/β-catenin signaling in PDAC is evolving. While mutations in CTNNB1, AXIN1 or APC are only rarely detected in PDAC (12–14), both pancreatic intraepithelial neoplasia (PanIN) and up to two-thirds of PDAC tumors have demonstrable Wnt/β-catenin activation based on the surrogate measure of increased nuclear and/or cytoplasmic β–catenin (15–17). Most in vitro and in vivo studies addressing the biologic effects of Wnt/β-catenin signaling in human PDAC cell lines indicate it is pro-tumorigenic (15, 18–20). While offering important insights into the biochemical, transcriptional and phenotypic consequences of Wnt/β-catenin signaling in PDAC, these studies commonly rely on a small number of PDAC cell lines and do not address the potentially confounding effects of variations in Wnt/β-catenin signal strength, duration or context. As an example, in vivo developmental and cancer models reveal Wnt/β-catenin signaling can mediate diverse and sometimes opposing phenotypic outcomes in the pancreas depending on its spatiotemporal context and relative levels of activation (16, 21). Moreover, even if the function of Wnt/β-catenin signaling is consistently pro-tumorigenic in PDAC, it is unclear to what extent the pathway is variably activated in human tumors or whether such variations impact clinical behavior.
In this study we examine variations in Wnt/β-catenin activation across a spectrum of PDAC cell lines and patient samples, identifying a subset of tumors that are distinguished by higher levels of Wnt/β-catenin transcriptional activation and more aggressive clinical behavior. We further describe an important relationship between WNT7B expression, Wnt/β-catenin signaling and anchorage-independent growth in PDAC cell lines with elevated pathway activation. We also conclude autocrine Wnt/β-catenin signaling in PDAC can be primarily initiated and regulated by a single Wnt ligand, WNT7B, acting alone or in conjunction with other Wnt ligands. These results are of significance for future attempts to successfully deploy Wnt-directed therapy in this or other cancers.
RESULTS
Wnt/β-catenin signaling varies widely across pancreatic cancer cell lines
In order to determine the variability of Wnt/β-catenin-dependent signaling across pancreatic cancer, Wnt/β-catenin-dependent luciferase-based reporter assays were performed on multiple PDAC cell lines. Cell lines were transiently transfected with plasmid containing BAR (β-catenin activated reporter) driving luciferase expression or fuBAR (found unresponsive β-catenin activated reporter) identical to BAR with the exception of a mutation in the TCF/LEF binding motif abrogating β-catenin responsiveness. This BAR system has been previously described and shown to have greater sensitivity and dynamic range for β-catenin transcriptional activity relative to TOPFlash or SuperTOPFlash (22). HPDE, a non-transformed pancreatic ductal epithelial cell line, and several PDAC cell lines were found to have minimal baseline Wnt/β-catenin reporter activity as reflected by BAR/fuBAR ratios of 0.5–2.0, hereafter referred to as “Low BAR” cell lines. In contrast, a subset of PDAC cell lines including AsPC-1, Capan-1 and HPAF-2 had far more significant baseline activation with BAR/fuBAR ratios of 5–20 (Figure 1A), hereafter referred to as “High BAR” cell lines. Expression of AXIN2, a widely documented endogenous transcriptional target of Wnt/β-catenin activation (23), was also increased in High BAR lines compared to Low BAR lines (Figure 1B). Treatment of cell lines with either WNT3A ligand or the small-molecule GSK3β inhibitor CHIR99021 significantly increased Wnt/β-catenin signaling as measured by BAR-luciferase reporter assays (Figure 1C) and endogenous AXIN2 expression (Figures 1D). However, the relative fold change observed after drug or ligand stimulation was greatest in Low BAR lines, reflecting their initial lower baseline activity. Therefore, while most PDAC cell lines had the capacity to respond to exogenous Wnt stimulation, only a subset demonstrated appreciable levels of cell autonomous Wnt/β-catenin activity.
Figure 1.
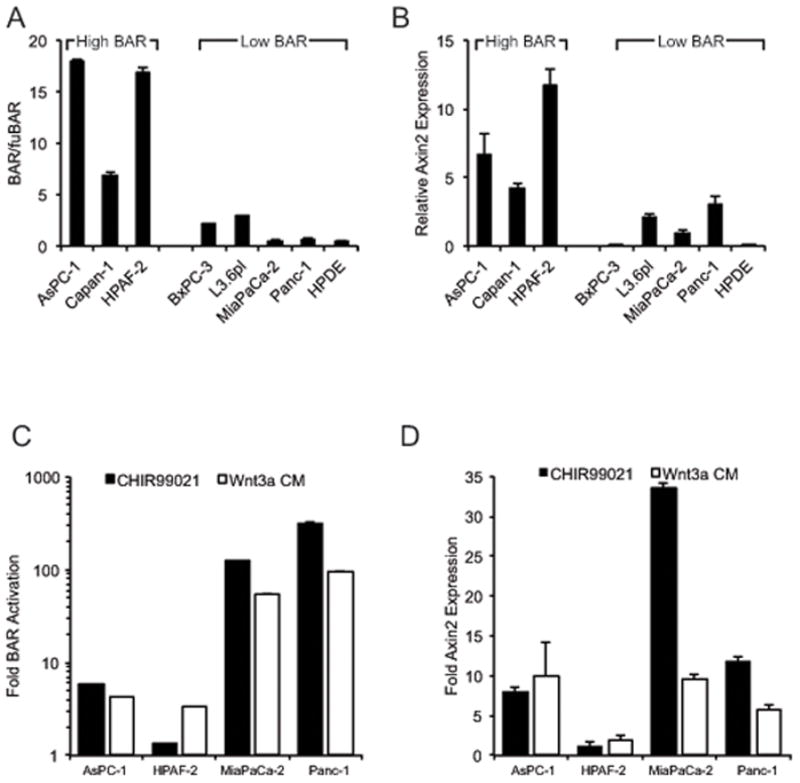
(a) BAR/fuBAR ratios for indicated cell lines as measured by dual luciferase assays 48 hours after transfection with control Renilla and either BAR-Luciferase or fuBAR-luciferase constructs. (b) AXIN2 expression for indicated cell lines by QPCR. (c) BAR-luciferase activity and (d) AXIN2 expression after treatment with CHIR990221 or Wnt3a conditioned media shown relative to vehicle alone (DMSO) or control L-cell conditioned media, respectively, as measured in cell lines with stable BAR-luciferase reporter. ACTB served as normalization control for QPCR.
High Wnt/β-catenin transcriptional signature predicts worse disease-specific survival
Given wide variation in baseline Wnt/β-catenin activation across PDAC cell lines, we next explored the consequences of such variations in patient tumor samples. To date the determination of Wnt/β-catenin signaling in patient tumors has been primarily addressed by immunohistochemistry (IHC) for nuclear and/or cytoplasmic β-catenin expression, a widely recognized surrogate for Wnt pathway activation (10). However, published data regarding β-catenin IHC in PDAC are hard to interpret given significant differences in the reported frequencies of nuclear and/or cytoplasmic β-catenin expression, as well as variable and contradictory results for β-catenin IHC and its correlation with survival and other clinicopathologic variables (16). We therefore evaluated a pancreatic cancer-specific Wnt/β-catenin transcriptional signature as an alternative measurement of Wnt/β-catenin activation in patient tumors. To define this signature, the High BAR PDAC line AsPC-1 was transfected with either control or CTNNB1 siRNA to generate gene expression profiles of Wnt/β-catenin activation and inhibition, respectively. These profiles were then used as a training set to statistically model pancreas-specific Wnt/β-catenin transcriptional activity in CREATE SIGNATURE, a publically available web-based application that employs Bayesian methods to determine and analyze gene expression signatures (24). The generated statistical model (Supplemental Table 3) assigned a relative Wnt/β-catenin activity score that ranged from 0 (inactive) to 1 (high activity). The model was then used to evaluate a published microarray dataset (5) of 41 PDAC patient tumors linked to clinicopathologic information and survival data (Supplemental Table 4). A variable and continuous range of low to high Wnt/β-catenin transcriptional activity was noted across the 41 PDAC tumors (Figure 2A).
Figure 2.
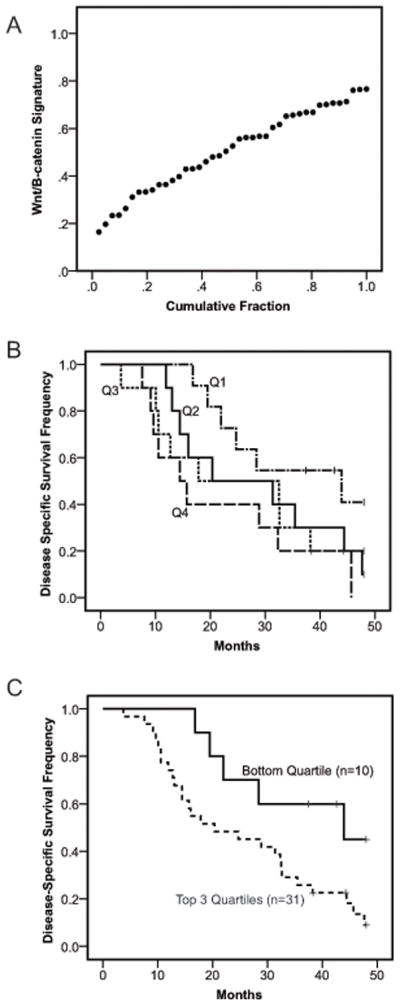
(a) Wnt/β-catenin transcriptional activity scores for primary human PDAC tumors (N=41) calculated using a model of pancreas-specific Wnt/β-catenin activation generated in CREATE SIGNATURE (24). (b–c) Kaplan-Meier analysis of disease-specific survival for PDAC patients grouped by Wnt/β-catenin transcriptional activity scores and separated into either (b) individual quartiles with Q1 to Q4 progressively indicating bottom to top or (c) bottom quartile (solid line) versus combined top three quartiles (dashed line). P value for log-rank test.
The potential clinical relevance of our defined pancreas-specific Wnt/β-catenin signature was next evaluated by stratifying tumors into quartiles based on their activity score. Tumors in the lowest quartile of activity showed a trend toward improved survival relative to the top three quartiles (Figure 2B). Thereafter, patients were divided into two groups encompassing either low (bottom quartile) or high (combined top three quartiles) Wnt/β-catenin transcriptional activity. These dichotomized groups showed no significant relationships with a variety of baseline clinicopathologic variables with the exception of a statistically significant correlation between high Wnt/β-catenin activity and the presence of lymphvascular invasion (P=0.01, Supplemental Table 5). In univariate analysis, high Wnt/β-catenin activity was associated with worse disease-specific survival (Hazard Ratio = 2.75, 95% CI 1.03–7.32, P=0.039) with lymphvascular invasion and increased tumor size also trending toward worse survival (Table 1). Multivariate Cox regression analysis indicated high Wnt/β-catenin activity was an independent predictor for worse disease-specific survival in a model with all other significant clinicopathologic variables retained after backward selection (Hazard Ratio = 3.16, 95% CI 1.19–8.40, P=0.021, Table 1). Kaplan-Meier analysis also verified high Wnt/β-catenin activity was correlated with reduced disease-specific survival in the patient cohort (Figure 2C, median disease-specific survival time 20.3 vs 43.9 months, log rank P = 0.03).
Table 1.
Cox proportional hazard models for prognostic factors
| Variable | Univariate analysis | Multivariate analysis | ||
|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI) | P | |
| Age (≥ 65/<65) | 1.45 (0.69–3.04) | 0.33 | 1.93 (0.87–4.25) | 0.10 |
| Sex (male/female) | 1.03 (0.73–1.47) | 0.85 | - | |
| Histologic grade (G3/G1+G2) | 1.28 (0.62–2.68) | 0.50 | - | |
| Tumor size (≥ 3cm/<3cm) | 1.90 (0.94–3.84) | 0.07 | 2.34 (1.11–4.94) | 0.03 |
| pT (pT3/pT1+pT2) | 1.74 (0.24–12.9) | 0.59 | - | |
| pN (pN1/pN0) | 1.44 (0.69–2.99) | 0.33 | - | |
| Margin (R1/R0) | 1.23 (0.47–3.22) | 0.67 | - | |
| LVI (positive/negative) | 2.11 (0.91–4.88) | 0.08 | - | |
| Wnt/β-catenin (high/low) | 2.75 (1.03–7.32) | 0.04 | 3.16 (1.19–8.40) | 0.02 |
Hazard ratio >1 indicates an increased risk of death for the first of the two groups indicated for each variable. P values are from Chi-square test using the Cox proportional hazards model. Only those variables retained in the model after backward selection are shown for the multivariate analysis. Abbreviations: HR, hazard ratio; CI, confidence interval; LVI, lymphvascular invasion.
Wnt/β-catenin activity in pancreatic cancer is mediated by autocrine Wnt ligand secretion
Wnt/β-catenin reporter activity was next measured in PDAC cell lines following treatment with either IWP-2 or XAV939, small molecule compounds that inhibit Wnt/β-catenin signaling by discrete mechanisms. IWP-2 inactivates the N-palmitoyltransferase PORCN to selectively inhibit the palmitylation and secretion of Wnt ligands (25). XAV939 is a tankyrase inhibitor that augments β-catenin degradation through its stabilization of AXIN (26). IWP-2 significantly inhibited Wnt/β-catenin reporter activity of High BAR lines at nanomolar concentrations (Figure 3A). XAV939 only partially inhibited Wnt/β-catenin reporter activity of High BAR lines at low micromolar concentrations, while higher concentrations were associated with increasing cytotoxicity (Figure 3B and data not shown). The reporter activities of Low BAR lines L3.6pl, MiaPaca-2 and PANC1 were unaffected by treatment with either IWP-2 or XAV939 (data not shown). Treatment with recombinant sFRP1, which can sequester Wnt ligands and prevent their binding to Fzd receptors, also significantly inhibited reporter activity in AsPC-1 cells in dose-dependent fashion (Figure 3C). The strong inhibitory effects of IWP-2 and sFRP-1 suggested that Wnt/β-catenin activation in High BAR lines required effective Wnt ligand processing, secretion and receptor engagement. To address this further, BAR reporter activity was measured following siRNA-mediated gene knockdown of CTNNB1 or WLS, a key gene involved in the trafficking and secretion of Wnt ligands (27). Quantitative real-time PCR (qPCR) verified CTNNB1 and WLS gene knockdown of 75–90% (data not shown). Inhibition of reporter activity seen with WLS knockdown was nearly identical to that observed with CTNNB1 knockdown (Figure 3D), indicating autocrine Wnt/β-catenin signaling in High BAR lines is heavily dependent upon Wnt ligand secretion.
Figure 3.
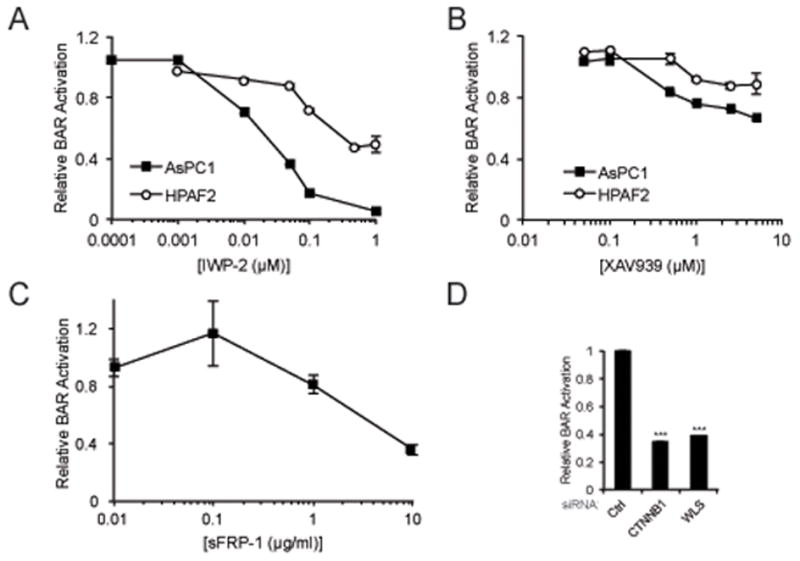
Dose-response curves for (a) IWP-2, (b) XAV939 or (c) recombinant sFRP-1 in cell lines with stable BAR-luciferase reporter. BAR-luciferase activity was measured 24–48 hours after treatment and normalized to treatments with an equivalent volume of vehicle alone (DMSO). (d) BAR-luciferase activity for AsPC-1 transfected with control, CTNNB1 or WLS siRNAs. *** P<0.001.
WNT7B mediates high levels of autocrine Wnt/β-catenin activity in pancreatic cancer
Given the link between Wnt/β-catenin activation and Wnt ligand secretion in PDAC cell lines, we sought to identify the specific ligands mediating this effect. Published RT-PCR and expression microarray analyses indicate nearly every one of the nineteen Wnt genes is expressed in at least subsets of pancreatic cell lines (5, 15, 28). Therefore, to determine which Wnt genes are most abundant in pancreatic cancer cells we performed in silico-based analysis of a published SAGE (serial analysis of gene expression) dataset of 24 human pancreatic tumors that excluded non-malignant cells through serial passage as in vitro cell lines or in vivo xenografts (6). WNT3, WNT4, WNT5A, WNT7A, WNT7B, WNT10A and WNT11 were determined to be the most abundant Wnt genes in pancreatic cancer cells on a per sequence tag basis (Supplemental Figure 1). Gene expression microarray analysis was next performed on High BAR and Low BAR PDAC cell lines to measure relative expression of Wnt genes. This revealed WNT7B expression to be the most highly and consistently increased Wnt gene in High BAR lines (Figure 4A). Further evaluation of WNT7B expression by qPCR confirmed it was elevated in High BAR lines compared to Low BAR lines (Figure 4B). Directly linking WNT7B expression and Wnt/β-catenin reporter activity, transient transfections of the High BAR lines AsPC-1 and HPAF2 with siRNAs targeting WNT7B significantly inhibited WNT7B gene expression (Figure 4C) with corresponding significant reductions in Wnt/β-catenin reporter activation (Figure 4D) and endogenous AXIN2 expression (Figure 4E). WNT7B knockdown inhibited Wnt/β-catenin activation to a similar or greater extent than that observed with CTNNB1 or WLS knockdown (Figure 3C). Additional knockdown of WLS in combination with WNT7B knockdown had no further effect on reporter activity or endogenous AXIN2 expression (Supplemental Figures 2A–B), suggesting other Wnt ligands provided no significant additive or complementary effects on WNT7B and its regulation of autocrine Wnt/β-catenin signaling in these cell lines. Furthermore, individual siRNA-mediated knockdowns of various other Wnt ligands, including WNT3, WNT3A, WNT4, WNT5A, WNT 10A and WNT16, did not significantly inhibit reporter activity in AsPC-1 (Supplemental Figure 2C).
Figure 4.
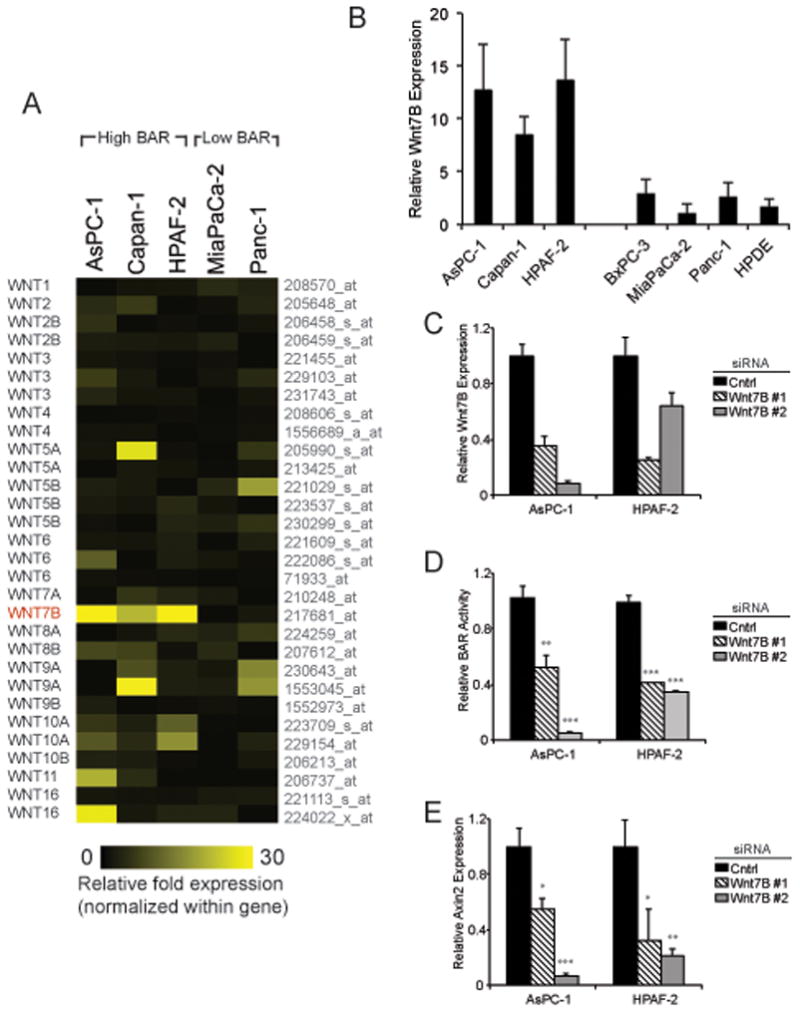
(a) Heatmap showing relative fold-change in gene expression for each Wnt ligand across PDAC cell lines as determined by Affymetrix U133 plus 2.0 oligonucleotide arrays. (b) Relative WNT7B expression for indicated cell lines by QPCR. (c–e) Cell lines with stable BAR-luciferase reporter were transiently transfected with control or WNT7B siRNAs. At 48 hours post-transfection, cells were analyzed by QPCR for (c) WNT7B or (e) AXIN2 expression, as well as (d) BAR-luciferase activity. ACTB served as normalization control for QPCR.
WNT7B promotes an anchorage-independent growth phenotype
To determine cellular phenotypes linked to WNT7B-dependent autocrine Wnt/β-catenin signaling, we stably depleted WNT7B in both AsPC-1 and HPAF2 cell lines using two separate lentiviral shRNAs (Figure 5A). Stable WNT7B depletion significantly inhibited Wnt/β-catenin transcriptional reporter activity in both cell lines (Figure 5B), but had no significant effect on cell viability or proliferation in standard monolayer culture as measured by MTT assay (Figure 5C) or BrdU incorporation (data not shown). In contrast, stable WNT7B depletion significantly inhibited colony formation in both monolayer culture (Figure 5D) and soft agar (Figure 5E). HPAF2 tumorsphere formation in serum-free, non-adherent conditions was also significantly inhibited by WNT7B depletion (Figure 5F), while AsPC-1 failed to form tumorspheres and were not evaluated. Further supporting Wnt ligand secretion and its role in autocrine signaling and anchorage-independent growth, IWP-2 also significantly inhibited the non-adherent growth of High BAR PDAC cell lines in both soft agar and tumorsphere assays (Figure 6A–B). Likewise, treatment of High BAR PDAC cell lines with anti-WNT7B antibody partially but significantly inhibited Wnt/β-catenin reporter activation and tumorsphere formation (Figure 6C–D).
Figure 5.

AsPC-1 and HPAF-2 cells stably transduced with either control or WNT7B shRNA lentiviral constructs were assayed for (a) WNT7B expression by QPCR, (b) BAR/fuBAR ratios by dual luciferase, (c) adherent cell growth by MTT, (d) clonogenicity in monolayer cultures, (e) colony formation in soft agar and (f) tumorsphere growth in non-adherent conditions. All data are shown relative to control shRNA for the corresponding cell line. Representative images of clonogenic, soft agar and tumorsphere assays show HPAF-2 cells. ACTB served as normalization control for QPCR. Scale Bar 100 μM. ** P<0.01, *** P<0.001.
Figure 6.
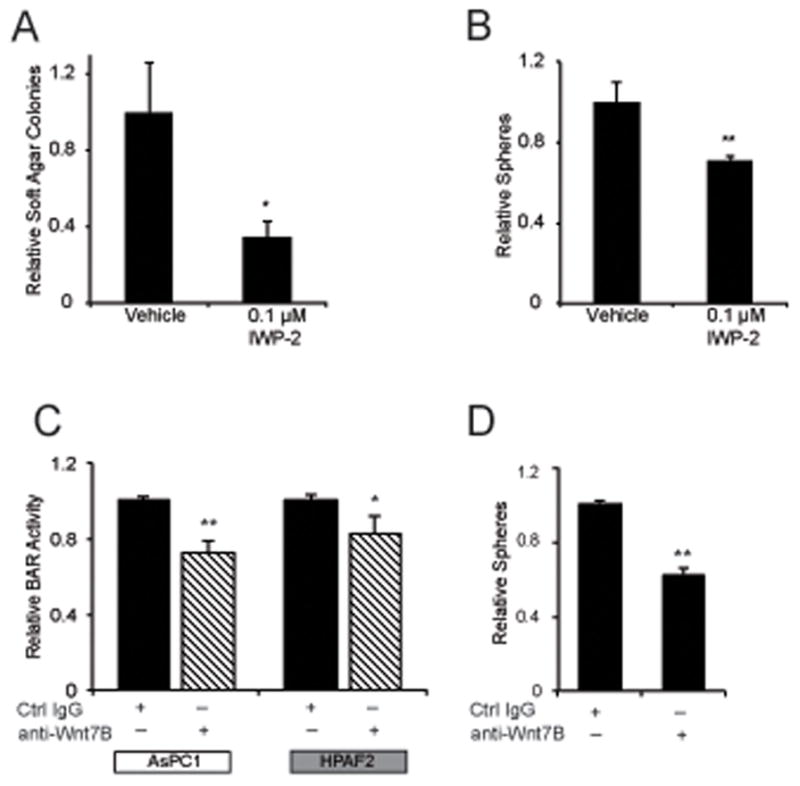
(a) AsPC-1 soft agar growth or (b) HPAF-2 tumorsphere growth was measured in the context of vehicle (DMSO) or 0.1 μM IWP-2 treatment every 72 hours. (c) BAR-luciferase reporter assays at 48 hours in indicated cell lines with stable BAR-luciferase reporter and (d) HPAF tumorsphere growth were measured following treatment every 48 hours with 20 μg/ml anti-WNT7B antibody or matched isotype control antibody. * P<0.05, ** P<0.01.
DISCUSSION
The origin and biological significance of heterogeneous levels of Wnt/β-catenin activation across patient PDAC tumors remains unclear. Here we have interrogated variable Wnt/β-catenin transcriptional activation across pancreatic tumors and cell lines as assessed by promoter-reporter assays, endogenous AXIN2 gene expression and an experimentally-defined Wnt/β-catenin transcriptional signature. Our results parallel those of previous reports surveying PDAC cell lines by β-catenin-dependent transcriptional reporter assays (15, 18, 19) or endogenous AXIN2 expression (29). We identify subsets of PDAC cell lines with either (i) minimal/low or (ii) higher levels of autocrine Wnt/β-catenin activation and hypothesize that these and other variations in the context and/or strength of pathway activation could significantly alter the biochemical and phenotypic effects of Wnt/β-catenin signaling in subsets of PDAC tumors. This variability may be a confounding factor when experimentally manipulating the Wnt/β-catenin pathway to study its function in PDAC. For instance, it is unclear whether loss-of-function approaches employed in cell lines without appreciable Wnt/β-catenin transcriptional activation (i.e., PANC-1, MiaPaCa-2) truly reflect biological activities linked to Wnt/β-catenin signaling, particularly in relation to in vitro studies examining cell autonomous signaling. Likewise, gain-of-function approaches that act to stabilize β-catenin will result in constitutively hyperactivated levels of Wnt/β-catenin signaling that are not typical of PDAC. Genetic mutations in APC, AXIN or CTNNB1 that hyperactivate Wnt/β-catenin signaling are rare in PDAC and instead occur at high frequencies in solid-pseudopapillary tumors, pancreatoblastomas and acinar carcinomas of the pancreas (16). Not surprisingly, transgenic mice expressing a pancreas-specific CTNNB1 mutation that stabilizes β-catenin develop solid-pseudopapillary tumors instead of PDAC. This mutation also blocks the formation of acinar-to-ductal metaplasia, mPanIN and PDAC when introduced into transgenic mice carrying oncogenic Kras (30, 31), indicating that high constitutive levels of Wnt/β-catenin may actually inhibit the initiation of PDAC. We propose that a more accurate view of Wnt/β-catenin signaling in PDAC requires in vitro and in vivo studies that better account for variations in Wnt dosage and spatiotemporal context during pancreatic tumor development and progression.
Wnt ligand secretion was required for autocrine signaling in High BAR lines as demonstrated by both WLS gene silencing and IWP-2 treatment. Both Low and High BAR cell lines were also able to respond to further exogenous Wnt ligand treatment, indicating paracrine sources of Wnt ligand originating from the tumor microenvironment could further impact pathway activation in vivo. In addition to paracrine-mediated Wnt ligand signaling, a variety of additional extracellular factors and signals have been previously shown to modulate Wnt/β-catenin signaling in PanIN and/or PDAC. Downregulation of secreted Wnt inhibitors from the tumor (20) or its associated stromal compartment (32, 33), as well as upregulation of sulfatases that potentiate Wnt ligand signaling (18), have been shown to promote Wnt/β-catenin signaling and pancreatic tumorigenesis. Additional extracellular signaling pathways that promote pancreatic tumorigenesis have also been shown to cross-talk with Wnt/β-catenin signaling in PDAC, including TGFβ, Hedgehog, Notch and fibroblast growth factor signaling (16). Beyond such extracellular mechanisms, tumor cell-associated changes in the expression, localization and/or activation of intracellular mediators of the pathway also facilitate Wnt/β-catenin activation in PDAC (7, 19). These various mechanisms for augmenting Wnt/β-catenin activation in PDAC appear to share the common requirement of a Wnt ligand/receptor-initiated signaling event.
Gene expression analysis and RNAi depletion studies presented here identify a significant role for WNT7B in PDAC cell lines with higher levels of autocrine Wnt/β-catenin signaling. WNT7B and possibly other Wnt ligands alone or in combination appear to be important determinants of differential Wnt activation seen across PDAC. Our array profiling studies of Wnt ligands combined with the observation that Wnt/β-catenin pathway activation in High BAR PDAC lines is readily inhibited by knockdown of a single Wnt ligand supports a model in which Wnt/β-catenin signaling can be regulated by the tissue-specific expression of specific ligands without appreciable ligand redundancy. This type of model would suggest that the targeting of an individual Wnt ligand may be sufficient to manipulate the pathway for therapeutic purposes if the ligand context is well understood (34). These cell lines offer a potential opportunity to further explore the function of WNT7B or other Wnt ligands that work individually or in synergy to initiate pathway activation. For example, individual Wnt ligands may possess unique specificities in terms of their patterns of downstream signaling and transcriptional targets. Future studies will need to address the potential complexity of responses mediated by different combinations of Wnt ligands and receptors.
Phenotypically, WNT7B was required for anchorage-independent cell growth and tumor sphere formation. Although we saw no significant effects on anchorage-dependent cell proliferation, our results are otherwise consistent with various reports that indicate Wnt/β-catenin facilitates anchorage-independent growth and in vivo tumorigenicity in PDAC (15, 18–20). Most recently, Wnt signaling has been implicated in pancreatic metastasis through its promotion of anchorage-independent growth and anoikis resistance (35). Wnt2 and a non-canonical Wnt signature are enriched in circulating tumor cells from mouse and human PDAC, while growth of human PDAC cell lines as tumorspheres variably induces the expression of multiple Wnt ligands, including WNT7B (35). Overall, accumulating data presented here and elsewhere in the literature indicate that both canonical and non-canonical Wnt signaling can facilitate anchorage-independent growth and metastasis in PDAC.
IHC for nuclear and/or cytoplasmic β-catenin is the most widely used surrogate for detecting Wnt/β-catenin activation in patient tumors. However, the literature is plagued by variable and confounding conclusions regarding β-catenin IHC staining in PDAC (16). As an alternative to measuring the status Wnt/β-catenin activation in primary human tumors, we adopted the novel approach of generating a pancreas-specific Wnt/β-catenin transcriptional signature using a publically available web-based application, CREATE SIGNATURE (24). We identified a continuous range of low to high Wnt/β-catenin transcriptional activity across tumors, suggesting that an ON/OFF model for Wnt signaling in PDAC is probably too simplistic and inadequate for describing its apparent range of activation and variable phenotypic consequences. Patients with a higher Wnt/β-catenin transcriptional signature had significantly worse disease-free survival and increased histologic evidence of lymphovascular invasion. This finding must be viewed cautiously as it involved the retrospective analysis of a small cohort of patients and does not account for possible heterogeneity of signaling within a tumor due to spatiotemporal differences or associated non-malignant cell types. However, the findings are compatible with the various aforementioned in vitro and in vivo studies linking Wnt/β-catenin to increased pancreatic tumorigenicity. As an example, the observed increase in lymphvascular invasion could be the consequence of augmented anchorage-independent growth and survival of circulating tumor cells. Future retrospective and prospective evaluation of larger patient cohorts using a refined Wnt/β-catenin transcriptional signature derived from multiple PDAC cell lines will be necessary to evaluate its potential clinical utility as a prognostic or predictive biomarker for PDAC.
The dependency on Wnt ligand (autocrine or paracrine) for initiating Wnt/β-catenin signaling in PDAC suggests these tumors may be especially responsive to therapeutic approaches that inhibit Wnt ligand or extracellular receptors. IWP-2, which blocks Wnt ligand secretion, was an effective inhibitor of Wnt/β-catenin activation and anchorage-independent growth in High BAR PDAC lines. Unfortunately, IWP-2 is not suitable for in vivo use and existing approaches for inhibiting Wnt/β-catenin in vivo are limited (16). However, the monoclonal antibody OMP-18R5, which binds to multiple FZD receptors to inhibit Wnt/β-catenin signaling, was recently shown to effectively block in vivo tumor growth and improve chemotherapeutic response of primary human xenografts derived from pancreas and breast adenocarcinomas (36). These preclinical data suggest promise for pharmacologic or genetic approaches that disrupt Wnt ligand interactions with their extracellular receptors. The role of Wnt/β-catenin signaling as a master regulator of cellular transcription and cell fate also raise the possibility that variations in pathway activation may regulate the response of PDAC cells to chemotherapeutic agents. Given the potential efficacy of approaches targeting the proximal arm of the Wnt pathway, future work will also need to focus on the development and optimization of predictive clinical biomarkers that identify subsets of tumors most likely to respond to Wnt/β-catenin inhibitors.
MATERIALS AND METHODS
Cell Lines and Reagents
All human pancreatic cancer cell lines were cultured as previously described (37). AsPC-1, BxPC-3, Capan-1, HPAF-2, MiaPaCa-2 and PANC-1 were obtained from the American Type Culture Collection (ATCC, Rockville, MD). Dr. Timothy Donahue (University of California, Los Angeles) provided L3.6pl. HPDE, an immortalized, non-transformed pancreatic ductal epithelial cell line was kindly provided by Dr. Ming-Sound Tsao (Princess Margaret Hospital, Toronto, Canada) and cultured as previously described (38). Control and Wnt3A-expressing L cells (ATCC) were cultured in Dulbecco’s Modified Eagle’s Medium + 10% fetal calf serum, with conditioned media collected at 96 hours. Recombinant human sFRP-1 (R&D Systems, Minneapolis, MN) and anti-WNT7B antibody (Q-13, sc-26363) and corresponding isotype control normal goat IgG antibody (sc-2028) (Santa Cruz Biotechnology, Santa Cruz, CA) were purchased and used as indicated.
Quantitative Real-time PCR
RNA extraction, cDNA synthesis and SYBR Green-based quantitative PCR were performed as previously described (37) using an ABI-Prism 7700 sequence detector (Applied Biosystems, Foster City, CA). Primers are listed in Supplemental Table 1.
Gene Knockdown
For transient gene knockdown, cell lines were transfected with 10–20 nM of duplexed siRNAs using Lipofectamine 2000 (Invitrogen/Life Technologies, Grand Island, NY). For stable WNT7B knockdown, cells were infected with lentiviral shRNA particles and selected in puromycin (1μg/ml). WNT7B shRNAs TRCN0000061873 (shWnt7B #73) and TRCN0000061877 (shWnt7B #77) in pLKO.1 backbone were purchased from Open Biosystems (Lafayette, CO). All RNAi sequences are listed in Supplemental Table 2.
MTT Assay
AsPC-1 and HPAF-2 cells were plated at 5,000 and 2,000 cells per well, respectively in 96-well plates under normal growth conditions. MTT assays (ATCC) were carried out per manufacturer’s instructions
Soft Agar Assay
Stably transduced AsPC-1 or HPAF-2 cells (5,000 or 15,000 cells per well of 24 well plate, respectively) were plated as single cell suspensions in 0.4% Noble Agar overlaid on a base layer of 0.8% Noble Agar. Media (RPMI-1640 + 10% FBS) was replaced every 3 days. After 2–3 weeks, colonies were visualized by incubation with 10% MTT Reagent for 5 hours. Colonies greater than 200 microns in diameter were counted.
Clonogenic Assay
Stably transduced AsPC-1 or HPAF-2 cells were plated at 300 cells per well in 6-well plates in RPMI-1640 and 10% FBS. After 14 days, cell colonies were fixed in methanol, stained with crystal violet and counted.
Sphere Assay
Indicated HPAF-2 cells were suspended in Neurobasal Media (Invitrogen/Life Technologies) containing Glutamax, bFGF (20 ng/ml), EGF (20ng/ml) N-2 and B27 at 10,000 cells/well in 6-well ultra-low attachment plates (Corning, Lowell, MA). After 8 days, tumor spheres were visualized and counted by phase contrast microscopy.
β-catenin activated reporter (BAR) assays
Cell lines were transiently transfected with plasmid (pGL3 backbone) or stably transduced with lentiviral construct containing a β-catenin activated reporter (BAR) driving luciferase expression and separate construct with constitutive EF1α promoter driving Renilla expression (normalization control). The BAR reporter construct has been previously described and is comprised of 12 TCF/LEF binding motifs (5′-AGATCAAAGG-3′), each separated by distinct 5–base pair linkers upstream of a minimal promoter and firefly luciferase open reading frame (22). Found unresponsive β-catenin activated reporter (fuBAR) is identical to BAR with the exception of a mutation in the TCF/LEF binding motifs that abrogates β-catenin responsiveness. Dual luciferase assays (Promega, Madison, WI) were performed to simultaneously measure firefly luciferase and Renilla expression.
Gene expression microarray and data analysis
Gene expression microarray analysis with Affymetrix U133 plus 2.0 oligonucleotide arrays was performed in the UCLA Clinical Microarray Core Facility. To determine relative expression of Wnt ligands across PDAC lines, total RNA was extracted from cells grown under normal conditions to 70–80% confluence. Microarray analysis was performed in the dChip Analysis software package using invariant set normalization. The signal intensity was summarized using the model-based expression index (MBEI) algorithm with mismatch probe option for background subtraction. For the pancreatic cancer-specific Wnt/β-catenin-dependent gene signature, gene expression microarrays were performed on total RNA extracted from AsPC-1 cells 48 hours post transfection with either control or CTNNB1 siRNAs to represent “Wnt activated” or “Wnt inhibited” training sets, respectively. These training sets were used to generate a statistical model in CREATE SIGNATURE, a publically available application utilizing Bayesian methods to process gene expression via a web-based interface in GenePattern (24). Default parameters were selected in CREATE SIGNATURE utilizing BinReg algorithm version 2 with both quantile and shiftscale normalization. A final statistical model of 50 genes and 1 metagene was generated and used to predict levels of pancreatic-specific Wnt/β-catenin transcriptional activation in a previously published (5) microarray dataset of 41 PDAC patient tumor samples (deposited in NCBI GEO, GSE32688).
Statistical Methods
SPSS 20.0 for Windows (IBM, Armonk, NY) was used for all statistical analyses. Continuous variables were evaluated using Student t-tests. Chi square tests were used to evaluate the significance of dichotomized patient groups in relation to baseline clinicopathologic factors. Kaplan-Meier survival curves were evaluated by log-rank test. Univariate Cox regression analyses were performed to determine the prognostic significance of individual clinicopathologic variables. Multivariate Cox regression analysis was performed in stepwise fashion with backward selection using the Akaike Information Criterion. The level of significance for all statistical tests was defined as α = 0.05.
Supplementary Material
Acknowledgments
DWD was supported by an American Cancer Society Research Scholar Grant, an American Association for Cancer Research/Pancreatic Cancer Action Network Career Development Award in memory of Seena Magowitz and a Research Services Research Fund Grant from the UCLA Department of Pathology. AJC was supported by a Career Development Award (1K08128565) from the NCI/NIH and Bridge Funding from the University of Washington Office of the Provost.
Footnotes
CONFLICT OF INTEREST
There are no competing financial interests in relation to this work.
References
- 1.Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011 Aug 13;378(9791):607–20. doi: 10.1016/S0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hong SM, Park JY, Hruban RH, Goggins M. Molecular signatures of pancreatic cancer. Arch Pathol Lab Med. 2011 Jun;135(6):716–27. doi: 10.5858/2010-0566-ra.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008;3:157–88. doi: 10.1146/annurev.pathmechdis.3.121806.154305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.American_Cancer_Society. Cancer Facts & Figures 2012. Cancer. New York: American Cancer Society; 2012. pp. 1–66. [Google Scholar]
- 5.Donahue T, Tran L, Hill R, Li Y, Kovochich A, Calvopina J, et al. Intergrative survival-based molecular profiling of human pancreatic cancer. Clinical Cancer Research. 2012;18:1352–63. doi: 10.1158/1078-0432.CCR-11-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008 Sep 26;321(5897):1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vincent A, Omura N, Hong SM, Jaffe A, Eshleman J, Goggins M. Genome-wide analysis of promoter methylation associated with gene expression profile in pancreatic adenocarcinoma. Clin Cancer Res. 2011 Jul 1;17(13):4341–54. doi: 10.1158/1078-0432.CCR-10-3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clevers H, Nusse R. Wnt/beta-Catenin Signaling and Disease. Cell. 2012 Jun 8;149(6):1192–205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 9.Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol. 2009 Jul;10(7):468–77. doi: 10.1038/nrm2717. [DOI] [PubMed] [Google Scholar]
- 10.Chien AJ, Conrad WH, Moon RT. A Wnt survival guide: from flies to human disease. J Invest Dermatol. 2009 Jul;129(7):1614–27. doi: 10.1038/jid.2008.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lucero OM, Dawson DW, Moon RT, Chien AJ. A re-evaluation of the “oncogenic” nature of Wnt/beta-catenin signaling in melanoma and other cancers. Curr Oncol Rep. 2010 Sep;12(5):314–8. doi: 10.1007/s11912-010-0114-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caca K, Kolligs FT, Ji X, Hayes M, Qian J, Yahanda A, et al. Beta- and gamma-catenin mutations, but not E-cadherin inactivation, underlie T-cell factor/lymphoid enhancer factor transcriptional deregulation in gastric and pancreatic cancer. Cell Growth Differ. 1999 Jun;10(6):369–76. [PubMed] [Google Scholar]
- 13.Gerdes B, Ramaswamy A, Simon B, Pietsch T, Bastian D, Kersting M, et al. Analysis of beta-catenin gene mutations in pancreatic tumors. Digestion. 1999 Nov-Dec;60(6):544–8. doi: 10.1159/000007704. [DOI] [PubMed] [Google Scholar]
- 14.Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012 Nov 15;491(7424):399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pasca di Magliano M, Biankin AV, Heiser PW, Cano DA, Gutierrez PJ, Deramaudt T, et al. Common activation of canonical Wnt signaling in pancreatic adenocarcinoma. PLoS One. 2007;2(11):e1155. doi: 10.1371/journal.pone.0001155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.White BD, Chien AJ, Dawson DW. Dysregulation of Wnt/beta-catenin signaling in gastrointestinal cancers. Gastroenterology. 2012 Feb;142(2):219–32. doi: 10.1053/j.gastro.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeng G, Germinaro M, Micsenyi A, Monga NK, Bell A, Sood A, et al. Aberrant Wnt/beta-catenin signaling in pancreatic adenocarcinoma. Neoplasia. 2006 Apr;8(4):279–89. doi: 10.1593/neo.05607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nawroth R, van Zante A, Cervantes S, McManus M, Hebrok M, Rosen SD. Extracellular sulfatases, elements of the Wnt signaling pathway, positively regulate growth and tumorigenicity of human pancreatic cancer cells. PLoS One. 2007;2(4):e392. doi: 10.1371/journal.pone.0000392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L, Heidt DG, Lee CJ, Yang H, Logsdon CD, Zhang L, et al. Oncogenic function of ATDC in pancreatic cancer through Wnt pathway activation and beta-catenin stabilization. Cancer Cell. 2009 Mar 3;15(3):207–19. doi: 10.1016/j.ccr.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhong Y, Wang Z, Fu B, Pan F, Yachida S, Dhara M, et al. GATA6 Activates Wnt Signaling in Pancreatic Cancer by Negatively Regulating the Wnt Antagonist Dickkopf-1. PLoS One. 2011;6(7):e22129. doi: 10.1371/journal.pone.0022129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris JPt, Wang SC, Hebrok M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer. 2010 Oct;10(10):683–95. doi: 10.1038/nrc2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biechele TL, Moon RT. Assaying beta-catenin/TCF transcription with beta-catenin/TCF transcription-based reporter constructs. Methods Mol Biol. 2008;468:99–110. doi: 10.1007/978-1-59745-249-6_8. [DOI] [PubMed] [Google Scholar]
- 23.Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002 Feb;22(4):1172–83. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang JT, Gatza ML, Lucas JE, Barry WT, Vaughn P, Nevins JR. SIGNATURE: a workbench for gene expression signature analysis. BMC Bioinformatics. 2011;12:443. doi: 10.1186/1471-2105-12-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen B, Dodge ME, Tang W, Lu J, Ma Z, Fan CW, et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol. 2009 Feb;5(2):100–7. doi: 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009 Oct 1;461(7264):614–20. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 27.Port F, Basler K. Wnt trafficking: new insights into Wnt maturation, secretion and spreading. Traffic. 2010 Oct;11(10):1265–71. doi: 10.1111/j.1600-0854.2010.01076.x. [DOI] [PubMed] [Google Scholar]
- 28.Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S, et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature. 2012 Jul 26;487(7408):510–3. doi: 10.1038/nature11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lowe AW, Olsen M, Hao Y, Lee SP, Taek Lee K, Chen X, et al. Gene expression patterns in pancreatic tumors, cells and tissues. PLoS ONE. 2007;2(3):e323. doi: 10.1371/journal.pone.0000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heiser PW, Cano DA, Landsman L, Kim GE, Kench JG, Klimstra DS, et al. Stabilization of beta-catenin induces pancreas tumor formation. Gastroenterology. 2008 Oct;135(4):1288–300. doi: 10.1053/j.gastro.2008.06.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morris JPt, Cano DA, Sekine S, Wang SC, Hebrok M. beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest. 2010 Jan 11; doi: 10.1172/JCI40045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Froeling FE, Feig C, Chelala C, Dobson R, Mein CE, Tuveson DA, et al. Retinoic Acid-Induced Pancreatic Stellate Cell Quiescence Reduces Paracrine Wnt-beta-Catenin Signaling to Slow Tumor Progression. Gastroenterology. 2011 Jun 24; doi: 10.1053/j.gastro.2011.06.047. [DOI] [PubMed] [Google Scholar]
- 33.Froeling FE, Mirza TA, Feakins RM, Seedhar A, Elia G, Hart IR, et al. Organotypic culture model of pancreatic cancer demonstrates that stromal cells modulate E-cadherin, beta-catenin, and Ezrin expression in tumor cells. Am J Pathol. 2009 Aug;175(2):636–48. doi: 10.2353/ajpath.2009.090131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chien AJ, Moon RT. WNTS and WNT receptors as therapeutic tools and targets in human disease processes. Front Biosci. 2007;12:448–57. doi: 10.2741/2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S, et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature. 2012 Jul 1; doi: 10.1038/nature11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gurney A, Axelrod F, Bond CJ, Cain J, Chartier C, Donigan L, et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci U S A. 2012 Jul 17;109(29):11717–22. doi: 10.1073/pnas.1120068109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kovochich AN, Arensman MD, Lay AR, Rao NP, Donahue T, Li X, et al. HOXB7 promotes invasion and predicts survival in pancreatic adenocarcinoma. Cancer. 2012 doi: 10.1002/cncr.27725. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu N, Furukawa T, Kobari M, Tsao MS. Comparative phenotypic studies of duct epithelial cell lines derived from normal human pancreas and pancreatic carcinoma. Am J Pathol. 1998 Jul;153(1):263–9. doi: 10.1016/S0002-9440(10)65567-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.