

Abstract
In the present study, the biophysical properties of His6-tagged Bacillus stearothermophilus aminopeptidase II (His6-tagged BsAmpII) are characterized in detail by gel-filtration, analytical ultracentrifugation, and various spectroscopic techniques. Using size-exclusion chromatography and analytical ultracentrifugation, we demonstrate that His6-tagged BsAmpII exists predominantly as a dimer in solution. The enzyme is active and stable at pHs ranging from 6.5 to 8.5. Far-UV circular dichroism analysis reveals that the secondary structures of His6-tagged BsAmpII are significantly altered in the presence of SDS, whereas the presence of 5–10% acetone and ethanol was harmless to the folding of the enzyme. Thermal unfolding of His6-tagged BsAmpII was found to be irreversible and led to the formation of aggregates. The native enzyme started to unfold beyond 0.6 M guanidine hydrochloride and had a midpoint of denaturation at 1.34 M. This protein remained active at concentrations of urea below 2.7 M but experienced an irreversible unfolding by >5 M denaturant. Taken together, this work lays a foundation for potential biotechnological applications of His6-tagged BsAmpII.
Keywords: Aminopeptidase, Bacillus stearothermophilus, Thermal denaturation, Guanidine hydrochloride, Urea
Introduction
Aminopeptidases (EC 3.4.11) selectively remove N-terminal amino-acid residues from peptide and protein substrates. These enzymes are widely distributed in nature [1–3] and play a key role in the biosynthesis of a variety of biologically active peptides [4–8]. The potential of aminopeptidases has been reported in various industrial processes, including the preparation of debittered hydrolysates [9] and the conversion of l-homophenylalanyl amide into l-homophenylalanine, the versatile intermediate for a class of angiotensin-l-converting enzyme inhibitors [10]. Besides, aminopeptidase S (PepS) from Streptococcus thermophilus is probably involved in both bacterial growth and the development of dairy product flavor [11].
Aminopeptidases with two metal ions in an active site are a structurally heterogeneous group and have been divided into clans MF, MG, MH, and MQ on the basis of protein folds, active-site architectures, identity of the metal ions and their substrates and inhibitors [12, 13]. Bacillus stearothermophilus aminopeptidase II (BsAmpII) and closely related peptidases do not resemble the standard metallopeptidases with a metal-binding motif of H-E-x-x-H or H-x-x-E-H and have therefore been grouped together in the new MEROPS peptidase clan MQ. Structural information on clan MQ peptidases has so far relied exclusively on the crystal structure of Staphylococcus aureus S (AmpS) [14] and Thermus thermophilus T (AmpT) [15]. The protein structure of AmpS shows a very elongated dimer with active sites at opposite ends of a large internal cavity that is entirely inaccessible from outside, whereas the molecular structure of AmpT contains five different protomers in an asymmetric unit and these protomers display fully closed to nearly open conformations so that the active site is almost directly accessible.
Experimental data for AmpT provide the strongest evidence to date for an aminopeptidase activity in this group of enzymes. The biochemical data indicate rather broad substrate specificity with a preference for hydrophobic residues, especially leucine, at the amino terminus of the substrate [16]. Functional data for BsAmpII provide more insight into clan MQ peptidases. The enzyme can be inactivated by metal-chelating agents EDTA and 1,10-orthophenanthroline and its activity is easily restored by the addition of exogenous cobalt ions [17]. Equilibrium dialysis further demonstrates that there are 2.2 bound cobalt ions per subunit of BsAmpII and the enzyme has a dissociation constant for cobalt ions of ~5 μM [17]. With the scope of industrial applications, we have previously performed the over-expression of His6-tagged BsAmpII in recombinant Escherichia coli M15 cells [18]. The recombinant enzyme exhibited a marked preference for leucine-p-nitroanilide (Leu-p-NA) and was sensitive to oxidative damage by hydrogen peroxide [19]. Site-directed mutagenesis was further conducted to identify residues essential for the catalytic activity of His6-tagged BsAmpII [20–22]. Although some researchers have focused their studies on the biochemical and structural characterization of clan MQ peptidases, the biophysical properties of this group of enzymes have not been explored before. In the present investigation, we deal with the biophysical characterization of His6-tagged BsAmpII using analytical ultracentrifugation, circular dichroism (CD), and intrinsic tryptophan fluorescence measure. Our results provide clues to the oligomeric state, the stability in different environmental conditions, and the chemical-induced unfolding of the protein molecule.
Materials and methods
Chemicals
Luria-Bertani (LB) medium for bacterial culture was purchased form Difco Laboratories (Detroit, MI, USA). Ni2 + -nitrilotriacetate (Ni2 + -NTA) resin was acquired from Qiagen Inc. (Valencia, CA, USA). Imidazole, guanidine hydrochloride, Leu-p-NA, p-nitroaniline (p-NA), ampicillin, and kanamycin were obtained from Sigma-Aldrich Chemical (St. Louis, MO, USA). All other chemicals were commercial products of analytical or molecular biological grade.
Expression and purification of the recombinant enzyme
The expression and purification of His6-tagged BsAmpII was carried out as described previously [18]. To produce His6-tagged BsAmpII, Escherichia coli M15 cells harboring pQE-LAPII were grown at 37 °C in LB broth supplemented with 100 μg/ml ampicillin and 25 μg/ml kanamycin. When the absorbance of the culture reached approximately 1.0 at 600 nm, isopropyl-β-d-thiogalactopyranoside (IPTG) was added to a final concentration of 0.5 mM and the incubation was continued at 28 °C for 12 h. Then, the cells were harvested by centrifugation at 6,000 × g for 10 min at 4 °C and the pellet was resuspended in 50 mM Tris-HCl buffer (pH 8.0), followed by sonication. The extracts were subjected to affinity chromatography on a Ni2 + -NTA resin column equilibrated with 20 mM Tris-HCl buffer (pH 7.9) containing 0.5 M NaCl and 5 mM imidazole. After a three-times wash with the same buffer, the adherent proteins were eluted from the column by a buffer containing 0.5 M imidazole, 0.5 M NaCl, and 20 mM Tris-HCl buffer (pH 7.9). The collected fractions were pooled and dialyzed against 1,000 volumes of 50 mM Tris-HCl buffer (pH 7.9) with a 10-kDa cutoff membrane to remove imidazole and salt.
Enzymatic activity assay
Aminopeptidase activity was assayed by monitoring the hydrolysis of Leu-p-NA [17]. The molar extinction coefficient ε = 9,620 (at 405 nm) for p-NA was used to calculate the enzymatic activity. The assay mixture contained 0.25 mM Leu-p-NA, 50 mM Tris-HCl buffer (pH 8.0), 1.0 mM CoCl2 and an appropriate amount of the purified enzyme in the final volume of 500 μl. The reaction mixture was incubated at 60 °C for 10 min and stopped by the addition of 500 μl of 10% (v/v) acetic acid. The resultant sample was thoroughly mixed and heated at 100 °C for 5 min, cooled immediately on ice and the absorbance at 405 nm was determined. The reaction mixture in the absence of an enzyme was used as a blank. One unit (U) of His6-tagged BsAmpII activity is defined as the amount of enzyme that releases 1 μmol of p-NA per min at 60 °C.
Size-exclusion HPLC
The molecular size of His6-tagged BsAmpII was determined on a GE Healthcare AKTA basic HPLC system with Superdex 200 column at 25 °C. The elution buffer contained 50 mM Tris-HCl buffer (pH 8.0) and 150 mM NaCl (recommended to prevent electrostatic binding of protein to the column matrix). The column was calibrated using the following marker proteins (GE Healthcare): thyroglobulin (669,000 Da), ferritin (440,000 Da), aldolase (158,000 Da), conalbumin (75,000 Da) and ovalbumin (44,000 Da). Fractions of 1.5 ml were collected at a flow rate of 1.5 ml/min. The protein was monitored using an in-line UV monitor set at 280 nm.
200 column at 25 °C. The elution buffer contained 50 mM Tris-HCl buffer (pH 8.0) and 150 mM NaCl (recommended to prevent electrostatic binding of protein to the column matrix). The column was calibrated using the following marker proteins (GE Healthcare): thyroglobulin (669,000 Da), ferritin (440,000 Da), aldolase (158,000 Da), conalbumin (75,000 Da) and ovalbumin (44,000 Da). Fractions of 1.5 ml were collected at a flow rate of 1.5 ml/min. The protein was monitored using an in-line UV monitor set at 280 nm.
Analytical ultracentrifugation (AUC)
Sedimentation velocity was performed in a Beckman-Coulter XL-A analytical ultracentrifuge. The sample (370 μl) with a protein concentration of 1.5 mg/ml in 50 mM Tris-HCl buffer (pH 7.9) was loaded into the double sector centerpiece separately and built up in a Beckman An-50Ti rotor. Experiments were carried out at 20 °C and a rotor speed of 42,000 rpm. The protein sample was monitored by UV absorbance at 280 nm in a continuous mode with a time interval of 8 min and a step size of 0.003 cm. Multiple scans at different time points were calculated by the SEDFIT program [23, 24]. The sedimentation data were fitted using a two-dimension distribution with respect to frictional ratio 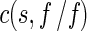 according to Lamm equation [24] (1):
according to Lamm equation [24] (1):
 |
1 |
with a(r,t) denoting the observed optical signal at radius r and time t; χ(s, D,r,t), the solution of the Lamm equation; and  , the dependence of diffusion coefficient (D) on sedimentation coefficient (s) and frictional ratio
, the dependence of diffusion coefficient (D) on sedimentation coefficient (s) and frictional ratio 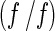 , where
, where
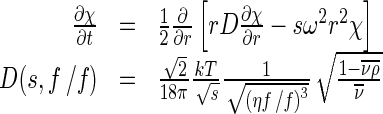 |
with ω denoting angular velocity, T the absolute temperature, k the Boltzmann constant, 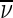 the partial specific volume, ρ the density of the solvent, and η the viscosity.
the partial specific volume, ρ the density of the solvent, and η the viscosity.
All two-dimensional distributions were solved and normalized to a confidence level of p = 0.95 by maximum entropy and a resolution N of 200 with sedimentation coefficients between 0.1 and 20 S. The anhydrous friction ratio is from 1.0 to 2.0 or 3.5 at a resolution of 10.
Circular dichroism (CD) and spectrofluorimetric analyses
Far-UV CD spectra of His6-tagged BsAmpII were recorded at 25 °C on a JASCO model J-815 spectropolarimeter (JASCO Inc., Japan) from 250 to 190 nm in cuvettes using a 1.0-nm bandwidth, 0.1-nm resolution, 0.1-cm path length, 1.0-s response time, and a 100-nm/min scanning speed. The photomultiplier absorbance was always kept below 600 V in the analyzed region. Each scanning was repeated ten times and the average was reported. Data were corrected for the buffer effect and the results were expressed as molar ellipticity [θ] in units of degrees cm2 decimol − 1 according to (2):
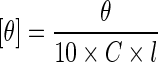 |
2 |
where l represents the light path length (cm), C is the molar concentration of protein (mol/l), and θ represents the observed ellipticity (mdeg).
Thermal denaturation experiments were performed by monitoring the ellipticity at 222 nm. The temperature was increased with heating rates of 0.5, 1.0, and 4.0 °C/min from 20 to 100 °C and the transition midpoint (Tm) was recorded. For the refolding experiment, the temperature was decreased by 0.5, 1, and 4 °C/min and measurements were taken once every minute. Turbidity (denote voltage) was recorded as described elsewhere [25] using a UV detector in a JASCO model J-815 spectropolarimeter equipped with thermoelectric temperature control.
His6-tagged BsAmpII was unfolded with different concentrations of GdnHCl or urea in 50 mM Tris-HCl buffer (pH 8.0) at room temperature. After 12-h incubation, the sample was analyzed in unfolding experiments. Fluorescence spectra of His6-tagged BsAmpII were measured at 30 °C in a Hitachi F-7000 fluorescence spectrophotometer with an excitation wavelength of 295 nm. All spectra were corrected for buffer absorption. The fluorescence emission spectra of protein samples with a concentration of 24 μM were recorded from 300 to 400 nm at a scanning speed of 240 nm/min. The maximal peak of the fluorescence spectrum and the change in fluorescence intensity were used in monitoring the unfolding processes of the enzyme. Both the red shift and the change in fluorescence intensity were analyzed together using the average emission wavelength (AEW) (λ) according to (3) [26]:
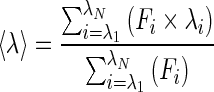 |
3 |
where Fi is the fluorescence intensity at the specific emission wavelength (λi).
Unfolding data analysis
The unfolding data of His6-tagged BsAmpII were treated with the following thermodynamic model by global fitting of the data. The two-state unfolding model (Scheme 1) was described by (4) [27].
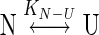 |
Scheme I
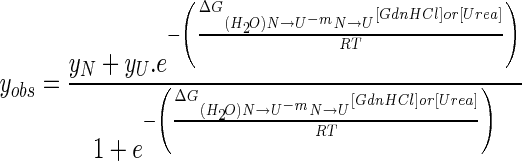 |
4 |
where yobs is the observed biophysical signal; yN and yU are the calculated signals of the native and unfolded states, respectively. [GdnHCl] and [Urea] are the concentrations of GdnHCl and urea respectively and ΔGN − U is the free energy change for the N  U process. The mN − U is the sensitivity to denaturant concentration.
U process. The mN − U is the sensitivity to denaturant concentration.
Results
Quaternary structure of the purified enzyme
To determine the biophysical properties of His6-tagged BsAmpII, the enzyme was purified from crude extract of IPTG-induced E. coli M15 (pQE-LAPII) to apparent homogeneity using Ni2 + -NTA resin. Size-exclusion chromatography on the Superdex 200 column exhibited only the elution of one A280 signal peak corresponding to the molecular mass of approximately 83.47 kDa (Fig. 1). Our previous study showed that the purified enzyme had a molecular mass of 44.5 kDa [18]. Together with the present observation of gel filtration, it can be concluded that the enzyme exists predominantly as a dimer under chromatographic conditions.
200 column exhibited only the elution of one A280 signal peak corresponding to the molecular mass of approximately 83.47 kDa (Fig. 1). Our previous study showed that the purified enzyme had a molecular mass of 44.5 kDa [18]. Together with the present observation of gel filtration, it can be concluded that the enzyme exists predominantly as a dimer under chromatographic conditions.
Fig. 1.

Size-exclusion chromatography of purified His6-tagged BsAmpII. The elution volumes for blue dextran (1), thyroglobulin (2), ferritin (3), aldolase (4), conalbumin (5), and ovalbumin (6) were used to calculate the K av values: K av = (V e − V o)/(V t V o), where V o is the void volume of the column, V t is the total volume of the column and V e is the elution volume of the protein. The K av values for His6-tagged BsAmpII were also plotted to determine their native molecular masses
Oligomeric analysis of His6-tagged BsAmpII by AUC
Among the analytical methods for defining macromolecular assemblies, AUC is experiencing a renaissance due largely to the availability of modern computer-based instrumentation and its resulting expanded applications [28, 29]. In this regard, the oligomeric state of His6-tagged BsAmpII was examined by this approach. The excellent matching of the experimental data points and the fitted curve (Fig. 2a), the homogeneous bitmap picture (data not shown) and the randomly distributed residual values (Fig. 2b) all indicate that a highly reliable model for sedimentation velocity experiments is obtained and AUC is a suitable biophysical probe for assessing the molecular structure of this enzyme. The His6-tagged BsAmpII molecule sediments at 5.71 ±0.18 S (Fig. 2c) corresponding to a species with a molar mass of 109,000 ± 4,000 Da (Fig. 2d), which is in good agreement with the molecular mass calculated from the amino acid sequence of a dimer (47,310 Da).
Fig. 2.

Sedimentation velocity experiments of His6-tagged BsAmpII. a A typical trace of absorbance at 280 nm of the enzyme during the sedimentation velocity experiment. The symbols are experimental data and the lines are computer-generated results by fitting the experimental data to the Lamm equation with the SEDFIT program. b The residuals of model fitting the data in the panel. c The continuous sedimentation coefficient distribution of the enzyme at a high protein concentration of 1.5 mg/ml. d The continuous molar mass distribution of the enzyme at the indicated concentration
pH-induced changes in the activity and structure of His6-tagged BsAmpII
His6-tagged BsAmpII was found to retain >80% of the enzymatic activity in the pH range 6.2–8.0 (Fig 3a). Irreversible inactivation occurred when the enzyme was pre-incubated at pH < 5.5 or pH > 9.5 for 30 min. His6-tagged BsAmpII stability was also examined by measuring intrinsic tryptophan fluorescence as a function of pH, following incubation of the samples at 25 °C. Maximum stability for the His6-tagged BsAmpII structure was observed at pH 7.5. Shift in the emission maximum and a reduction in the fluorescence intensity of the enzyme occurred at both acidic and basic pH conditions (Fig. 3b), indicating that His6-tagged BsAmpII was unfolded under such conditions.
Fig. 3.

Effects of pH on the enzymatic activity a and fluorescence b of His6-tagged BsAmpII. The enzyme activity was assayed in 50 mM sodium-citrate buffer (pH 4–7), 50 mM Tris-HCl buffer (pH 7–9), 50 mM NaHCO3-NaOH (pH 7–11),and 50 mM KCl-NaOH buffer (pH 12–13). Buffer systems used for fluorescence analysis were 50 mM KCl-HCl buffer (pH 1.0), 50 mM glycine-HCl buffer (pH 3.5), 50 mM Tris-HCl buffer (pH 7.5) and 50 mM KCl-NaOH buffer (pH 12 and 13). The results are the mean of three individual experiments
Effects of chemicals and organic solvents on the His6-tagged BsAmpII structure
Effects of SDS on the enzymatic activity and secondary structures of His6-tagged BsAmpII were investigated. SDS, an anionic surfactant, has been demonstrated to unfold and inactivate creatine kinase [30] and aminoacylase [31]. As shown in Fig. 4a, His6-tagged BsAmpII activity was appreciably affected by the addition of SDS, with less than 1.2% of the activity remaining. Considering that SDS could bind to the His6-tagged BsAmpII protein with more than one binding site, both the modified structure and altered charge distribution induced by the specific binding of this surfactant might be responsible for the inhibition. Effects of acetone and ethanol on the enzymatic activity of His6-tagged BsAmpII were also evaluated. After 30-min incubation at 25° C, the residual activity of the treated enzyme was monitored under standard assay conditions (Fig. 4a). Apparently, His6-tagged BsAmpII was stable in the presence of 5–10% acetone or ethanol. To analyze the secondary structures of His6-tagged BsAmpII, far-UV CD measurements were performed under various conditions. The CD spectrum of His6-tagged BsAmpII displays strong peaks of negative ellipticity at 215 nm, indicative of substantial α-helical content (Fig. 4b). A high concentration of SDS reduced the activity of His6-tagged BsAmpII but it is unexpected that the secondary content of this enzyme was significantly increased in the presence of this detergent. Since the addition of SDS to proteins invariably leads to the loss of their biological activity, it is often mistakenly believed that this surfactant completely unfolds proteins. In fact, SDS has been shown to induce and stabilize the secondary structure, particularly β-strands with helical propensity and α-helices in peptides [32–36]. Interestingly, no significant alteration in the CD spectra was observed with the addition of 5–10% acetone and ethanol (Fig. 4b), reflecting that His6-tagged BsAmpII is stable in the presence of organic solvents. Organic-solvent-tolerant enzymes appear to be quite attractive for biotechnological applications [37].
Fig. 4.

Influence of various solvents on enzymatic activity a, CD spectra b and fluorescence c of His6-tagged BsAmpII
As shown in Fig. 4c, SDS had an appreciable effect on the tertiary structure of His6-tagged BsAmpII. This might be the reason for the BsAmpII inactivation by 5 and 10% SDS. Shifts in the emission maximum and a significant decrease in the fluorescence intensity of His6-tagged BsAmpII were also observed in the presence of acetone and ethanol (Fig. 4c), reflecting that the tertiary structure of His6-tagged BsAmpII has been changed upon the addition of these two solvents.
Thermal unfolding of His6-tagged BsAmpII
Thermal denaturation of His6-tagged BsAmpII was followed by monitoring ellipticity at 222 nm under constant heating rates. Figure 5 shows the transition curves obtained from His6-tagged BsAmpII solutions at heating rates of 0.5, 1.0 and 4.0 °C/min, respectively. As compared with the heating rates of 1.0 and 4 °C/min, the thermal transition for 0.5 °C/min appeared at a lower temperature. This resembles a common result for transitions under kinetic control [5, 38, 39]. Also, the lower heating rate resulted in larger changes in the far-UV CD transition, probably indicating an aggregation of the enzyme. The turbidity changes in His6-tagged BsAmpII clearly reflect the coincidence between the onset of thermal unfolding and aggregation (Fig. 5a and b). The impact of aggregation and heating rates on the apparent transition temperatures has also been described for several other cases [40–43]. To further explore if the unfolding reaction was reversible or not, the sample was heated at a constant heating rate. After the thermal denaturation transitions went to completion, the protein solutions were cooled down to 30 °C at the same scan speed. Figure 5 shows the cooling curve obtained with His6-tagged BsAmpII solution at a heating rate of 0.5 °C/min. It is clear that the secondary structures of the native polypeptide were not recovered after the sample was cooled down to the indicated temperature. These results indicate that thermal denaturation of His6-tagged BsAmpII is highly irreversible. The irreversible process seems to be the more common case for most multi-domain proteins [44].
Fig. 5.

Thermal unfolding and aggregation of His6-tagged BsAmpII in 50 mM Tris-HCl buffer (pH 8.0). a Thermal unfolding curves. Transitions were obtained by recording the ellipticity of the sample at 222 nm. b Turbidity (dynode voltage) melting curves recorded at 285 nm. A reduction in V(T) of the sample upon heating >80 °C reflects the precipitation of heat-treated protein
It has been reported that the molecular structure of proteins may be stabilized or destabilized by buffer species [45, 46], thereby making the selection of a buffer for protein formulation a formidable challenge. To evaluate the influence of buffer systems on the thermal denaturation of His6-tagged BsAmpII, we individually used Tris-HCl, HEPES-NaOH, and K2HPO4-KH2PO4 buffers at pH 8.0 as a core buffer to prepare the elution solution for enzyme purification. The purified enzyme was adjusted to a protein concentration of 0.1 mg/ml and then subjected to thermal unfolding by CD signal at 222 nm. As shown in Table 1, the Tm values at a heating rate of 0.5 °C/min for Tris-HCl, HEPES-NaOH and K2HPO4-KH2PO4 buffers were 62.08, 69.31, and 64.08 °C, respectively. The transition points were increased up to 76.42 °C at a heating rate of 4.0 °C/min. No marked difference was observed for the Tm values when either Tris-HCl or K2HPO4-KH2PO4 was used as the core buffer. However, there was a profound increase in the Tm value for the HEPES/NaOH-based sample. These observations indicate that the buffer species used will definitely have a significant affect on the thermal denaturation of His6-tagged BsAmpII.
Table 1.
Effects of heating rate and buffer species on the thermal denaturation of His6-tagged BsAmpII
| Heating rate (°C/min) | T m (°C) | ||
|---|---|---|---|
| Tris-HCl buffer (50 mM, pH 8.0) | HEPES-NaOH buffer (50 mM, pH 8.0) | K2HPO4-KH2PO4 buffer (50 mM, pH 8.0) | |
| 0.5 | 62.08 | 69.31 | 64.07 |
| 1.0 | 67.57 | 74.31 | 68.71 |
| 4.0 | 76.42 | 77.80 | 71.78 |
Molecular properties of His6-tagged BsAmpII associated with GdnHCl- and urea-induced unfolding
The effects of GdnHCl- and urea-induced changes on the structural and functional properties of His6-tagged BsAmpII were studied. Although urea and GdnHCl are believed to have a similar mode of action [26], GdnHCl is a monovalent salt that has both ionic and chaotropic effects [47] but urea has only a chaotropic effect. Thus, urea is an ideal control agent for distinguishing between the ionic and chaotropic effects of GdnHCl. Time-dependent changes in the structural properties and enzymatic activity of His6-tagged BsAmpII in 0–6 M GdnHCl and 0–8 M urea were monitored to standardize the incubation time required to achieve equilibrium. Enzymatic activity can be regarded as the most sensitive probe to study the changes in enzyme conformation during various treatments, because it reflects the conformational change at an active site, enabling very small conformational variations of an enzyme structure to be detected. The effect of increasing concentrations of GdnHCl and urea on the enzymatic activity of His6-tagged BsAmpII is summarized in Fig. 6a. The enzyme treated with GdnHCl below 0.4 M retained 80% of the aminopeptidase activity. An increase in concentration up to 1.2 M resulted in 40% of the remaining activity, whereas the enzyme was completely inactivated after 3 M GdnHCl treatment. However, more than 80% of the aminopeptidase activity was retained at the concentration of urea up to 3 M. A sharp decrease in enzyme activity was observed when urea concentrations were set between 3 and 5 M and the activity was abolished completely at concentrations greater than 6 M.
Fig. 6.

GdnHCl- or urea-induced denaturation of His6-tagged BsAmpII. a Change in enzymatic activity of His6-tagged BsAmpII with increasing concentrations of GdnHCl or urea. The enzyme was incubated with the desired concentration of GdnHCl or urea for 4 h at 4 °C and the enzyme activity was measured under standard assay conditions. His6-tagged BsAmpII activity observed in the absence of a denaturant was taken as 100%. b GdnHCl- or urea-induced changes in the secondary structures of His6-tagged BsAmpII as monitored by negative ellipticity of the sample at 222 nm. c GdnHCl- or urea-induced changes in the AWE value of His6-tagged BsAmpII
GdnHCl- and urea-induced unfolding of His6-tagged BsAmpII was conducted to explore the effect of this denaturant on its secondary structures. The effect of increasing GdnHCl or urea concentrations on the ellipticity of His6-tagged BsAmpII at 222 nm is illustrated in Fig. 6b. Obviously, the secondary elements of BsAmpII were very sensitive to GdnHCl treatment. A large decrease in negative ellipticity was observed at GdnHCl concentrations between 0.5 and 2 M, indicating a significant disruption in the secondary structures of the protein under the experimental conditions. A slight decrease (approximately 8%) in negative ellipticity occurred when the urea concentration was increased up to 2.2 M. However, a rapid decrease in negative ellipticity was observed at urea concentrations between 2.4 and 5 M. By fitting the data with Eq. 4, His6-tagged BsAmpII showed [urea]0.5,N − U and [GdnHCl]0.5,N − U of 4.51 and 1.34 M, respectively, corresponding to a free energy change of 6.86 and 3.21 kcal/mol for the N→U process.
AEW, which reports on the changes in both fluorescence wavelength and intensity, was further used to calculate the thermodynamic parameters of the unfolding process. For His6-tagged BsAmpII, the AEW values in the absence of GdnHCl and at GdnHCl concentrations > 3.2 M were 345 nm and 357 nm, respectively. The transition occurred at 2.84 M GdnHCl (Fig. 6c)., A tryptophan emission λ max of 358 nm was observed with treatment of BsAmpII with 6 M GdnHCl. Normally exposed tryptophan in the unfolded protein shows emission λ max between 340 and 356 nm [48], indicating that incubation of His6-tagged BsAmpII with a higher concentration of GdnHCl leads to significant unfolding of the protein molecule. The urea-induced unfolding of His6-tagged BsAmpII was also demonstrated by tryptophan fluorescence studies by increasing its concentrations. As shown in Fig. 6c, the fluorescence signal of urea-induced His6-tagged BsAmpII followed a monophasic process and the enzyme started to unfold at 2.1 M denaturant with [urea]0.5 N − U of 4.99 M. A low concentration of urea did not induce a change in the AEW value, whereas the tryptophan residues in the protein were highly exposed to the buffer environment at values above 8 M, allowing us to consider the protein completely unfolded.
Taken together, BsAmpII is stable against the unfolding action of urea but not against GdnHCl. It has been reported that GdnHCl is ~2 times more effective as a denaturant than urea in the unfolding of proteins [49]. Consistently, GdnHCl is ~1.6 times more effective than urea in the unfolding of His6-tagged BsAmpII.
Conclusions
In summary, a biophysical investigation of His6-tagged BsAmpII provides more information on the inherent quaternary structure of clan MQ peptidases and contributes to a better understanding of their enzymatic properties, especially a fuller appreciation of the structure–activity relationship at a molecular level. Besides, the ability of enzymes being active in the presence of organic solvents has received much attention in biotechnological applications [50]. There are numerous advantages of using biocatalysts in organic media, including the increased solubility of apolar substrates and the elimination of microbial contamination in the reaction mixture. In this regard, His6-tagged BsAmpII could be useful for practical applications in biotechnology-based processes that will be performed in the presence of organic solvents.
Acknowledgements
The authors thank the anonymous reviewers for their valuable comments and suggestions to improve the quality of the manuscript. Financial support (NSC 100-2313-B-415-003-MY3) from the National Science Council of Taiwan is also acknowledged.
Contributor Information
Tzu-Fan Wang, Email: catalpa0905@gmail.com.
Long-Liu Lin, Email: llin@mail.ncyu.edu.tw.
References
- 1.Gonzales T, Robert-Baudouy J. Bacterial aminopeptidases: properties and functions. FEMS Microbiol. Rev. 1996;18:319–344. doi: 10.1111/j.1574-6976.1996.tb00247.x. [DOI] [PubMed] [Google Scholar]
- 2.Sträter N, Sherratt DJ, Colloms SD. Leucyl aminopeptidase (animal and plant) In: Barrett AJ, Rawlings ND, Woessner JF, editors. Handbook of Proteolytic Enzymes. New York: Academic Press; 1998. pp. 1384–1389. [Google Scholar]
- 3.Walling LL, Gu Y. Plant aminopeptidase: occurrence, function and characterization. In: Taylor A, editor. Aminopeptidases. Austin, TX: Landes Publishing; 1970. pp. 173–219. [Google Scholar]
- 4.Terenius L, Sandin J, Sakurada T. Nociceptin/orphanin FQ metabolism and bioactive metabolites. Peptides. 2000;21:919–922. doi: 10.1016/S0196-9781(00)00228-X. [DOI] [PubMed] [Google Scholar]
- 5.Cappiello M, Lazzarotti A, Buono F, Scaloni A, D’Ambrosio C, Amodeo P, Mendez BL, Pelosi P, Del Corso A, Mura U. New role for leucyl aminopeptidase in glutathione turnover. Biochem. J. 2004;378:35–44. doi: 10.1042/BJ20031336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsumoto H, Nagasaka T, Hattori A, Rogi T, Tsuruoka N, Mizutani S, Tsujimoto M. Expression of placental leucine aminopeptidase/oxytocinase in neuronal cell and its action on neuronal peptides. Eur. J. Biochem. 2001;268:3259–3266. doi: 10.1046/j.1432-1327.2001.02221.x. [DOI] [PubMed] [Google Scholar]
- 7.Goldberg AT, Cascio P, Saric T, Rock KL. The importance of the proteasome and subsequent proteolytic steps in the generation of antigenic peptides. Mol. Immunol. 2002;39:147–164. doi: 10.1016/S0161-5890(02)00098-6. [DOI] [PubMed] [Google Scholar]
- 8.Mitsui T, Nomura S, Itakura A, Mizutani S. Aminopeptidase in health and diseases: role of aminopeptidase in the blood pressure regulation. Biol. Pharmaceut. Bull. 2004;27:768–771. doi: 10.1248/bpb.27.768. [DOI] [PubMed] [Google Scholar]
- 9.Rao MB, Tanksale AM, Ghatge MS, Desphande VV. Molecular and biotechnological aspects of microbial proteases. Microbiol. Mol. Biol. Rev. 1998;62:597–635. doi: 10.1128/mmbr.62.3.597-635.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kamphuis J, Meijer EM, Boesten WHJ, Broxterman QB, Kaptein B, Hermes HFM, Schoemaker HE. Production of natural and synthetic l- and d-amino acids by aminopeptidases and amino amidases. In: Rozzell JD, Wagner F, editors. Biocatalytic Production of Amino Acids and Derivatives. New York: Wiley; 1992. pp. 178–206. [Google Scholar]
- 11.Fernandez-Espla MD, Rul F. PepS from Streptococcus thermophilus: a new member of the aminopeptidase T family of thermophilic bacteria. Eur. J. Biochem. 1999;263:502–510. doi: 10.1046/j.1432-1327.1999.00528.x. [DOI] [PubMed] [Google Scholar]
- 12.Rawlings ND, O’Brien E, Barrett AJ. MEROPS: the protease database. Nucleic Acids Res. 2002;30:343–346. doi: 10.1093/nar/30.1.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rawlings ND, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2012;40:343–350. doi: 10.1093/nar/gkr987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Odintsov SG, Sabata I, Bourenkov G, Rybin V, Bochtler M. Staphylococcus aureus aminopeptidase S is a founding member of a new peptidase clan. J. Biol. Chem. 2005;280:27792–27799. doi: 10.1074/jbc.M502023200. [DOI] [PubMed] [Google Scholar]
- 15.Odintsov SG, Sabata I, Bourenkov G, Rybin V, Bochtler M. Substrate access to the active sites in aminopeptidase T, a representative of a new metallopeptidase clan. J. Mol. Biol. 2005;354:403–412. doi: 10.1016/j.jmb.2005.09.042. [DOI] [PubMed] [Google Scholar]
- 16.Minagawa E, Kaminogawa S, Matsuzawa H, Ohta T, Yamauchi K. Isolation and characterization of a thermostable aminopeptidase (aminopeptidase T) from Thermus aquaticus YT-1, an extremely thermophilic bacterium. Agric. Biol. Chem. 1988;52:755–763. doi: 10.1271/bbb1961.52.1755. [DOI] [Google Scholar]
- 17.Stoll E, Weder HG, Zuber H. Aminopeptidase II from Bacillus stearothermophilus. Biochim. Biophys. Acta. 1976;438:212–220. doi: 10.1016/0005-2744(76)90237-0. [DOI] [PubMed] [Google Scholar]
- 18.Kuo LY, Hwang GY, Lai YJ, Yang SL, Lin LL. Overexpression, purification, and characterization of the recombinant leucine aminopeptidase II of Bacillus stearothermophilus. Curr. Microbiol. 2003;47:40–45. doi: 10.1007/s00284-002-3950-z. [DOI] [PubMed] [Google Scholar]
- 19.Kuo LY, Hwang GY, Yang SL, Hua YW, Chen W, Lin LL. Inactivation of Bacillus stearothermophilus leucine aminopeptidase II by hydrogen peroxide and site-directed mutagenesis of methionine residues on the enzyme. Protein J. 2004;23:295–302. doi: 10.1023/B:JOPC.0000027854.56051.e4. [DOI] [PubMed] [Google Scholar]
- 20.Hwang GY, Kuo LY, Tsai MR, Yang SL, Lin LL. Histidines 345 and 378 of Bacillus stearothermophilus leucine aminopeptidase II are essential for the catalytic activity of the enzyme. Antonie van Leeuwenhoek. 2005;87:355–359. doi: 10.1007/s10482-004-5777-z. [DOI] [PubMed] [Google Scholar]
- 21.Yang SL, Chen RS, Chen W, Lin LL. Identification of glutamate residues important for catalytic activity of Bacillus stearothermophilus leucine aminopeptidase II. Antonie van Leeuwenhoek. 2006;90:195–199. doi: 10.1007/s10482-006-9068-8. [DOI] [PubMed] [Google Scholar]
- 22.Lin LL, Chen YP, Yang JC, Hua YW, Wang WC, Kuo LY. Significance of the conserved Tyr352 and Asp380 residues in the catalytic activity of Bacillus stearothermophilus aminopeptidase II as evaluated by site-directed mutagenesis. Protein J. 2008;27:215–222. doi: 10.1007/s10930-008-9127-2. [DOI] [PubMed] [Google Scholar]
- 23.Brown PH, Schuck P. Macromolecular size-and-shape distributions by sedimentation velocity analytical ultracentrifugation. Biophys. J. 2006;90:4651–4661. doi: 10.1529/biophysj.106.081372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benjwal S, Verma S, Röhm KH, Gursky O. Monitoring protein aggregation during thermal unfolding in circular dichroism experiments. Protein Sci. 2006;15:635–639. doi: 10.1110/ps.051917406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Royer CA, Mann CJ, Matthews CR. Resolution of the fluorescence equilibrium unfolding profile of trp aporepressor using single tryptophan mutants. Protein Sci. 1993;2:1844–1852. doi: 10.1002/pro.5560021106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pace CN. Measuring and increasing protein stability. Trends Biotechnol. 1990;8:93–98. doi: 10.1016/0167-7799(90)90146-O. [DOI] [PubMed] [Google Scholar]
- 28.Hensley P. Defining the structure and stability of macromolecular assemblies in solution: the re-emergence of analytical ultracentrifugation as a practical tool. Structure. 1996;4:367–373. doi: 10.1016/S0969-2126(96)00042-1. [DOI] [PubMed] [Google Scholar]
- 29.Laue TM, Statford WF. Modern applications of analytical ultracentrifugation. Annu. Rev. Biophys. Biomol. Struct. 1999;28:75–100. doi: 10.1146/annurev.biophys.28.1.75. [DOI] [PubMed] [Google Scholar]
- 30.Wang ZF, Huang MQ, Zou XM, Zhou HM. Unfolding, conformational change of active sites and inactivation of creatine kinase in SDS solutions. Biochim. Biophys. Acta. 1995;1251:109–114. doi: 10.1016/0167-4838(95)00088-C. [DOI] [PubMed] [Google Scholar]
- 31.He B, Zhang Y, Zhang T, Wang HR, Zhou HM. Inactivation and unfolding of aminoacylase during denaturation in sodium dodecysulphate solution. J. Protein Chem. 1995;14:349–357. doi: 10.1007/BF01886792. [DOI] [PubMed] [Google Scholar]
- 32.Zhong L, Johnson WC., Jr Environment affects amino acid preference for secondary structure. Proc. Natl. Acad. Sci. U.S.A. 1992;89:4462–4465. doi: 10.1073/pnas.89.10.4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Papavoine CH, Konings RN, Hilbers CW, van de Ven FJ. Location of M13 coat protein in sodium dodecyl sulfate micelles as determined by NMR. Biochemistry. 1994;33:12990–12997. doi: 10.1021/bi00248a007. [DOI] [PubMed] [Google Scholar]
- 34.Pervushin KV, Orekhov VY, Popov AI, Musina LY, Arseniev AS. Three-dimensional structure of (1-71) bacterioopsin solubilized in methanol/chloroform and SDS micelles determined by 15N-1H heteronuclear NMR spectroscopy. Eur. J. Biochem. 1994;219:571–583. doi: 10.1111/j.1432-1033.1994.tb19973.x. [DOI] [PubMed] [Google Scholar]
- 35.Micelli S, Meleleo D, Picciarelli V, Stoico MG, Gallucci E. Effect of nanomolar concentrations of sodium dodecyl sulfate, a catalytic inductor of α-helices, on human calcitonin incorporation and channel formation in planar membranes. Biophys. J. 2004;87:1065–1075. doi: 10.1529/biophysj.103.037200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Montserret R, McLeich M, Bockmann A, Geourjon C, Penin F. Involvement of electrostatic interaction in the mechanism of peptide folding induced by sodium dodecyl sulfate binding. Biochemistry. 2000;39:8362–8373. doi: 10.1021/bi000208x. [DOI] [PubMed] [Google Scholar]
- 37.Gupta MN, Roy I. Enzymes in organic media: forms, functions and applications. Eur. J. Biochem. 2004;271:2575–2583. doi: 10.1111/j.1432-1033.2004.04163.x. [DOI] [PubMed] [Google Scholar]
- 38.Freire E, van Osdol WW, Mayorga OL, Sanchez-Ruiz JM. Calorimetrically determined dynamics of complex unfolding transitions in proteins. Annu. Rev. Biophys. Biophys. Chem. 1990;19:159–188. doi: 10.1146/annurev.bb.19.060190.001111. [DOI] [PubMed] [Google Scholar]
- 39.Sanchez-Ruiz JM. Theoretical analysis of Lumry–Eyring models in differential scanning calorimetry. Biophys. J. 1992;61:921–935. doi: 10.1016/S0006-3495(92)81899-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galisteo ML, Mateo PL, Sanchez-Ruiz JM. Kinetic study on the irreversible thermal denaturation of yeast phosphoglycerate kinase. Biochemistry. 1991;3:2061–2066. doi: 10.1021/bi00222a009. [DOI] [PubMed] [Google Scholar]
- 41.Lepock JR, Ritchie KP, Kolios MC, Rodahl AM, Heinz KA, Kruuv J. Influence of transition rates and scan rate on kinetic simulations of differential scanning calorimetry profiles of reversible and irreversible protein denaturation. Biochemistry. 1992;31:12706–12712. doi: 10.1021/bi00165a023. [DOI] [PubMed] [Google Scholar]
- 42.Plaza del Pino IM, Ibarra-Molero B, Sachez-Ruiz JM. Lower kinetic limit to protein thermal stability: a proposal regarding protein stability in vivo and its relation with misfolding diseases. Proteins. 2000;40:58–70. doi: 10.1002/(SICI)1097-0134(20000701)40:1<58::AID-PROT80>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 43.Vogl T, Jatzke C, Hinz HJ, Benz J, Huber R. Thermodynamic stability of Annexin V E17G: equilibrium parameters from an irreversible unfolding reaction. Biochemistry. 1997;36:1657–1668. doi: 10.1021/bi962163z. [DOI] [PubMed] [Google Scholar]
- 44.Fitter J. The perspectives of studying multi-domain protein folding. Cell. Mol. Life Sci. 2009;66:1672–1681. doi: 10.1007/s00018-009-8771-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng JY, Janis LJ. Influence of pH, buffer species, and storage temperature on physiochemical stability of a humanized monoclonal antibody LA298. Int. J. Pharm. 2006;308:46–51. doi: 10.1016/j.ijpharm.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 46.Katayama DS, Nayar R, Chou DK, Valente JJ, Cooper J, Henry C, Vander Velde DG, Villarete L, Liu CP, Manning MC. Effect of buffer species on the thermally induced aggregation of interferon-tau. J. Pharm. Sci. 2006;95:1212–1226. doi: 10.1002/jps.20471. [DOI] [PubMed] [Google Scholar]
- 47.Monera OD, Kay CM, Hodges RS. Protein denaturation with guanidine hydrochloride or urea provides a different estimate of stability depending on the contributions of electrostatic interactions. Protein Sci. 1994;3:1984–1991. doi: 10.1002/pro.5560031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2. New York: Kluwer Academic/Plenum Publishers; 1999. [Google Scholar]
- 49.Del Vecchio P, Granziano G, Granata V, Barone G, Mandrich L, Rossi M, Manco G. Denaturing action of urea and guanidine hydrochloride towards two thermophilic esterases. Biochem. J. 2002;367:857–863. doi: 10.1042/BJ20020695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karan R, Capes MD, DasSarma S. Function and biotechnology of extremophilic enzymes in low water activity. Aquat. Biosys. 2012;8:4. doi: 10.1186/2046-9063-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]