

Abstract
The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that mediates the toxic and biological effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, dioxin) and a wide variety of structurally diverse ligands through its ability to translocate into the nucleus and bind to a specific DNA recognition site (the dioxin-responsive element [DRE]) adjacent to responsive genes. Although the sequence of the DRE is well defined, several reports suggested that the nucleotide specificity of AhR DNA binding may vary depending on the structure of its bound ligand. Given the potential toxicological significance of this hypothesis, an unbiased DNA-selection-and-PCR-amplification approach was utilized to directly determine whether binding and activation of the AhR by structurally diverse agonists alter its nucleotide specificity of DNA binding. Guinea pig hepatic cytosolic AhR activated in vitro by equipotent concentrations of TCDD, 3-methylcholanthrene, β-naphthoflavone, indirubin, L-kynurenine, or YH439 was incubated with a pool of DNA oligonucleotides containing a 15-base pair variable region consisting of all possible nucleotides. The AhR-bound oligonucleotides isolated by immunoprecipitation were PCR amplified and used in subsequent rounds of selection. Sequence analysis of a total of 196 isolated oligonucleotides revealed that each ligand-activated AhR:ARNT complex only bound to DRE-containing DNA oligonucleotides; no non-DRE-containing DNA oligonucleotides were identified. These results demonstrate that the binding and activation of the AhR by structurally diverse agonists do not appear to alter its nucleotide specificity of DNA binding and suggest that stimulation of gene expression mediated by direct DNA binding of ligand-activated AhR:ARNT complexes is DRE dependent.
Key Words: Ah Receptor, TCDD, DRE, dioxin.
The aryl hydrocarbon receptor (AhR) is a ligand-activated basic-helix-loop-helix-Per/ARNT/SIM (bHLH-PAS) containing transcription factor that has been classically regarded as a xenobiotic sensor. In this role, lipid soluble environmental chemicals bind to the cytosolic AhR protein complex and stimulate its translocation into the nucleus where the AhR binds the aryl hydrocarbon receptor nuclear translocator (ARNT) protein. This ligand:AhR:ARNT heterodimer binds with high affinity to specific DNA recognition sites known as dioxin-responsive elements (DREs) to stimulate transcription of downstream genes including those involved in xenobiotic metabolism (Denison et al., 1998a, 2011). The best characterized and highest affinity ligands for the AhR are the halogenated aromatic hydrocarbons (HAHs), including 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, dioxin), some polycyclic aromatic hydrocarbons (PAHs) and PAH-like chemicals, such as 3-methylcholanthrene (3MC), benzo(a)pyrene, and a variety of flavonoids (ie, β-naphthoflavone [βNF] and others). These chemicals are designated “classical” AhR ligands due to similarities in their physiochemical properties (planar, hydrophobic) and ability to bind to and activate the AhR and AhR signaling pathway (Denison and Nagy, 2003; Denison et al., 1998b).
In contrast to the classical AhR ligands, the nonclassical AhR ligands are a group of disparate, structurally diverse (often non-planar) chemicals that have also been shown to bind and/or activate the AhR and AhR signal transduction (Denison and Nagy, 2003; Denison et al., 1998b). Recent studies propose that the ligand binding promiscuity of the AhR may result from differential binding of these structurally diverse ligands within the binding cavity of the AhR ligand-binding domain (LBD) (DeGroot et al., 2011; Whelan et al., 2010; Xing et al., 2012; Zhao et al., 2010), suggesting additional mechanisms by which ligands can bind to the AhR to initiate biological and/or toxicological effects. Although some studies report ligand-specific effects on AhR functionality such as the differential recruitment of coactivator proteins to the AhR:ARNT:DRE complex (Hestermann and Brown, 2003; Zhang et al., 2008), others suggest more dramatic differences. It has been proposed that some nonclassical ligands relax the nucleotide specificity of DNA binding such that the ligand:AhR:ARNT complex can bind to DNA that does not contain the 5 base pair invariant core sequence 5′-GCGTG-3′ of the canonical DRE. For example, Matikainen et al. (2001) reported that a dihydrodiol metabolite of dimethylbenz[a]anthracene (DMBA), but not TCDD, induced AhR-dependent gene expression through 2 degenerate DREs in the murine Bax promoter. In a second study, 3MC and the polyphenol quercetin were reported to stimulate AhR:ARNT DNA binding to and reporter gene expression from a GC-rich DRE-like DNA response element present in the human paraoxonase 1 (PON1) promoter that was unresponsive to TCDD (Gouedard et al., 2004). In contrast to the Bax AhR-responsive element, the PON1 recognition site was only a 4/5 consensus fit with the invariant DRE core sequence, indicating its uniqueness as a potential AhR:ARNT DNA binding site. Although altered nucleotide specificity in the DNA binding of the AhR:ARNT heterodimer has been reported by others, the differential responsiveness of these genes to various ligands either did not occur (DiNatale et al., 2010) or was unexamined (Kinehara et al., 2009). More recently, the use of ChIP-chip and ChIP-Seq analysis has suggested the ability of TCDD:AhR:ARNT complexes to bind to DNA lacking a DRE core sequence (Dere et al., 2011; Lo and Matthews, 2012). Together, the above studies suggest that AhR-dependent regulation of gene expression is complex and may involve multiple distinct mechanisms.
Given the potential biological and toxicological implications of AhR and AhR:ARNT complex binding to non-DRE-responsive elements, a systematic evaluation of ligand-specific changes in the nucleotide specificity of DNA binding was warranted. To date, most studies have only examined the effect of single nucleotide substitutions on ligand-dependent AhR DNA binding (Bank et al., 1992; Shen and Whitlock, 1992; Yao and Denison, 1992), but this approach has limited ability to identify novel AhR DNA binding sequences. In contrast, Swanson and coworkers (1995) utilized a DNA binding site selection and amplification protocol as a more global approach to define the DRE nucleotides (consensus and nonconsensus) that appeared critical for AhR:ARNT DNA binding. Although their studies identified key nucleotides that appeared important in AhR:ARNT DRE DNA binding, the AhR used was a truncated, constitutively active AhR protein that did not require a ligand to bind DNA (Swanson et al., 1995). Thus, the effect of ligands and ligand structure on the nucleotide specificity of AhR:ARNT DNA binding remains unresolved. In this report, we utilize an unbiased DNA binding site selection and amplification methodology to identify any DNA sequences to which the AhR and AhR:ARNT complex can bind when it is bound and activated by 6 structurally diverse ligands that have been reported to differentially interact with the AhR LBD.
MATERIALS AND METHODS
Chemicals.
TCDD was provided by Dr Steven Safe (Texas A&M University). The AhR ligands used in these studies are shown in Figure 1. 3MC, L-kynurenine (LK), βNF, and dimethyl sulfoxide (DMSO) were from Sigma-Aldrich (St Louis, Missouri). Indirubin (IR) was from AmplaChem (Carmel, Indiana), and isopropyl-2-(1,3-dithietane-2-ylidene)-2-[N-(4-methylthiazol- 2-yl)carbamoyl]acetate (YH439) was a gift from Yuhan Research Institute (Republic of Korea). All chemical stocks and dilutions were made in DMSO.
FIG. 1.
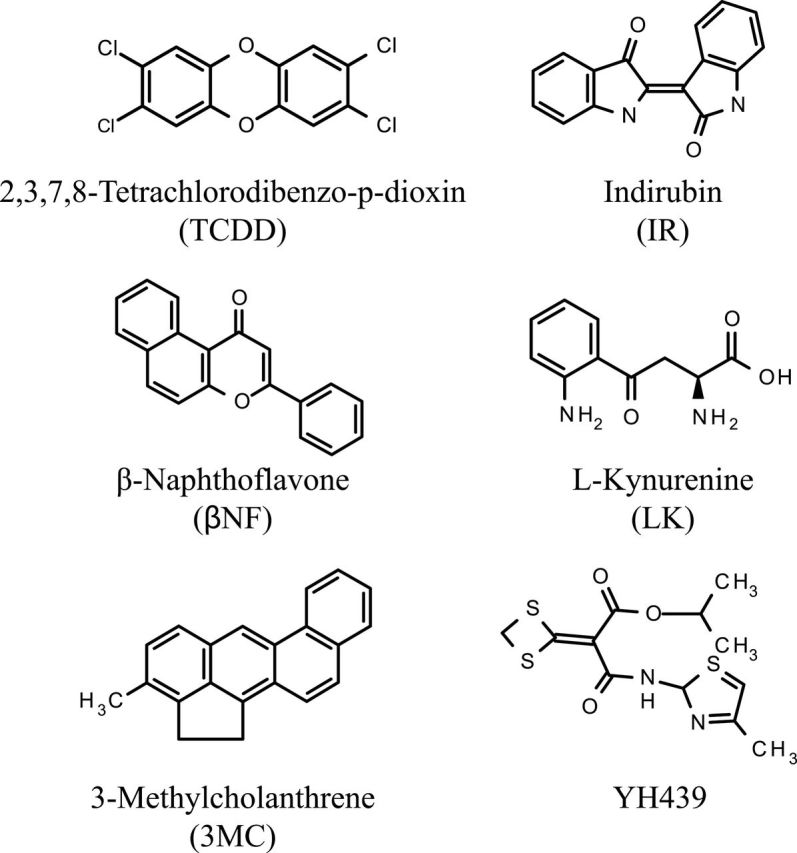
Structures of the 6 structurally diverse aryl hydrocarbon receptor agonists used for AhR DNA binding site selection and amplification analysis.
Oligonucleotides.
Complementary oligonucleotides containing a single copy of the murine DRE3 (5′-GATCCGGAGTTGCGTGAGAAGAGCCA-3′ and 5′-GATCTGGCTCTTCTCACGCAACTCCG-3′) or complementary oligonucleotides containing a single base mutation in the DRE3 sequence were synthesized by Operon Biotechnologies (Huntsville, Alabama) or using an Applied Biosystems DNA synthesizer and purified as previously described (Denison et al., 1988). The oligonucleotides were annealed and end labeled using T4 polynucleotide kinase (New England Biolabs, Ipswich, Massachusetts) and [γ-32P]ATP (Perkin Elmer, Waltham, Massachusetts) for use as probes in elecrophoretic mobility shift assays (EMSA) (Denison et al., 2002) or phosphorylated and subcloned into luciferase or chloramphenicol acetyltransferase (CAT) reporter gene plasmids for transient transfections.
Plasmids.
DRE-luciferase reporter gene plasmids were created by subcloning a single copy of the wild-type DRE3 or indicated mutant DRE oligonucleotide into the BglII site of pGudLuc11.0. This plasmid is identical to that of pGudLuc7.0 (Rogers and Denison, 2000) except that a 31 base pair Acc65I-XhoI fragment containing a putative DRE-like DNA sequence (Ochs et al., 2012) was deleted from the plasmid’s multiple cloning site. The DRE-CAT reporter gene plasmids were created by subcloning a single copy of the wild-type DRE3 or indicated mutant DRE oligonucleotide into the BglII site of pMcat5 (Jones et al., 1986). All constructs were sequence verified.
Preparation of cytosol.
The procedure for preparing cytosol has been previously described (Denison et al., 2002). Male Hartley guinea pigs (250–300g) were purchased from Charles River Laboratories (Wilmington, Massachusetts) and maintained in a 12-h light:12-h dark cycle with ad libitum access to food and water. Guinea pig hepatic cytosol was prepared in HEDG buffer (25mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (Hepes) (pH 7.5), 1mM EDTA, 1mM dithiothreitol (DTT), 10% (vol/vol) glycerol) and protein concentration determined as previously described (Denison et al., 2002). Samples were stored at −80°C until use.
EMSA.
EMSA analysis of the ligand-activated AhR:ARNT complex was carried out as previously described in detail (Denison et al., 2002). Briefly, guinea pig hepatic cytosol (8mg/ml in HEDG) was incubated for 2h at room temperature with DMSO (2% final solvent concentration) or the indicated test chemical at the concentrations indicated in the text. An aliquot of this reaction was mixed with poly[dI–dC] and [32P]-labeled DRE3 oligonucleotide (100 000 cpm) and the protein:DNA complexes resolved by nondenaturing polyacrylamide gel electrophoresis (PAGE). Visualization and quantitation of the ligand:AhR:ARNT:DRE complex formation were determined using a Fuji PhosphorImager and Multi-Gauge software (Tokyo, Japan).
AhR:ARNT DNA binding site selection.
DNA selection and amplification were performed using a modification of the method of Wright et al. (1991). A synthetic 56-mer oligonucleotide was synthesized (Operon Biotechnologies) to contain a 15-base variable region (each base represented at each position) flanked by 2 invariant sequences containing PCR primers (Fig. 2) and designated here as the “randomer.” The complementary strand was made by annealing a 5-fold molar excess of the reverse (lower) primer (5′-GCTAGATCTCAGCTCAGGGCC-3′) to 5 µg of randomer and incubating with Taq polymerase at 72°C for 25min. The double-stranded randomer was then purified by PAGE, eluted, and ethanol precipitated. For the first round of AhR DNA binding site selection, the AhR was transformed into its high-affinity DNA binding heterodimer (AhR:ARNT) by incubating guinea pig hepatic cytosol (8mg/ml) with each test compound for 2h at room temperature. An aliquot of this reaction was incubated with poly[dI–dC] for 15min at room temperature, and then approximately 14fmol (0.5ng) of the double-stranded, purified randomer was added to this reaction (10 µg of total protein) and allowed to incubate for an additional 15min at room temperature. The final DNA binding conditions were 25mM HEPES, 1mM EDTA, 1mM DTT, 10% (vol/vol) glycerol, and 37.5ng of poly[dI–dC]. An aliquot of the DNA binding reaction was mixed with 1.5 µg of anti-AhR antibody (DiNatale et al., 2010) and incubated for 2h at room temperature with mixing, followed by the addition of 20 µl of this reaction to a vial containing 10 µl of Dynabeads Protein A (Life Technologies, Grand Island, New York) and further incubation with mixing for 10min at room temperature.
FIG. 2.
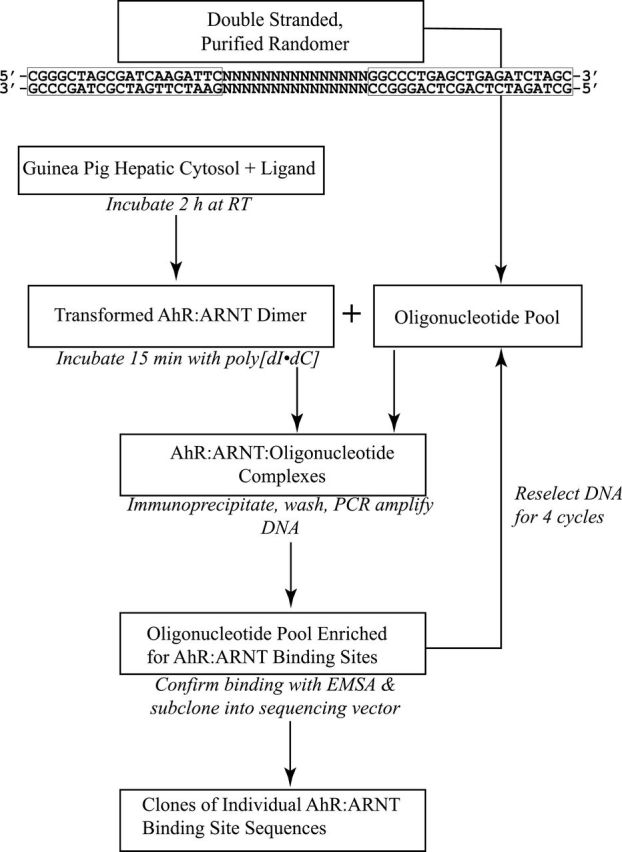
Experimental flow for the DNA binding site selection and amplification procedure. The invariant primer sequences flanking the 15-base variable region in the randomer are shown at the top. See text for details.
Bead:antibody:AhR:ARNT:DNA complexes were isolated by magnetic separation and washed thrice with 500 µl wash buffer (25mM HEPES, 1mM EDTA, 150mM NaCl, 10% (vol/vol) glycerol, 1% (vol/vol) IGEPAL CA-630) to remove unbound oligonucleotide. Following a fourth buffer exchange wash with 1X PCR buffer, the beads were then resuspended in 50 µl of the same buffer. One fifth of the bead solution (10 µl) was added to a PCR reaction mix containing 20mM tris(hydroxymethyl)aminomethane (Tris-HCl), 50mM KCl, 1.5mM MgCl2, 0.2mM of each deoxyribonucleotide triphosphate (dNTP), 2.5 units of Taq polymerase (Life Technologies) and forward (5′-CGGGCTAGCGATCAAGATTC-3′) and reverse primers at a final concentration of 1µM each. Following an initial protein denaturing step at 100°C for 5min, the PCR conditions were 95°C (1min), 55°C (1min), and 72°C (1min) for 20 cycles. The PCR product was visualized by ethidium bromide staining and the concentration estimated using DNA standards. For subsequent rounds of binding site selection, approximately 250 pg of PCR-amplified DNA from the previous round was used in the initial AhR:ARNT:DNA binding reaction. Following 4 rounds of selection, the resulting oligonucleotides were directly subcloned into pGEM-T according to the manufacturer’s instructions (Promega, Madison, Wisconsin). Individual clones were sequenced, the sequences were aligned and compared, and sequence logos depicting the prevalence of each base at each position were generated using WebLogo 3.1 (Crooks et al., 2004).
Cell culture, transfection, and luciferase and CAT activity measurement.
Mouse hepatoma (hepa1c1c7 cells) were cultured under standard conditions (37°C, 5% CO2, and 85% humidity) in alpha Minimum Essential Medium (MEM) (Gibco, Carlsbad, California) supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, Georgia). For luciferase expression analysis, cells were plated at 25 000 cells per well in a 96-well plate and allowed to attach overnight, followed by transfection with 200ng of test plasmid using Lipofectamine 2000 (Life Technologies) per the manufacturer’s instructions. The medium was changed after 4h, and the cells were allowed to recover overnight followed by treatment with DMSO (0.1% [vol/vol]) or TCDD (1nM) for 4h. The treatment medium was removed, and the cells were washed twice with PBS and lysed with 20 µl of Cell Culture Lysis Reagent (Promega). Luciferase activity was measured following the addition of 100 µl of Luciferase Assay Reagent (Promega), with a 2-s read delay and 10-s read integration using an Orion microplate luminometer (Berthold Technologies, Oak Ridge, Tennessee). To assay for CAT activity, subconfluent plates (6cm) of hepa1c1c7 cells were transfected with 7.5 µg of the test plasmid as described (Denison et al., 1988). Following transfection, the cells were allowed to recover for 48h and then treated with DMSO or TCDD as described above except that treatment was for 24h. The cells were harvested by scraping and CAT activity determined as previously described (Gorman et al., 1982).
RESULTS
Structurally Diverse Chemicals Stimulate AhR DRE DNA Binding
In addition to the classical high affinity ligands, the AhR is known to bind to a wide variety of different compounds that share little structural similarity to each other or to its prototypical ligand, TCDD (Denison and Nagy, 2003). Based on previously published results (Gouédard et al., 2004; Matikainen et al., 2001), it has been suggested that differences in binding of structurally diverse chemicals (ie, agonists) within the AhR ligand binding cavity can result in unique conformational changes in AhR structure that alter its nucleotide specificity of DNA binding. Accordingly, a collection of structurally diverse AhR agonists representing a range of chemicals with reported differences in their ability to bind to and activate the AhR were identified for binding site selection studies and include TCDD, YH439, βNF, 3MC, indirubin, and L-kynurenine (Fig. 1) (DeGroot et al., 2011; Goryo et al., 2007; Opitz et al., 2011; Whelan et al., 2010; Xing et al., 2012; Zhao et al., 2010). For optimal DNA binding site selection analysis, a comparable level of DNA binding stimulated by each ligand was desired, and as such, the concentration-dependent ability of each compound to stimulate AhR:ARNT transformation and DNA binding was confirmed in preliminary studies using EMSA (data not shown). The concentrations of each compound that could stimulate AhR DNA binding to a level comparable to that produced by a maximally activating concentration of TCDD were identified and confirmed by EMSA (Fig. 3), and these equipotent concentrations were used for subsequent DNA binding site selection analysis.
FIG. 3.
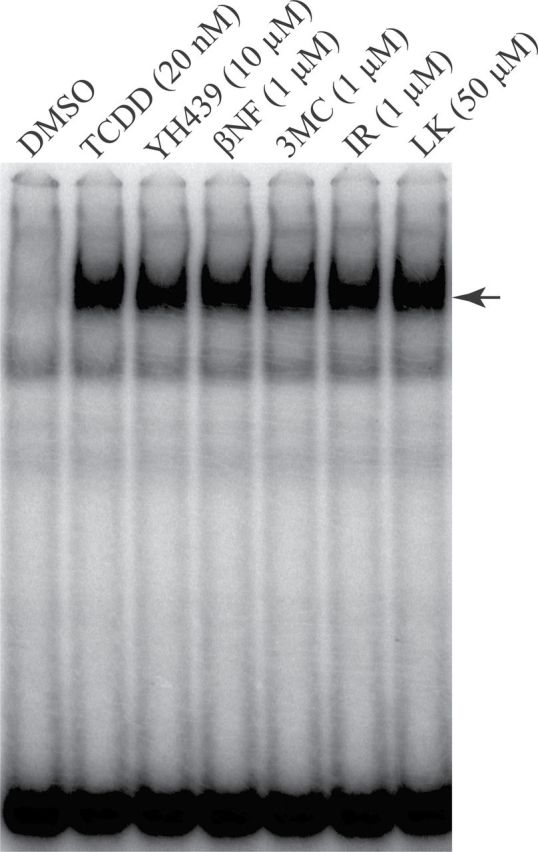
Equipotent concentrations of structurally diverse AhR ligands stimulate comparable levels of AhR:ARNT transformation and DNA binding. Guinea pig hepatic cytosol (8mg/ml) was incubated with the indicated compound and concentration (2% final DMSO solvent concentration) for 2h at 20°C, followed by the addition and incubation with [32P]DRE3. Samples were subjected to EMSA, and protein-DNA complexes were visualized by PhosphorImager analysis. The arrow shows the position of the induced ligand:AhR:ARNT complex. Abbreviations: AhR, aryl hydrocarbon receptor; ARNT, aryl hydrocarbon receptor nuclear translocator; DMSO, dimethyl sulfoxide; DRE, dioxin-responsive element; EMSA, elecrophoretic mobility shift assays.
Structurally Diverse Ligands Promote AhR:ARNT Binding to the Canonical DRE
To determine whether ligand-specific alterations in the nucleotide specificity of DNA binding exist, we examined the ability of structurally diverse AhR agonists to stimulate AhR DNA binding and isolated and sequenced all AhR-bound DNA oligonucleotide sequences using the DNA selection and amplification protocol presented in Figure 2. This method provides a relatively unbiased approach for determining the nucleotide specificity of transcription factor binding sites (Wright et al., 1991) and has been used successfully with a constitutively active AhR:ARNT heterodimer (Swanson et al., 1995). Ligand-activated AhR complexes were incubated with oligonucleotides containing a 15-base pair variable region (all nucleotides represented at each position), and the resulting ligand:AhR:ARNT:oligonucleotide complexes were isolated by immunoprecipitation with an AhR antibody. The selected/isolated DNA was PCR amplified and used in the next round of binding site selection and repeated for a total of 4 rounds of selection. Oligonucleotides from each round were radiolabeled and analyzed using EMSA to confirm enrichment of the oligonucleotide pool for AhR:ARNT DNA binding sites and an example of such binding enhancement with TCDD as the AhR ligand is shown in Figure 4. The oligonucleotides from the 4 rounds of selection were subcloned into pGEM-T, sequenced, and manually aligned for sequence comparisons. The final alignment results (from between 27 and 46 individual DNA sequences isolated for each ligand) were entered into WebLogo 3.1, which graphically illustrates the nucleotide sequence preferences for all ligand-activated AhR:ARNT-bound DNA binding sites. The results (Fig. 5A) reveal a dramatic preference of ligand:AhR:ARNT binding to DNA sequences that contain the canonical DRE (5′-GCGTG-3′) regardless of the activating ligand and are in agreement with a previous report that failed to demonstrate ligand-specific differences in the guinea pig AhR:ARNT DNA binding site using a limited number of targeted mutant DREs (Bank et al., 1992). Strikingly, no sequence variation was observed in positions 10–13 (5′-CGTG-3′) in any DNA sequence for any ligand tested. Although the DRE is widely reported in the literature to be invariant in the 5 “core” nucleotides (positions 9–13), our results confirm other studies that have reported that the AhR:ARNT DNA binding complex can tolerate sequence variation at the first nucleotide of the 5 base pair DRE core sequence (ie, position 9 in the DRE oligonucleotides) (Shen and Whitlock, 1992; Yao and Denison, 1992). Although the overall pattern of nucleotides flanking the core sequence was similar for the different ligands, some minor ligand-specific differences were observed, such as no sequence variation from a T at position 7 for 3MC and LK activated AhRs or from an A at position 16 for TCDD. Given the similarity of the AhR:ARNT DNA binding sequence for each ligand, all of the sequences produced from the DNA selection and amplification assay were combined to generate an overall AhR:ARNT DNA binding consensus sequence in WebLogo 3.1 (Fig. 5B). All of the DNA sequences isolated in the ligand-dependent DNA selection and amplification approach and a table of the variation of each base at each position of the DRE oligonucleotide are presented in Supplementary Figures S1 and S2.
FIG. 4.
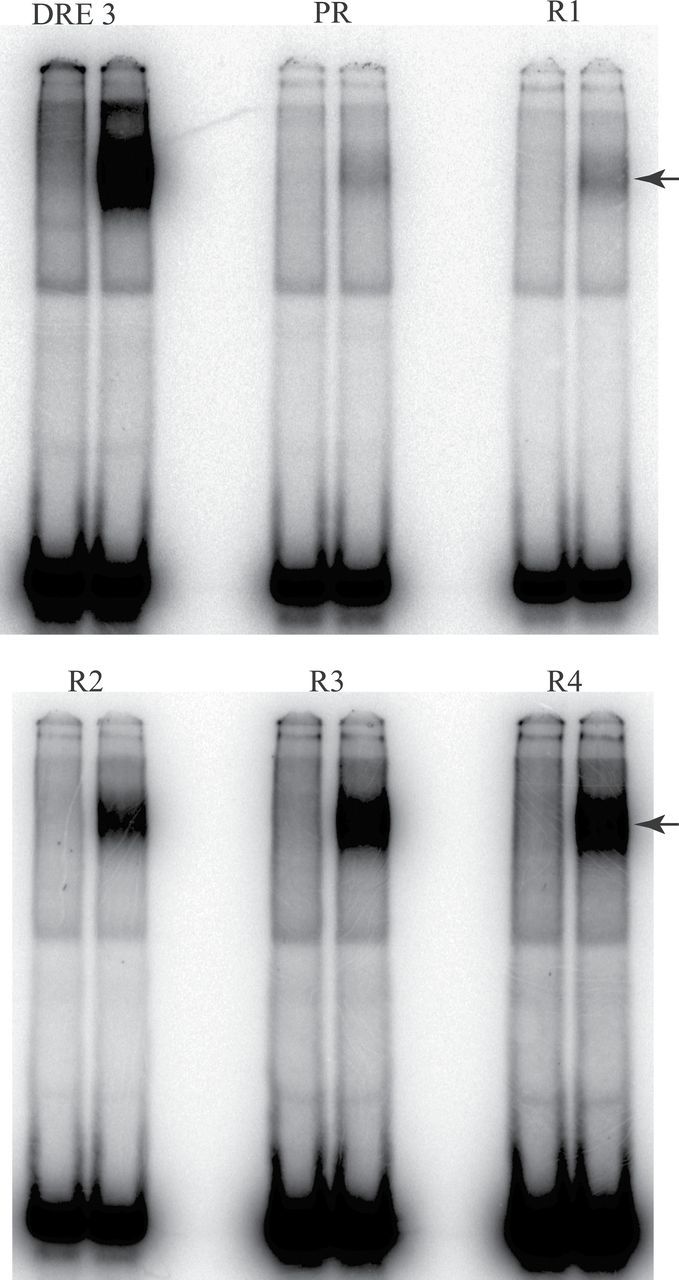
Progressive enrichment for AhR:ARNT binding sites within the random oligonucleotide pool during repeated cycles of the DNA selection and amplification procedure. Guinea pig hepatic cytosol (8mg/ml) was incubated with 20nM TCDD for 2h at 20°C, followed by the addition and incubation with 32P-labeled oligonucleotides from each round of the DNA binding site selection and amplification procedure. The matched DMSO solvent control (2% vol/vol) is shown for each selection round. Samples were subjected to EMSA, and protein-DNA complexes were visualized by PhosphorImager analysis. The arrow shows the position of the induced ligand:AhR:ARNT complex. PR indicates incubation with the purified randomer; R indicates the number of rounds of selection. The DRE3-containing oligonucleotide was used as a positive control. Abbreviations: AhR, aryl hydrocarbon receptor; ARNT, aryl hydrocarbon receptor nuclear translocator; DMSO, dimethyl sulfoxide; DRE, dioxin-responsive element; EMSA, elecrophoretic mobility shift assays; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin.
FIG. 5.
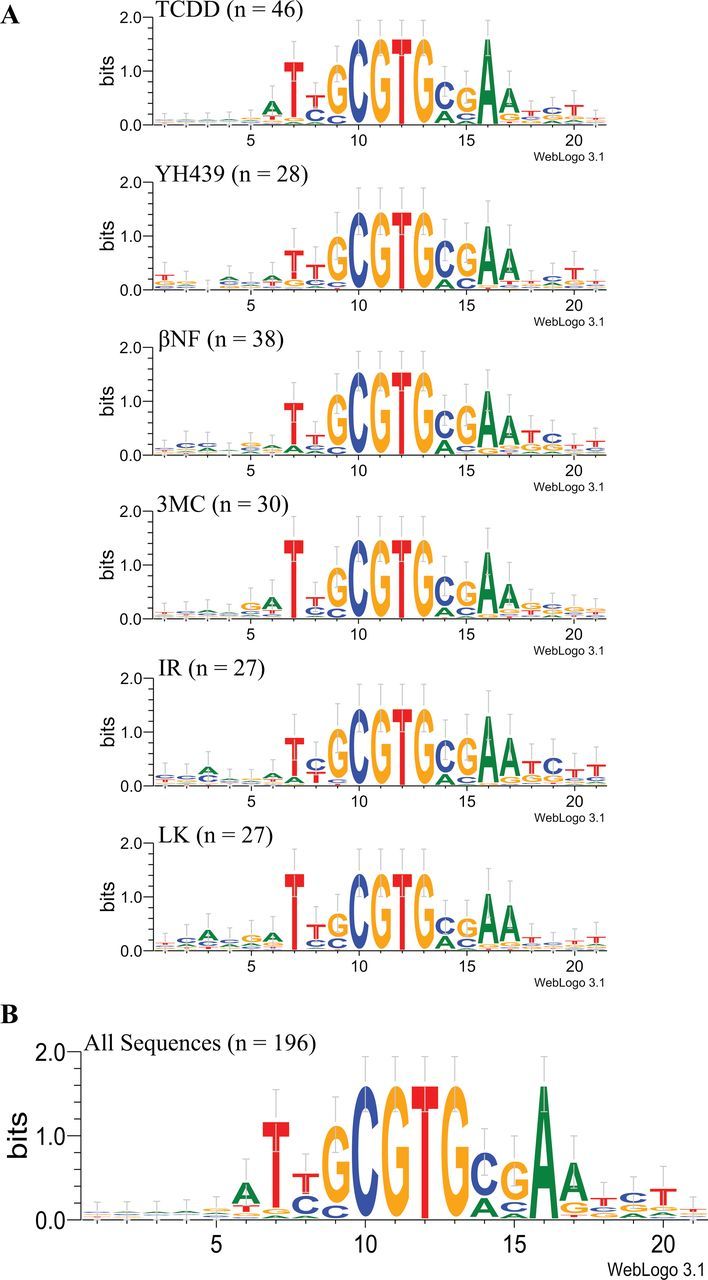
Nucleotide specificity of DNA binding of guinea pig hepatic AhR:ARNT complexes activated by structurally diverse AhR agonists. Guinea pig hepatic cytosol was incubated with equipotent concentrations of the indicated ligand for 2h at 20°C, and the resulting ligand:AhR:ARNT complex was used in the DNA selection and amplification protocol as described in Materials and Methods section. Individual DNA oligonucleotides were isolated, subcloned, and sequenced. A, DRE DNA binding consensus sequences were derived by manual alignment and nucleotide preferences in each ligand group presented using WebLogo 3.1. The number of individually isolated DNA sequences used to derive each sequence is indicated in parentheses after the compound name. B, The overall nucleotide preferences (ie, consensus sequence) for ligand-dependent AhR:ARNT DRE DNA binding were determined by alignment of all 196 isolated DNA sequences and presented using WebLogo 3.1 (color image available in online version). Abbreviations: AhR, aryl hydrocarbon receptor; ARNT, aryl hydrocarbon receptor nuclear translocator; DRE, dioxin-responsive element.
In Vitro Binding of AhR:ARNT Complexes to Variant DRE Sequences is Not Ligand Specific
Although the DNA selection and amplification assay results indicate that 4 bases of the DRE core consensus sequences are absolutely conserved in all DNA oligonucleotides bound by ligand-activated AhR:ARNT complexes, they do not eliminate the potential that the minor changes identified by this analysis could contribute to ligand-specific differences in DNA binding. Accordingly, to assess whether the variation in selected nucleotides we identified within and flanking the core DRE consensus affected ligand specificity of AhR:ARNT DNA binding, we exhibited AhR DNA binding to a series of oligonucleotides containing single nucleotide mutations within the wild-type mouse DRE DNA sequence (Table 1) (Denison et al., 1988; Yao and Denison, 1992). We decided on this approach, rather than utilizing specific sequences pulled down in the DNA selection and amplification assay, in order to both standardize our results to our previous studies and to avoid having to test sequences containing more than one mutation, which would confound interpretation of the results. Although the DNA selection sequence results we obtained were the initial basis for determining which nucleotide substitutions to test, we refined our selection by several factors, namely whether the specific substitution/mutation was untested in previous studies and whether the substitution was present in or adjacent to nucleotides known to be important in AhR DNA binding and/or the relative frequency of the nucleotide substitution (Table 1). This evaluation identified 8 substitutions of interest, and mutant DRE3 oligonucleotides containing single mutations of each base were radiolabeled and subjected to EMSA analysis using each ligand to examine whether these substitutions affected AhR:ARNT binding in a ligand-specific manner. The results of these analyses (Fig. 6) revealed that all the ligands tested were able to stimulate comparable levels of AhR:ARNT binding to all 8 mutated DRE3 oligonucleotides, and no mutation was able to abrogate any induced ligand:AhR:ARNT complex. These results indicate that the small variation in flanking nucleotide specificity observed from the DNA selection and amplification analysis had little or no effect on ligand-specific binding of the AhR:ARNT complex to DRE-containing DNA.
TABLE 1.
DNA Sequences of the Wild-Type DRE3 (WT) and Mutant (M) Oligonucleotides Containing Specific Substitutions Found by Binding Site Selection
| Oligo | DRE Oligonucleotide Sequence | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Position | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 |
| WT | C | G | G | A | G | T | T | G | C | G | T | G | A | G | A | A | G | A | G |
| M1 | C | G | G | A | G | A | T | G | C | G | T | G | A | G | A | A | G | A | G |
| M2 | C | G | G | A | G | T | A | G | C | G | T | G | A | G | A | A | G | A | G |
| M3 | C | G | G | A | G | T | G | G | C | G | T | G | A | G | A | A | G | A | G |
| M4 | C | G | G | A | G | T | T | C | C | G | T | G | A | G | A | A | G | A | G |
| M5 | C | G | G | A | G | T | T | G | C | G | T | G | T | G | A | A | G | A | G |
| M6 | C | G | G | A | G | T | T | G | C | G | T | G | A | C | A | A | G | A | G |
| M7 | C | G | G | A | G | T | T | G | C | G | T | G | A | G | G | A | G | A | G |
| M8 | C | G | G | A | G | T | T | G | C | G | T | G | A | G | A | G | G | A | G |
Note. The specific mutations are indicated in bold and underlined type.
FIG. 6.

No ligand structure–dependent differences in the nucleotide specificity of DNA binding were observed with DNA oligonucleotides that contained the DRE nucleotide substitutions identified in the binding site selection assay. Guinea pig hepatic cytosol (8mg/ml) was incubated with equipotent concentrations of the indicated compound for 2h at 20°C, followed by the addition of the indicated 32P-labeled wild-type (WT) or mutant (M) DRE oligonucleotide and the protein-DNA complexes resolved by EMSA. Only the ligand-activated AhR:ARNT:DNA complexes are shown. The specific nucleotide substitution in each mutant DRE oligonucleotide is indicated in Table 1. Abbreviations: AhR, aryl hydrocarbon receptor; ARNT, aryl hydrocarbon receptor nuclear translocator; DRE, dioxin-responsive element.
Binding Site Selected Variant DRE Sequences Can Mediate AhR-Dependent Reporter Gene Expression
Although the above results revealed no significant difference in the DNA binding of ligand-transformed AhR complex to DRE3 oligonucleotides containing mutations of variant bases identified by binding site selection, whether these substitutions can alter the ability of these sequences to confer TCDD/AhR responsiveness upon an adjacent promoter and gene remained to be determined. To examine this, mutant DRE oligonucleotides (Table 1) were cloned as single elements (and in the same orientation) upstream of a heterologous promoter and a firefly luciferase reporter gene and its ligand (TCDD) responsiveness determined. Transfected mouse hepatoma cells were incubated with DMSO or 1nM TCDD for 4h after which luciferase activity was determined. TCDD was utilized as the inducing ligand in order to exclude potential confounding effects by metabolic breakdown products of the other ligands. Although all mutant DRE sequences tested were able to confer TCDD-/AhR:ARNT-dependent responsiveness upon the reporter gene, the overall luciferase activity varied among the different oligonucleotides (Fig. 7). All DRE mutations present 5′-ward of the CGTG core reduced the magnitude of luciferase reporter gene activity to ~40% of the wild-type DRE3 sequence; mutations present 3′-ward of the DRE core sequence had a less pronounced effect. Interestingly, mutation 8 had no significant effect on the magnitude of TCDD-inducible luciferase reporter gene activity, whereas mutation 6 significantly enhanced TCDD-inducible luciferase activity above that observed with the wild-type DRE3.
FIG. 7.

TCDD stimulates AhR:ARNT-dependent reporter gene expression from mutant DRE oligonucleotides. Hepa1c1c7 cells were transiently transfected with the pGudLuc11.0 luciferase reporter gene plasmid containing a single wild-type (WT) or mutant (M) DRE (see Table 1 for specific mutations). Cells were incubated with DMSO (0.1%) or 1nM TCDD for 4h followed by measurement of luciferase activity. Luciferase activity was expressed as the mean ± SD of 4 (DMSO) or 8 (TCDD) individual incubations and measurements, and all activity normalized to that obtained with TCDD-treated cells transfected with pGudLuc11.0 containing the WT DRE. Luciferase activity of TCDD incubations from all oligonucleotides was significantly greater than DMSO (p < .005), and TCDD incubations from the mutant oligonucleotides were found to be significantly different than TCDD incubations from the wild-type oligonucleotide (*p < .001; **p < .05) as determined by the Student’s t test. Abbreviations: AhR, aryl hydrocarbon receptor; ARNT, aryl hydrocarbon receptor nuclear translocator; DMSO, dimethyl sulfoxide; DRE, dioxin-responsive element; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin.
The combination of the above results with previously unpublished studies examining the effects of other targeted single nucleotide DRE mutations on TCDD-stimulated AhR:ARNT DRE binding (Yao and Denison, 1992) and the ability of these same mutant oligonucleotides to confer TCDD- and AhR:ARNT-dependent responsiveness upon CAT reporter gene expression is presented in Table 2 and Supplementary Figure S3, respectively. Consistent with a lack of DNA binding, nucleotide transversion substitutions within the highly conserved 4 base pair DRE core sequence eliminated transcriptional activity (mutant ME through MH). Mutant MI, a transition substitution immediately adjacent to the core sequence, also eliminated AhR:ARNT DNA binding and functional activity. Interestingly, although an A to C substitution at position 16 (mutant MK) only moderately reduced the binding of the AhR:ARNT complex and completely abrogated TCDD-induced CAT reporter gene activity, an A to G substitution in this position (mutant M7) was still able to confer TCDD-responsiveness although the overall response was significantly reduced (Fig. 7, Table 2). Combining the results of these studies along with those from previously published DRE mutagenesis experiments (Supplementary Table S1) allowed generation of an overall functional DRE consensus sequence (Table 3). These sequence analyses not only revealed a consensus sequence similar to that previously reported for functional DREs (Denison et al., 1998a), but they indicate that the variability in nucleotides in the regions flanking the 5′-CGTG-3′ core is much greater than previously reported, and they demonstrate the critical nature of both the DRE core sequence as well as nucleotides flanking the core in overall DRE functional activity.
TABLE 2.
Effect of Site-Directed DRE Mutagenesis on TCDD Inducible Reporter Gene Activity in Transfected Mouse Hepatoma Cells
| Oligo | DRE Oligonucleotide Sequence | Fold Induction | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Position | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | |
| WT | C | G | G | A | G | T | T | G | C | G | T | G | A | G | A | A | G | A | G | 3.8* |
| MA | T | G | G | A | G | T | T | G | C | G | T | G | A | G | A | A | G | A | G | 2.3 |
| MB | C | G | G | A | A | T | T | G | C | G | T | G | A | G | A | A | G | A | G | 2.1* |
| MC | C | G | G | A | G | G | T | G | C | G | T | G | A | G | A | A | G | A | G | 2.7* |
| M1 | C | G | G | A | G | A | T | G | C | G | T | G | A | G | A | A | G | A | G | 2.9* |
| M2 | C | G | G | A | G | T | A | G | C | G | T | G | A | G | A | A | G | A | G | 2.4* |
| M3 | C | G | G | A | G | T | G | G | C | G | T | G | A | G | A | A | G | A | G | 2.0* |
| MD | C | G | G | A | G | T | T | T | C | G | T | G | A | G | A | A | G | A | G | 2.0* |
| M4 | C | G | G | A | G | T | T | C | C | G | T | G | A | G | A | A | G | A | G | 1.7* |
| ME | C | G | G | A | G | T | T | G | A | G | T | G | A | G | A | A | G | A | G | 1.1 |
| MF | C | G | G | A | G | T | T | G | C | T | T | G | A | G | A | A | G | A | G | 1.4 |
| MG | C | G | G | A | G | T | T | G | C | G | G | G | A | G | A | A | G | A | G | 1.1 |
| MH | C | G | G | A | G | T | T | G | C | G | T | T | A | G | A | A | G | A | G | 1.3 |
| MI | C | G | G | A | G | T | T | G | C | G | T | G | G | G | A | A | G | A | G | 0.8 |
| M5 | C | G | G | A | G | T | T | G | C | G | T | G | T | G | A | A | G | A | G | 3.3* |
| MJ | C | G | G | A | G | T | T | G | C | G | T | G | A | T | A | A | G | A | G | 1.6* |
| M6 | C | G | G | A | G | T | T | G | C | G | T | G | A | C | A | A | G | A | G | 4.4* |
| MK | C | G | G | A | G | T | T | G | C | G | T | G | A | G | C | A | G | A | G | 1.2 |
| M7 | C | G | G | A | G | T | T | G | C | G | T | G | A | G | G | A | G | A | G | 2.4* |
| M8 | C | G | G | A | G | T | T | G | C | G | T | G | A | G | A | G | G | A | G | 3.1* |
| ML | C | G | G | A | G | T | T | G | C | G | T | G | A | G | A | A | G | A | T | 3.2* |
Notes. Wild-type DRE3 (WT) and mutant (M) DRE3-containing oligonucleotides are shown with the specific substitutions indicated in bold type. Fold induction of reporter gene activity was obtained in transiently transfected mouse hepatoma (hepa1c1c7) cells incubated in the presence of TCDD (1nM) compared with cells incubated with DMSO (0.1%). Transcriptional activity was assayed using either luciferase (mutants M1–M8 [in italic bold]) or chloramphenicol acetyltransferase reporter gene plasmids (mutants MA–ML). An asterisk indicates TCDD-inducible reporter gene activity that was significantly greater than that of DMSO (Student’s t test; *p < .05).
TABLE 3.
Derivation of a Fully Functional DRE Consensus Sequence
| Position | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DRE3 wild type | C | G | G | A | G | T | T | G | C | G | T | G | A | G | A | A | G | A | G |
| Less functional | . | . | . | . | A* | A | A | C | . | . | . | . | T | T | G* | . | A | . | T |
| . | . | . | . | . | G | T | . | . | . | . | . | . | T | . | . | . | |||
| Nonfunctional | T* | . | . | . | . | C | . | A | . | . | . | . | G | A | C | . | . | . | . |
| Consensus | N | N | N | N | N | T | T | G | C | G | T | G | A | G | A | N | N | N | N |
| G | C | C | T | C | G | ||||||||||||||
| A | A | T | C | T | T | ||||||||||||||
| G | |||||||||||||||||||
Notes. Data from this study were combined with previously published mutagenesis results (Supplementary Table S1) to yield an overall DRE consensus sequence that is able to stimulate AhR:ARNT-dependent reporter gene expression. Nucleotides in bold are those found to be fully functional in mutagenesis experiments (ie, can bind ligand-activated AhR and stimulate ligand-dependent gene expression). The cytosine residue at position 14 [underlined] has not been tested in mutagenesis experiments but is very commonly present in DRE sequences. An asterisk (*) indicates the specific nucleotide where there is conflicting data on the functionality of a DRE containing that residue. N indicates any nucleotide.
DISCUSSION
In addition to classical ligands, the AhR is well documented to bind to a wide variety of structurally diverse compounds, and an increasing number of studies are suggesting ligand-specific changes in AhR function that are mechanistically distinct from simple differences in metabolic transformation or AhR binding affinity (Denison et al., 2011; Murray et al., 2011; Zhang et al., 2008). One of these mechanisms suggests that the nucleotide specificity of DNA binding by the AhR:ARNT heterodimer is dependent upon the structure of the bound ligand with certain compounds such as 3MC and a dihydrodiol metabolite of dimethylbenz(a)anthracene, stimulating AhR-dependent transcription from DRE-like response elements that are unresponsive to TCDD (Gouedard et al., 2004; Matikainen et al., 2001). Because most of the previous work that defined the DRE consensus sequence utilized TCDD, we sought to further refine the nucleotide sequence of AhR:ARNT DNA binding using a series of 6 structurally different ligands. It is important to note that although some studies have previously reported AhR binding to nonconsensus DRE sites, ARNT was not shown to be part of the high affinity DNA binding complex (Huang and Elferink, 2012; Oesch-Bartlomowicz et al., 2005; Vogel et al., 2007), and these undoubtedly represent distinct mechanisms by which the AhR can produce effects beyond the classical DRE pathway (Denison et al., 2011). In this report, our focus was the nucleotide specificity of DNA binding of the AhR:ARNT heterodimer.
Using a relatively unbiased DNA selection and amplification approach, we have shown that 6 structurally diverse ligands, including 3 classical AhR activators (TCDD, 3MC, and βNF) and 3 nonclassical activators (IR, YH439, and LK) all stimulated AhR:ARNT binding to the DRE. The inability of our binding site selection approach to identify and isolate an AhR or AhR:ARNT DNA binding site that completely lacks a DRE or partial DRE sequence suggests that these elements either have extremely low affinity, are unstable, and/or require factors that are not present in our guinea pig cytosol experimental system. Our results (Fig. 5B) are more consistent with a DRE consensus sequence derived from AhR:ARNT binding sites found in vivo (Denison et al., 1998a) than with an in vitro consensus sequence derived using a DNA selection and amplification approach with the primary differences located in nucleotides flanking the DRE core (Swanson et al., 1995). In the latter study, the AhR used for binding analysis was a constitutively active (ligand independent) truncated form that lacked the LBD, and this may account for the discrepancy between the outcome of our studies and theirs. Other reports have defined the AhR:ARNT DNA binding sequence as 5′-GCGTGNNA/TNNNC/G-3′, indicating the importance of not only the core nucleotides but also those nucleotides 3′ of the core (Yao and Denison, 1992). Our data have confirmed the requirement for the core sequence, 5′-CGTG-3′, in AhR:ARNT DNA binding, and although not surprising, the unequivocal selection for these bases was striking. This sequence contains the 5′-GTG-3′ half-site that has been shown to be the binding site for ARNT, and these results support previous studies that suggest ARNT as the primary DNA binding component of the AhR:ARNT complex (Bacsi et al., 1995; Elferink et al., 1990). Although ARNT has also been shown to contact bases 3′ of the DRE core, our single nucleotide mutations in these flanking positions were unable to eliminate AhR:ARNT DNA binding and support the idea that these are relatively weak interactions (Bacsi et al., 1995) and are consistent with the requirement for multiple protein-DNA interactions. Our DNA selection and amplification assay also demonstrate AhR:ARNT DNA interactions that are consistent with a requirement for the AhR binding portion of the DRE half-site, 5′-T(C/T)GC-3′, (Swanson et al., 1995) although as demonstrated in our EMSA analysis of mutant DRE oligonucleotides and predicted from previous mutagenesis experiments, these positions can tolerate significantly more variability than the 3′ half of the DRE (Yao and Denison, 1992). Interestingly, the 5′ thymine in this half-site, although strongly selected in our study and that of others, has not been shown to directly contact either AhR or ARNT leaving an open question as to what factors may dictate its preference in the DRE consensus sequence (Bacsi et al., 1995; Swanson et al., 1995). More recently, a variety of chromatin immunoprecipitation (ChIP), gene expression analysis, and high throughput sequencing approaches have been used for genomewide mapping of AhR:ARNT DNA binding sites (Dere et al., 2011; Lo and Matthews, 2012; Sartor et al., 2009). Although these studies identified DRE and/or DRE-like elements present in the majority of the AhR-bound DNA fragments, DRE sequences were not always present, suggesting that the AhR could bind to DNA lacking a DRE consensus. Although the binding site selection analysis reported here suggests a very high degree of nucleotide specificity of DNA binding by the in vitro transformed guinea pig hepatic cytosolic ligand:AhR:ARNT complex (ie, all AhR-bound DNA identified contained a core DRE consensus motif), it is possible that the nucleotide specificity of DNA binding of the in vivo transformed nuclear ligand:AhR:ARNT complexes determined by ChIP analysis could appear more varied. ChIP analysis involves formaldehyde crosslinking of proteins and DNA and allows isolation of DNA that is crosslinked to a target protein(s) or protein complex. Accordingly, this methodology not only results in isolation of DRE-containing DNA to which the nuclear AhR complex was bound with high affinity, but also that of non-DRE- or partial DRE-containing DNA to which the AhR was bound nonspecifically, with low affinity and/or through an interaction with another nuclear protein that is directly bound to the DNA. Because the specific nucleotide sequence to which the ligand:AhR:ARNT complex binds within the majority of the non-DRE-containing DNA fragments cannot be identified by ChIP and the AhR responsiveness of these fragments has yet to be determined, the AhR DNA binding consensus sequence derived from these studies has been predominantly the result of computational analysis. Future studies directly examining AhR responsiveness of and DNA binding to selected non-DRE DNA fragments identified by ChIP and gene expression analysis are certain to provide greater insights into the nucleotide specificity of binding of nuclear ligand:AhR:ARNT complexes.
Although the DRE core sequence is also necessary for AhR:ARNT-dependent transcriptional activation, it is not sufficient, and nucleotides flanking the core sequence are known to play a key role in determining whether AhR:ARNT DNA binding will induce transcription of downstream genes (Denison et al., 1988). The results of our mutagenesis studies help to further define a DRE consensus sequence important in AhR:ARNT functional activity and indicate that this sequence is more flexible with regards to acceptable nucleotide substitutions than previously reported. However, despite this reduced nucleotide specificity, there remain key single nucleotide mutations in these flanking nucleotides that can eliminate the transcriptional activity of the AhR:ARNT complex (Table 3). The effect of these mutations correlate well with previously reported DRE sequences in the promoter regions of genes such as pS2 and cathepsin D that have been shown to be unresponsive to TCDD, yet interact with the AhR:ARNT complex. In each case, the reported DRE sequence contains at least one substitution in the flanking nucleotides that was shown to be nonfunctional in targeted DRE mutagenesis experiments (Gillesby et al., 1997; Krishnan et al., 1995). A more recent report of a nonconsensus DRE in the promoter region of the IL-6 gene, containing 2 nonfunctional mutations 3′ of the core, was reported to be weakly responsive to benzo(a)pyrene; however, the sequence was not tested as a single DNA element but as 3 linked elements, and the response could have resulted from interactions between these multiple AhR complexes to facilitate gene transcription (DiNatale et al., 2010).
The presence of multiple mutations within the flanking regions of the DRE consensus sequence raises an important question. To what extent can the AhR:ARNT complex tolerate substitutions in these flanking sequences before losing the ability to bind and/or activate transcription? In this study and others, DNA binding and transcriptional activation have been primarily assessed using single nucleotide mutations, and only a few oligonucleotides containing double or triple mutants within the wild-type DRE3 have been tested (Neuhold et al., 1989; Saatcioglu et al., 1990; Yao and Denison, 1992). Often, however, DRE core sequences found in gene promoters are flanked by several less than ideal substitutions. As an example, a DRE (ie, site C) found in the promoter region of the prototypical mouse CYP1A1 gene binds the AhR:ARNT complex but fails to mediate a functional response (Lusska et al., 1993). This site contains 2 substitutions (positions 7 and 16 described here) that when tested in isolation mediate transcription, and a third cytosine substitution (position 14), that although not directly tested in mutagenesis experiments, is commonly found in the DREs of classical AhR-responsive genes (Denison et al., 1998a). Thus, despite our further characterization of a functional DRE consensus sequence, it remains difficult to predict how a nonconsensus DRE site will function without experimental evidence. A significant question that arises from this study is whether a combination of mutations in these flanking sequences can mediate AhR:ARNT transcription by ligands other than TCDD. One limitation of our study is that we tested single mutations placed within the wild-type DRE3 rather than ligand-specific sequences pulled down in the DNA selection and amplification assay. Although all of the mutants were responsive to TCDD, we did not characterize these sequences with respect to other ligands.
In conclusion, although we and others have demonstrated that structurally diverse AhR agonists can differentially bind within the AhR LBD, ligand-dependent transformation and DNA binding of the ligand:AhR:ARNT complex are highly conserved. The results presented here strongly suggest that currently identified agonists of the AhR function essentially as an “on/off” switch to stimulate the AhR signal transduction pathway, and this activation is mediated through an interaction of the ligand-activated AhR with the DRE, with core nucleotides primarily involved in high affinity AhR:ARNT:DRE complex formation and flanking residues modulating the interaction. Although the mechanism of action appears to be predominantly mediated through the classical DRE-dependent AhR pathway, ligand-specific differences in AhR response are likely due to alternative mechanisms, such as ligand-selective interaction of nuclear AhR complexes with coactivators and other nuclear regulatory factors. Further studies examining ligand-selective alterations in AhR structure and function as well as in-depth analysis of subsequent interactions of these diverse ligand:AhR:ARNT complexes with coactivators are necessary to gain insights into ligand-specific differences in the toxic and biological effects resulting from activation of the AhR and AhR-dependent gene expression.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institutes of Environmental Health Sciences (ES012498, ES007685, ES004699 to M.S.D.); National Institutes of Environmental Health Sciences Toxicology Training Grant (T32 ES007059 to D.E.D.); the Schwall Fellowship in Medical Research (to D.E.D.); the California Agriculture Experiment Station and the generosity of the American taxpayers.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge Dr Gary H. Perdew (The Pennsylvania State University) for providing the AhR antibody.
REFERENCES
- Bacsi S. G., Reisz-Porszasz S., Hankinson O. (1995). Orientation of the heterodimeric aryl hydrocarbon (dioxin) receptor complex on its asymmetric DNA recognition sequence. Mol. Pharmacol. 47, 432–438 [PubMed] [Google Scholar]
- Bank P. A., Yao E. F., Phelps C. L., Harper P. A., Denison M. S. (1992). Species-specific binding of transformed Ah receptor to a dioxin responsive transcriptional enhancer. Eur. J. Pharmacol. 228, 85–94 [DOI] [PubMed] [Google Scholar]
- Crooks G. E., Hon G., Chandonia J. M., Brenner S. E. (2004). WebLogo: A sequence logo generator. Genome Res. 14, 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGroot D., He G., Fraccalvieri D., Bonati L., Pandini A., Denison M. S.(2011). AHR Ligands: Promiscuity in binding and diversity in response. In The AH Receptor in Biology and Toxicology (Pohjanvirta R., Ed.), pp. 63–79 John Wiley & Sons, Inc., Hoboken, NJ [Google Scholar]
- Denison M. S., Elferink C. F., Phelan D. (1998a). The Ah receptor signal transduction pathway. In Toxicant-Receptor Interactions in the Modulation of Signal Transduction and Gene Expression (Denison M. S., Helferich W. G., Eds.), pp. 3–33 Taylor and Francis, Philadelphia, PA [Google Scholar]
- Denison M. S., Fisher J. M., Whitlock J. P., Jr (1988). The DNA recognition site for the dioxin-Ah receptor complex. Nucleotide sequence and functional analysis. J. Biol. Chem. 263, 17221–17224 [PubMed] [Google Scholar]
- Denison M. S., Nagy S. R. (2003). Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 43, 309–334 [DOI] [PubMed] [Google Scholar]
- Denison M. S., Rogers J. M., Rushing S. R., Jones C. L., Tetangco S. C., Heath-Pagliuso S. (2002). Analysis of the aryl hydrocarbon receptor (AhR) signal transduction pathway. In Current Protocols in Toxicology (Maines M. D., Costa L. G., Reed D. J., Sassa S., Sipes I. G., Eds.), pp. 4.8.1–4.8.45 John Wiley & Sons, Inc., New York, NY: [DOI] [PubMed] [Google Scholar]
- Denison M. S., Seidel S. D., Rogers W. J., Ziccardi M., Winter G. M., Heath-Pagliuso S. (1998b). Natural and synthetic ligands for the Ah receptor. In Molecular Biology of the Toxic Response (Puga A., Wallace K. B., Eds.), pp. 393–410 Taylor & Francis, Philadelphia, PA [Google Scholar]
- Denison M. S., Soshilov A. A., He G., DeGroot D. E., Zhao B. (2011). Exactly the same but different: Promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol. Sci. 124, 1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dere E., Lo R., Celius T., Matthews J., Zacharewski T. R. (2011). Integration of genome-wide computation DRE search, AhR ChIP-chip and gene expression analyses of TCDD-elicited responses in the mouse liver. BMC Genomics 12, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNatale B. C., Schroeder J. C., Francey L. J., Kusnadi A., Perdew G. H. (2010). Mechanistic insights into the events that lead to synergistic induction of interleukin 6 transcription upon activation of the aryl hydrocarbon receptor and inflammatory signaling. J. Biol. Chem. 285, 24388–24397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elferink C. J., Gasiewicz T. A., Whitlock J. P., Jr (1990). Protein-DNA interactions at a dioxin-responsive enhancer. Evidence that the transformed Ah receptor is heteromeric. J. Biol. Chem. 265, 20708–20712 [PubMed] [Google Scholar]
- Gillesby B. E., Stanostefano M., Porter W., Safe S., Wu Z. F., Zacharewski T. R. (1997). Identification of a motif within the 5’ regulatory region of pS2 which is responsible for AP-1 binding and TCDD-mediated suppression. Biochemistry 36, 6080–6089 [DOI] [PubMed] [Google Scholar]
- Gorman C. M., Moffat L. F., Howard B. H. (1982). Recombinant genomes which express chloramphenicol acetyltransferase in mammalian cells. Mol. Cell. Biol. 2, 1044–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryo K., Suzuki A., Del Carpio C. A., Siizaki K., Kuriyama E., Mikami Y., Kinoshita K., Yasumoto K., Rannug A., Miyamoto A., et al. (2007). Identification of amino acid residues in the Ah receptor involved in ligand binding. Biochem. Biophys. Res. Commun. 354, 396–402 [DOI] [PubMed] [Google Scholar]
- Gouédard C., Barouki R., Morel Y. (2004). Dietary polyphenols increase paraoxonase 1 gene expression by an aryl hydrocarbon receptor-dependent mechanism. Mol. Cell. Biol. 24, 5209–5222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hestermann E. V., Brown M. (2003). Agonist and chemopreventative ligands induce differential transcriptional cofactor recruitment by aryl hydrocarbon receptor. Mol. Cell. Biol. 23, 7920–7925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G., Elferink C. J. (2012). A novel nonconsensus xenobiotic response element capable of mediating aryl hydrocarbon receptor-dependent gene expression. Mol. Pharmacol. 81, 338–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P. B., Durrin L. K., Galeazzi D. R., Whitlock J. P., Jr (1986). Control of cytochrome P1-450 gene expression: Analysis of a dioxin-responsive enhancer system. Proc. Natl. Acad. Sci. U.S.A. 83, 2802–2806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinehara M., Fukuda I., Yoshida K., Ashida H. (2009). Aryl hydrocarbon receptor-mediated induction of the cytosolic phospholipase A(2)alpha gene by 2,3,7,8-tetrachlorodibenzo-p-dioxin in mouse hepatoma Hepa-1c1c7 cells. J. Biosci. Bioeng. 108, 277–281 [DOI] [PubMed] [Google Scholar]
- Krishnan V., Porter W., Santostefano M., Wang X., Safe S. (1995). Molecular mechanism of inhibition of estrogen-induced cathepsin D gene expression by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in MCF-7 cells. Mol. Cell. Biol. 15, 6710–6719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo R., Matthews J. (2012). High-resolution genome-wide mapping of AHR and ARNT binding sites by ChIP-Seq. Toxicol. Sci. 130, 349–361 [DOI] [PubMed] [Google Scholar]
- Lusska A., Shen E., Whitlock J. P., Jr (1993). Protein-DNA interactions at a dioxin-responsive enhancer. Analysis of six bona fide DNA-binding sites for the liganded Ah receptor. J. Biol. Chem. 268, 6575–6580 [PubMed] [Google Scholar]
- Matikainen T., Perez G. I., Jurisicova A., Pru J. K., Schlezinger J. J., Ryu H. Y., Laine J., Sakai T., Korsmeyer S. J., Casper R. F., et al. (2001). Aromatic hydrocarbon receptor-driven Bax gene expression is required for premature ovarian failure caused by biohazardous environmental chemicals. Nat. Genet. 28, 355–360 [DOI] [PubMed] [Google Scholar]
- Murray I. A., Flaveny C. A., Chiaro C. R., Sharma A. K., Tanos R. S., Schroeder J. C., Amin S. G., Bisson W. H., Kolluri S. K., Perdew G. H. (2011). Suppression of cytokine-mediated complement factor gene expression through selective activation of the Ah receptor with 3’,4’-dimethoxy-α-naphthoflavone. Mol. Pharmacol. 79, 508–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhold L. A., Shirayoshi Y., Ozato K., Jones J. E., Nebert D. W. (1989). Regulation of mouse CYP1A1 gene expression by dioxin: Requirement of two cis-acting elements during induction. Mol. Cell. Biol. 9, 2378–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs S. D., Liu J., Fernando T. M., Fecher R. A., Sulentic C. E. (2012). A dioxin response element in the multiple cloning site of the pGL3 luciferase reporter influences transcriptional activity. Toxicol. In Vitro 26, 979–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesch-Bartlomowicz B., Huelster A., Wiss O., Antoniou-Lipfert P., Dietrich C., Arand M., Weiss C., Bockamp E., Oesch F. (2005). Aryl hydrocarbon receptor activation by cAMP vs. dioxin: Divergent signaling pathways. Proc. Natl. Acad. Sci. U.S.A. 102, 9218–9223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz C. A., Litzenburger U. M., Sahm F., Ott M., Tritschler I., Trump S., Schumacher T., Jestaedt L., Schrenk D., Weller M., et al. (2011). An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197–203 [DOI] [PubMed] [Google Scholar]
- Rogers J. M., Denison M. S. (2000). Recombinant cell bioassays for endocrine disruptors: Development of a stably transfected human ovarian cell line for the detection of estrogenic and anti-estrogenic chemicals. In Vitr. Mol. Toxicol. 13, 67–82 [PubMed] [Google Scholar]
- Saatcioglu F., Perry D. J., Pasco D. S., Fagan J. B. (1990). Aryl hydrocarbon (Ah) receptor DNA-binding activity. Sequence specificity and Zn2+ requirement. J. Biol. Chem. 265, 9251–9258 [PubMed] [Google Scholar]
- Sartor M. A., Schnekenburger M., Marlowe J. L., Reichard J. F., Wang Y., Fan Y., Ma C., Karyala S., Halbleib D., Liu X., et al. (2009). Genomewide analysis of aryl hydrocarbon receptor binding targets reveals an extensive array of gene clusters that control morphogenetic and developmental programs. Environ. Health Perspect. 117, 1139–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen E. S., Whitlock J. P., Jr (1992). Protein-DNA interactions at a dioxin-responsive enhancer. Mutational analysis of the DNA-binding site for the liganded Ah receptor. J. Biol. Chem. 267, 6815–6819 [PubMed] [Google Scholar]
- Swanson H. I., Chan W. K., Bradfield C. A. (1995). DNA binding specificities and pairing rules of the Ah receptor, ARNT, and SIM proteins. J. Biol. Chem. 270, 26292–26302 [DOI] [PubMed] [Google Scholar]
- Vogel C. F., Sciullo E., Li W., Wong P., Lazennec G., Matsumura F. (2007). RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol. Endocrinol. 21, 2941–2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan F., Hao N., Furness S. G., Whitelaw M. L., Chapman-Smith A. (2010). Amino acid substitutions in the aryl hydrocarbon receptor ligand binding domain reveal YH439 as an atypical AhR activator. Mol. Pharmacol. 77, 1037–1046 [DOI] [PubMed] [Google Scholar]
- Wright W. E., Binder M., Funk W. (1991). Cyclic amplification and selection of targets (CASTing) for the myogenin consensus binding site. Mol. Cell. Biol. 11, 4104–4110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y., Nukaya M., Satyshur K. A., Jiang L., Stanevich V., Korkmaz E. N., Burdette L., Kennedy G. D., Cui Q., Bradfield C. A. (2012). Identification of the Ah-receptor structural determinants for ligand preferences. Toxicol. Sci. 129, 86–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao E. F., Denison M. S. (1992). DNA sequence determinants for binding of transformed Ah receptor to a dioxin-responsive enhancer. Biochemistry 31, 5060–5067 [DOI] [PubMed] [Google Scholar]
- Zhang S., Rowlands C., Safe S. (2008). Ligand-dependent interactions of the Ah receptor with coactivators in a mammalian two-hybrid assay. Toxicol. Appl. Pharmacol. 227, 196–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B., Degroot D. E., Hayashi A., He G., Denison M. S. (2010). CH223191 is a ligand-selective antagonist of the Ah (Dioxin) receptor. Toxicol. Sci. 117, 393–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.