

Abstract
With the desire to understand the contributions of multiple cellular elements to the development of a complex tissue; such as the numerous cell types that participate in regenerating tissue, tumor formation, or vasculogenesis, we devised a multi-colored cellular transplant model of tumor development in which cell populations originate from different fluorescently colored reporter gene mice and are transplanted, engrafted or injected in and around a developing tumor. These colored cells are then recruited and incorporated into the tumor stroma. In order to quantitatively assess bone marrow derived tumor stromal cells, we transplanted GFP expressing transgenic whole bone marrow into lethally irradiated RFP expressing mice as approved by IACUC. 0ovarian tumors that were orthotopically injected into the transplanted mice were excised 6-8 weeks post engraftment and analyzed for bone marrow marker of origin (GFP) as well as antibody markers to detect tumor associated stroma using multispectral imaging techniques. We then adapted a methodology we call MIMicc- Multispectral Interrogation of Multiplexed cellular compositions, using multispectral unmixing of fluoroprobes to quantitatively assess which labeled cell came from which starting populations (based on original reporter gene labels), and as our ability to unmix 4, 5, 6 or more spectra per slide increases, we've added additional immunohistochemistry associated with cell lineages or differentiation to increase precision. Utilizing software to detect co-localized multiplexed-fluorescent signals, tumor stromal populations can be traced, enumerated and characterized based on marker staining.1
Keywords: Medicine, Issue 79, Immunology, Medicine, Cellular Biology, Molecular Biology, Genetics, Anatomy, Physiology, Biomedical Engineering, Immunohistochemistry (IHC), Microscopy, Fluorescence, Regeneration, Cellular Microenvironment, Tumor Microenvironment, Cell Biology, Investigative Techniques, Biological Phenomena, Mesenchymal stem cells (MSC), Tumor/Cancer associated fibroblasts (TAF/CAF), transgenic mouse model, regenerative medicine, wound healing, cancer
Introduction
Understanding tissue development and repair is significant to elucidating participating cellular components in wound healing,2,3 regenerative medicine, developmental biology and tumor biology. Under circumstances of repair, numerous cell types infiltrate the surrounding microenvironment to aid in vascularization, ECM deposition, proliferation and tissue restructuring. Cellular factors and phenotypes can be identified based on multiparameter, multiplexed markers that can identify the localization, differentiation status and interaction between cellular components within the investigated microenvironment. Herein, we describe tumor development as a prototypical example for this multicolor-multicellular transplant model followed by multispectral imaging and spectral unmixing methodology.
Tumor progression is a multistep process that is marked by several acquired capabilities that include enhanced proliferation, antiapoptotic, invasive and angiogenic properties.4 Tumor development is facilitated by non-neoplastic cells that are recruited into the surrounding environment to provide growth factors, structural matrices, vascular networks and immune modulation.1,5,6 This microhabitat consists of cells derived from local, neighboring tissues such as adipose, and blood vessels and distant sources such as bone marrow derived cells 1. The extent of non-neoplastic cell incorporation depends on the demand from the tumor, which often corresponds with the stage/grade of the tumor. To comprehend the role of the tumor supportive microenvironment, one must understand the origin and the differentiation potential of the non-neoplastic cell populations.
This protocol has been designed to aid in the interpretation of tumor progression through the visualization of both the bone marrow derived cellular components, and the local tissue derived cells. Utilizing fluorescent reporter gene-expressing transgenic mice, we transplanted GFP (green-fluorescent protein) bone marrow into a lethally irradiated RFP (red-fluorescent protein) mouse. Following successful bone marrow engraftment, a syngeneic tumor cell line is injected orthotopically and allowed to engraft for 4-8 weeks. The resulting tumor is excised from the mouse and processed for immunofluorescent (IF) staining to visualize the stromal components. Multiplexing IF markers is a commonly used technique that involves significant optimization7-9, however using a multispectral imaging/unmixing platform improves the potential for fluorescent marker combinations that possess spectral overlap. Herein we present a technique we call MIMicc- multispectral interrogation of Multiplexed cellular compositions to stain and analyze up to eight markers within a tumor section on a single slide in order to analyze the cellular origins, cellular differentiation status and cell-cell interactions of components within the tumor microenvironment. This simplified example has the potential to be expanded upon in order to analyze five, six, or more markers utilizing antibodies or intracellular promoter-driven fluorescent expression. Table 1 lists potential fluorescent antibody staining combinations with appropriate species taken into account.
Protocol
Syngeneic Murine Bone Marrow Transplantation (BMT) as approved by institutional IACUC protocols (Note: Success of this protocol requires utilization of any labeled cell type(s) as the target for multispectral analysis, and to facilitate the downstream analysis, one can exploit any genetic or stable cellular labeling. We suggest that any cell, tissue, or organ-to-be can be applied to a transplant model and that the transplant can contain as many unique labeled populations as the research deems useful. Additionally, multi-labeled cellular systems, such as those found in transgenic mice containing multiple lineage restricted-fluorescently labeled populations -can be designed to eliminate transplant complications for the study of certain pathological states.
Lethally irradiate BMT recipient mice with a single dose of 9.5 Gy four hours before reconstitution with donor bone marrow. (BMT GFP recipients should be 6-10 weeks of age).
Wet the mouse pelt with 70% ethanol before clipping and peeling it back to expose the hind limbs with sterile surgical scissors. Remove the entire leg including the iliac crest at the sacroiliac joint. Discard the foot by cutting just above the ankle and then thoroughly remove all muscle, ligaments and excess tissue from bone. Flush marrow from the tibia, fibia and iliac crest with a 23 G needle using 2% sterile FBS in PBS (PBS-2). (BMT RFP donors should be sacrificed according to institutional standards.)
Slice and gently crush the remaining bone into fragments using a scalpel and forceps and then place into 50 ml conical tube with 3 μg/ml collagenase I for 1 hr at 37 °C at 300 rpm.
Top off the tube with PBS-2 and collect supernatant into a fresh tube and combine with flushed cells. Spin down at 250rpm for 5 min at 4 °C.
Resuspend cells in PBS-2 and filter through 40 nm cell filter. At this point, the cells can be labeled for fluorescent activated cell sorting (FACS) if one wants to manipulate specific populations for the BMT.
Resuspend 2 x 106 cells per 100 μl PBS, per mouse. Use 29 G needle to systemically inject into irradiated GFP recipient mice by tail or retro-orbital vein-bevel-side-up. Inject one mouse with 100 μl PBS only as engraftment control. Use a heat lamp to dilate vessels prior to injection and use a tail vein injection restraint upon injection. Have sterile gauze available to apply light pressure to the tail post injection.
Three weeks post BMT, collect blood retro-orbitally and analyze by flow cytometry to confirm the presence of GFP circulating cells and the absence of RFP circulating cells. The control mouse will have died at this point.
2. Tumor Engraftment According to Institutional IACUC Guidelines
Grow ID8 ovarian murine tumor cells in vitro prior to in vivo injection (1 x 106 cells per mouse). Day of injection, trypsinize cells, wash with PBS and resuspend in PBS at 1 x 106 cells per 100 μl.
Inject tumor cells into intaperitoneal cavity of the mouse, needle should be bevel-side-up. Sterilize injection sight with 70% ethanol. Inject into the lower left or lower right quadrant of the abdomen (cranial and slightly medial to the inferior set of abdominal nipples of female mouse), avoid hitting the organs. Restrain mouse by grabbing by the scruff and holding the tail with the pinky or ring finger. Mice can be anesthetized with isoflurane for this procedure.
Sacrifice the mice when signs of tumor engraftment, such as slight abdominal bloating, are apparent. This will occur over 4-12 weeks.
Excise tumors from IP cavity. Tumors within the cavity will look like white nodules on and around the surface of the organs. With a scalpel, remove the tumors and place in a tube/jar of 10% formalin for 24 hr fixation. Histological preparation can be out-sourced to pathology/histology core if reagents and materials are not readily available for paraffin embedding and slide preparation. Section tissue into 5-8 μm slices on glass slides for subsequent staining procedures.
3. Multi-parameter Immunofluorescent Staining
Prepare Single color control slides for every fluorescent probe utilized in the experiment. In this scenario, four colors are used. Therefore, four slides for each individual color as well as one slide for background noise control and finally one slide for the full combination staining. (Total slides = 6) All slides will be processed side by side in an identical fashion to minimize experimental variation.
Bake paraffin sections at 56 °C for 1 hr.
Wash slides 3x 10 min in xylene.
Wash 2x 5 min 100% ethanol.
Wash 1x 3 min 95%ethanol
Wash 1x 2 min90%ethanol.
Wash 1x 2 min70%ethanol.
Wash 1x 2 min in water. Slowly immerse the staining rack in and out of the water 10x.
During the last wash series, pre-boil sodium citrate buffer for 2 min (microwave on high). (Can replace with tris-EDTA buffer depending on the antibodies in use)
Boil the slides for 20 min in sodium citrate buffer (microwave on 10% power or in a pressure cooker). If buffer boils, turn off microwave and let sit.
Remove from heat source and let the slides rest for 30 min in the sodium citrate buffer to cool down.
Rinse the slides in water for 5 min.
Mark around tumor sections with Pap Pen and put 2% BSA/1% FBS maleic acid blocking buffer for 1 hr. Prepare the slides individually as to not let them dry out during this step.
Prepare primary antibodies at optimal dilution in blocking buffer. (If the antibodies in use are all of different species (or IgG chain variation-ex: IgG1 vs IgG2a of same species) they may be used in combination during a single incubation period. If antibody species overlap, then incubations must be carried out sequentially to minimize antibody cross-reaction.)
At this point, leave two slides with blocking buffer only. These two slides will serve as unstained control and nuclear stain control. The other single color controls will be incubated with individual primary antibodies. Only the analysis slide will have a combination of primary antibodies.
Incubate slides with primary antibody in a moisture chamber box on slow-rotating shaker at 4 ° overnight (~16-20 hr) (Note: when working with 4+ fluorophores, sequential staining or direct conjugation may be necessary to minimize antibody cross-reaction. Under these circumstances repeat steps 1.16-1.21 until all antibodies are applied to the slides.)
Wash slides for 10 min in PBST (2x) gently on shaker
While wash is going, prepare Alexafluor 488, 594 and 647 secondary antibodies for a final dilution of 1:1,000 in blocking buffer. (Make sure these antibodies and dilutions are kept at 4 °C and in the dark at all times to preserve fluorescence across batches and over time.)
On the single color control slides, place single Alexafluor dilutions matching the species of the primary antibody used. All secondary antibodies will be placed on the analysis slide. The unstained and the nuclear stained control slides will remain with blocking buffer throughout this step.
Incubate in a moisture chamber box (covered with aluminum foil to avoid light exposure) on slow rotating shaker at RT for 2 hr.
Wash slides for 5 min in PBST (2x) gently on shaker. Cover with aluminum foil to protect from light.
Add DAPI (1:10,000) for 1 min to the DAPI-only slide and the analysis slide. Cover with aluminum foil to protect from light. The other slides may be left covered in the wash containers.
Wash the two DAPI-incubated slides in a separate container for the first 5 min wash as to not contaminate the other slides with DAPI stain. For the second 5 min wash, the slides can be combined in PBST (all washes should be conducted on a shaker). Cover with aluminum foil to protect from light.
Briefly dip slides in water before cover-slipping (1.5 thickness with aqueous mounting media to preserve fluorescence)
Let dry 3 hr in dark at RT.
Seal the edges of the cover slip with nail hardener. (Do not use nail polish as the acetone may quench the fluorescent signal).
Analyze slides immediately or store at 4 °C for future analysis.
4. Multispectral Imaging
Collect multispectral Images utilizing a Nuance EX camera (containing a liquid crystal tunable filter) on an Olympus X-63 upright microscope with Nuance software for data collection.
Collect images using an oil objective to increase the resolution.
- Initial set-up includes setting the spectral range for each fluorophore in use.
- For 4 color:
- DAPI: 420-480nm
- AF488: 490-580nm
- AF594: 590-640nm
- AF647: 650-720nm
Take an initial image of the analysis slide to obtain the proper exposure times for each of the spectral ranges.
- Create "spectral library" that the software will use to identify and separate the signal-to-noise ratio of the fluorophores from each other and background signal. (Note: Each fluorochrome must have its own single color control slide in order to assemble a spectral library with appropriate wavelength curves to ensure proper analysis of the data set. Example: 3 fluorophores = 4 control slides; 5 fluorophores = 6 control slides).
- Image the unstained slide first. This slide will be designated within the spectral library as background.>
- Image the DAPI-only slide second. This slide will be used to designate the DAPI-stained nuclei with the "draw" tool and will be saved within the spectral library as DAPI.>
- Image the AF488-only slide third. This slide will be used to designate the AF488-stained regions with the "draw" tool and will be saved within the spectral library as AF488.>
- Image the AF594-only slide fourth. This slide will be used to designate the AF594-stained regions with the "draw" tool and will be saved within the spectral library as AF594.>
- Image the AF647-only slide fifth. This slide will be used to designate the AF647-stained regions with the "draw" tool and will be saved within the spectral library as AF647.>
- Following the collection of each individual spectral component, save the spectral library and the protocol.>
Once the spectral library is complete, find regions of interest on the analysis slide on the microscope and collect/save images.>
5. Quantitative Analysis of Multispectral Images
Import a representative assortment of acquired Nuance images using InForm Software.
Open the saved spectral library as directed by the program
Identify tissue regions of interest to be analyzed. (e.g. Tumor tissue versus adipose tissue).
Identify individual cells based on DAPI-stain. Cytoplasm will be defined automatically based on the shape of the nucleus.
Pick the fluorescent intensity read-out of choice for each fluorescent parameter (e.g. mean, standard deviation)
Batch the acquired images and let the software process using the proposed algorithm.
When the image processing is finished, merge the result files.
Open the results in Excel (or any other analytical software package) and plot two fluorescent intensities against each other on XY axis. Each fluorescent intensity is given per cell. The double positive cells will appear in the upper right quadrant. User must define the baseline fluorescent intensities.
Representative Results
Using a multispectral imaging technique to analyze tumors engrafted in our transgenic BMT mouse model, we are able to discern stromal tumor components that are of bone marrow origin. The initial BMT was confirmed three weeks post-transplant by flow cytometry (Figure 1). Orthotopically injected unlabeled ovarian tumors following BMT engraftment confirmations were excised and formalin fixed 6 weeks following initial tumor injection. Paraffin embedded tumor sections were sectioned in <8 μm slices and stained. Appropriate single color control slides were made for each fluorophore including one for background autofluorescence. Table 1 includes potential specie and fluorochrome combinations for 2-7 parameters. Following the staining procedure, images were acquired using a Nuance camera attachment and Nuance Software. After creating a spectral library with single color control slides (Figure 2A), we were able to "unmix" the images of multi-labeled ovarian tumor sections that were taken showing the expression of GFP (Antibody #1) along with nuclear DAPI and two other markers (antibodies #2 and #3). (Figure 2B) Addition of more markers is possible and Figures 2C-D shows the image of a tumor section with six markers plus a nuclear stain. Finally, all acquired images were imported into InForm (CaliperLS, Hopkinton, MA) software for analysis. Nuance (CaliperLS, Hopkinton, MA) software is compatible with other packages like MetaMorph (Molecular Devices) or IPLab (Scanalytics, BD Biosciences). Furthermore, images and spectral datasets can also be saved as TIFF files and viewed in any program that reads this file format. Using the InForm Software, we were able to classify the tissue regions of interest within the tumor sections (Figures 3A and E) and set algorithms for the fluorescent intensities of each fluorochrome per cell based on DAPI nuclear stain (Figures 3B-C). Finally, we were able to plot each individual cell based on the fluorescent intensities of two markers, AF488 and AF647 (Figure 3D). We established a double positive cell population based on a determined fluorescent intensity threshold (Figure 4). As the data set becomes more complicated with multiple parameters, there is potential for the data to be analyzed using SPADE software10 (spanning-tree progression analysis of density normalized events), although this has yet to be performed. This software enables the visualization of co-expression trends over large data sets and can imply association/relationships between clusters of co-expressed markers within analyzed tissue sections.
Table 1. Immunofluorescent staining control preparation for 2-7 color multiplex staining. (A) Includes up to a four-color option. (B) includes up to a seven-color option utilizing an antibody that breaches the near infrared portion of the light spectrum whereas (C) includes a seven color option within the visible light spectrum.
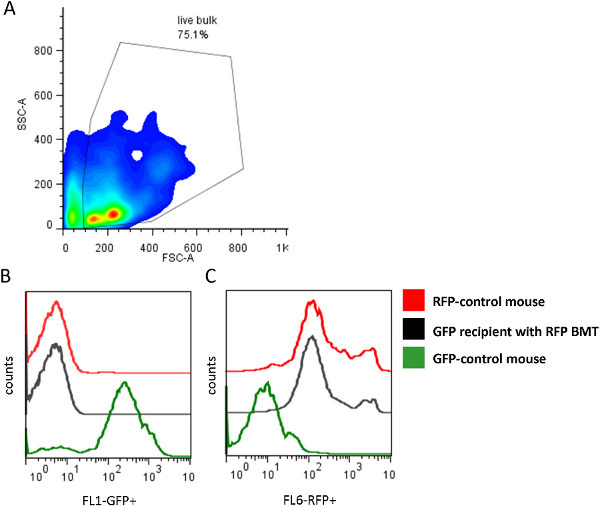 Figure 1. Bone marrow transplant confirmation by flow cytometry. (A) Dot plot of the circulating hematopoietic cells population within the GFP transgenic recipient mouse which is positive (B) for RFP+ hematopoietic cells compared to control RFP+ and control GFP+ mice and negative (C) for GFP+ cells.
Figure 1. Bone marrow transplant confirmation by flow cytometry. (A) Dot plot of the circulating hematopoietic cells population within the GFP transgenic recipient mouse which is positive (B) for RFP+ hematopoietic cells compared to control RFP+ and control GFP+ mice and negative (C) for GFP+ cells.
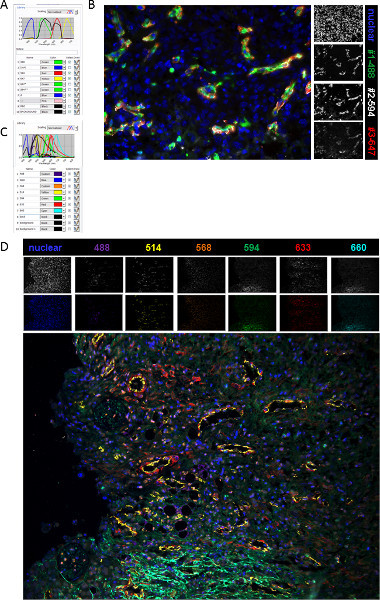 Figure 2. Spectrally unmixed image process. (A) Screenshot of the 4 color spectral library created for the analysis of the ovarian tumor sections. (B) Multispectral image a murine ovarian tumor section showing fluorescently labeled cells; #1: AF488 (ex 495 nm/em 510 nm); #2: AF594 (ex590/em617 nm); #3: AF647(ex 650/em 668 nm) and nuclear: DAPI(ex 350/em 470 nm). Single colors are shown in white. (C) Screenshot of the 7 color spectral library created for the analysis of the ovarian tumor sections. (D) Multispectral image a murine ovarian tumor section showing fluorescently labeled cells; nuclear: DAPI (ex350/em470 nm); AF488 (ex 495/em 519 nm); AF514 (ex518/em540 nm); AF568 (ex 578/em 603 nm); AF594 (ex 590/em 617 nm); AF633(ex 632/em 647 nm); AF660 (ex 663/em 690 nm) and. Single colors are shown in white as well as the matching composite color. Click here to view larger figure.
Figure 2. Spectrally unmixed image process. (A) Screenshot of the 4 color spectral library created for the analysis of the ovarian tumor sections. (B) Multispectral image a murine ovarian tumor section showing fluorescently labeled cells; #1: AF488 (ex 495 nm/em 510 nm); #2: AF594 (ex590/em617 nm); #3: AF647(ex 650/em 668 nm) and nuclear: DAPI(ex 350/em 470 nm). Single colors are shown in white. (C) Screenshot of the 7 color spectral library created for the analysis of the ovarian tumor sections. (D) Multispectral image a murine ovarian tumor section showing fluorescently labeled cells; nuclear: DAPI (ex350/em470 nm); AF488 (ex 495/em 519 nm); AF514 (ex518/em540 nm); AF568 (ex 578/em 603 nm); AF594 (ex 590/em 617 nm); AF633(ex 632/em 647 nm); AF660 (ex 663/em 690 nm) and. Single colors are shown in white as well as the matching composite color. Click here to view larger figure.
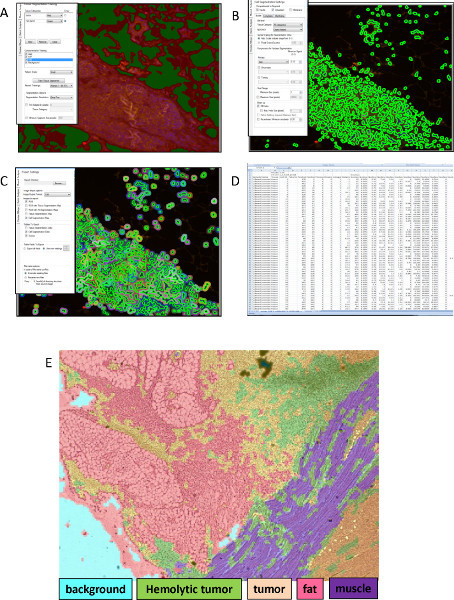 Figure 3. MIMicc Depiction of quantitative Inform analysis. (A) First image represents the classification of tissue segments followed by (B) the segmentation of the cells based on nuclear stained DAPI. (C) The quantification of fluorescent label can be measured in the nucleus and the cytoplasm and exported into (D) excel for further analysis. (E) The tissue segmentation algorithm is capable of further identifying tissue subtypes based on tissue architecture and not solely on fluorochrome identification providing a more robust analytical tool to classify staining in distinct tissue regions. In this image, the computer has been trained to identify 5 sub-regions based on architectural patterns within the tissue: background is defined by blank space, hemolytic tumor is defined by abundance of red blood cells within the tissue, tumor is defined by densely populated cells, fat is defined by large, symmetrical, empty cellular compartments, and muscle is defined by a striated pattern. Click here to view larger figure.
Figure 3. MIMicc Depiction of quantitative Inform analysis. (A) First image represents the classification of tissue segments followed by (B) the segmentation of the cells based on nuclear stained DAPI. (C) The quantification of fluorescent label can be measured in the nucleus and the cytoplasm and exported into (D) excel for further analysis. (E) The tissue segmentation algorithm is capable of further identifying tissue subtypes based on tissue architecture and not solely on fluorochrome identification providing a more robust analytical tool to classify staining in distinct tissue regions. In this image, the computer has been trained to identify 5 sub-regions based on architectural patterns within the tissue: background is defined by blank space, hemolytic tumor is defined by abundance of red blood cells within the tissue, tumor is defined by densely populated cells, fat is defined by large, symmetrical, empty cellular compartments, and muscle is defined by a striated pattern. Click here to view larger figure.
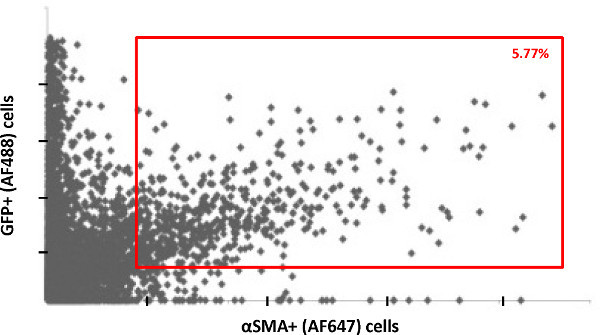 Figure 4. Plot of individual cells from 20 acquired images show the double positive cells based on fluorescent intensities of GFP labeled with AF488 and αSMA labeled with AF647.
Figure 4. Plot of individual cells from 20 acquired images show the double positive cells based on fluorescent intensities of GFP labeled with AF488 and αSMA labeled with AF647.
Supplementary Document. Tips and Tricks for Multiplexing 5 or more probes.
Discussion
Herein we describe the application of multispectral imaging we describe as MIMicc- multispectral interrogation of Multiplexed cellular compositions, to analyze the bone marrow derived stromal components of the tumor microenvironment, however this methodology and concept can be applied to deciphering other cellular elements that compose a complex tissues such as those seen during wound healing reactions or during a regenerative tissue. In these experiments we utilize transgenic mice to fluorescently distinguish the bone marrow derived cells from the host derived cells within an engrafted tumor microenvironment, however this technique lends itself to use of any reporter gene labeled cells, and is particularly amendable to cells expressing gene labels that are easily discernible by immunohistochemistry, such as LacZ, glucuronidase, His(6X)-, HA, FLAG-tag labeled cells, etc.11.
In our model, following the sacrifice of transplanted mice, the tumor sections were processed and stained with antibodies to identify the origin of the cell; RFP+ bone marrow donor derived or GFP+ host tissue derived. In Figure 2, we show two markers co-expressing with host derived GFP+ cells within the tumor microenvironment. However, we show the potential for seven color staining using the same system in Figure 2D.
These data are a representation of what is capable utilizing our transplant and multispectral imaging model. This system is extremely flexible and can accommodate additional parameters including: 1-altered BMT populations/transgenic mouse models; 2-alternate tumor/disease models; or 3-alternate markers (e.g. surface, intracellular, phospho-proteins). Usually, with increasing numbers of immunohistochemical stains on the same slide, data analysis becomes arduous, however using a multispectral imaging platforms and associated unmixing software enables efficient, reliable and reproducible results. We provide potential antibody species and fluorescent label combinations in Table 1. Furthermore, this technique allows for versatility in probe/antibody selection because of the potential to use both frozen and paraffin embedded tissue sections making this technique applicable to clinical tissue samples in addition to basic research models.
Disclosures
FCM, CMB, and ELS have no conflicts and nothing to disclose.
Acknowledgments
We are grateful to the discussions, guidance and support from Drs. Michael Andreeff MD, PhD., and Jared Burks PhD. from the MD Anderson Flow Cytometry and Cellular Imaging Core Facility. This work was supported in part by grants from the National Cancer Institute (RC1-CA146381, CA-083639, R01NS06994, CA116199 and CA109451 for FCM. ELS is also supported by the Army Department of Defense (BC083397).
References
- Kidd S, et al. Origins of the tumor microenvironment: quantitative assessment of adipose-derived and bone marrow-derived stroma. PLoS ONE. 2012;7:e30563. doi: 10.1371/journal.pone.0030563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintavalla J, et al. Fluorescently labeled mesenchymal stem cells (MSCs) maintain multilineage potential and can be detected following implantation into articular cartilage defects. Biomaterials. 2002;23:109–119. doi: 10.1016/s0142-9612(01)00086-2. [DOI] [PubMed] [Google Scholar]
- Sasaki M. Mesenchymal stem cells are recruited into wounded skin and contribute to wound repair by transdifferentiation into multiple skin cell type. Journal of immunology. 2008;180:2581–2587. doi: 10.4049/jimmunol.180.4.2581. [DOI] [PubMed] [Google Scholar]
- Spaeth EL. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE. 2009;4:e4992. doi: 10.1371/journal.pone.0004992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wels J, Kaplan RN, Rafii S, Lyden D. Migratory neighbors and distant invaders: Tumor-associated niche cells. Genes and Development. 2008;22:559–574. doi: 10.1101/gad.1636908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat. Med. 2011;17:1359–1370. doi: 10.1038/nm.2537. [DOI] [PubMed] [Google Scholar]
- Bataille F. Multiparameter immunofluorescence on paraffin-embedded tissue sections. Applied immunohistochemistry & molecular morphology : AIMM / official publication of the Society for Applied Immunohistochemistry. 1097;14:225–228. doi: 10.1097/01.pai.0000162009.31931.10. [DOI] [PubMed] [Google Scholar]
- van Vlierberghe RL, et al. Four-color staining combining fluorescence and brightfield microscopy for simultaneous immune cell phenotyping and localization in tumor tissue sections. Microscopy research and technique. 2005;67:15–21. doi: 10.1002/jemt.20181. [DOI] [PubMed] [Google Scholar]
- Robertson D, Savage K, Reis-Filho JS, Isacke CM. Multiple immunofluorescence labelling of formalin-fixed paraffin-embedded (FFPE) tissue. BMC cell biology. 2008;9:13. doi: 10.1186/1471-2121-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu P, et al. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotechnol. 2011;29:886–891. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belizario JE, Akamini P, Wolf P, Strauss B, Xavier-Neto J. New routes for transgenesis of the mouse. Journal of applied. 2012;53:295–315. doi: 10.1007/s13353-012-0096-y. [DOI] [PubMed] [Google Scholar]