

Abstract
Hosts have evolved two distinct defence strategies against parasites: resistance (which prevents infection or limit parasite growth) and tolerance (which alleviates the fitness consequences of infection). However, heritable variation in resistance and tolerance and the genetic correlation between these two traits have rarely been characterized in wild host populations. Here, we estimate these parameters for both traits in Leuciscus burdigalensis, a freshwater fish parasitized by Tracheliastes polycolpus. We used a genetic database to construct a full-sib pedigree in a wild L. burdigalensis population. We then used univariate animal models to estimate inclusive heritability (i.e. all forms of genetic and non-genetic inheritance) in resistance and tolerance. Finally, we assessed the genetic correlation between these two traits using a bivariate animal model. We found significant heritability for resistance (H = 17.6%; 95% CI: 7.2–32.2%) and tolerance (H = 18.8%; 95% CI: 4.4–36.1%), whereas we found no evidence for the existence of a genetic correlation between these traits. Furthermore, we confirm that resistance and tolerance are strongly affected by environmental effects. Our results demonstrate that (i) heritable variation exists for parasite resistance and tolerance in wild host populations, and (ii) these traits can evolve independently in populations.
Keywords: host–parasite interaction, quantitative genetics, molecular-based pedigree, genetic correlation, long-term survey
1. Introduction
To limit the fitness costs imposed by parasites, hosts have evolved a series of defensive responses that can be broadly categorized into two strategies: resistance and tolerance [1–3]. Host resistance prevents infection, limits parasite growth or clears an infection, through behavioural, morphological and/or immunological mechanisms [3]. Unlike resistance, host tolerance does not limit infection, but reduces or compensates parasite-induced damages through reduced immunopathology, increased wound repair mechanisms and a greater resilience to tissue injuries [3–6]. The expression of both defence responses results from the interactions between the host and parasite genomes and the environment [7,8].
Several studies suggested that these two strategies have different evolutionary consequences on host–parasite interactions, depending on whether resistance or tolerance is the trait under selection [1,9]. In particular, because resistance reduces parasite fitness, its evolution is expected to be governed by negative frequency-dependent selection [1]. When parasite prevalence is high, resistance spreads through a host population, causing the decline of parasite prevalence, and then, provided that resistance is costly in the absence of parasitism, a reduction in the frequency of resistant host genotypes is expected. By contrast, because tolerance does not reduce parasite fitness, its evolution is expected to result in a positive feedback whereby parasite infection selects for tolerant hosts, which in turn increases parasite prevalence. As a result, most theoretical models predict the maintenance of genetic variation in resistance but the fixation of tolerance along the course of host–parasite coevolution [2,7,9] (but see [10]).
Scientists have long recognized resistance and tolerance as two distinct defence strategies used by plants in response to biotic and abiotic pressures, including herbivory, pathogens and herbicides [3]. By contrast, the concept of tolerance has only recently been explored in animal systems, and studies on animal defence strategies have almost exclusively focused on resistance mechanisms [3–6]. However, recent observations by Råberg et al. [11] of variation in mice resistance and tolerance to malaria have prompted a surge of studies showing that tolerance plays an important role in animal defence against parasites [11–15].
Although these studies generated a significant body of knowledge on animal tolerance, they mostly used either unnatural host–parasite combinations, or natural systems under controlled laboratory conditions, such that little is known about tolerance variation in the wild [3]. Furthermore, it is still unclear whether host tolerance is enabled by factors that can be transmitted across generations (i.e. if these traits are heritable or not). Disentangling transmitted (heritable) versus non-transmitted (non-heritable) variation in defence strategies is, however, a prerequisite for assessing their evolutionary potential, and for predicting the coevolutionary outcomes of host–parasite interactions in natural settings [16]. To the best of our knowledge, no studies have yet quantified the proportion of transmitted variance explaining phenotypic variation observed in tolerance and resistance in animals (see [17,18] for plant systems).
Traditionally, the potential for evolutionary changes depends on the extent of the additive genetic variance in phenotypic traits [19], and phenotypic variance is hence generally split into genetic versus non-genetic components. However, recent studies have argued that phenotypic variance should rather be split into its transmitted versus non-transmitted variance [20]. Such a dichotomy is particularly relevant when investigating the immune system of vertebrates, which includes innate (i.e. transmitted) and acquired (i.e. non-transmitted) components. Transmitted variance encompasses additive (and non-additive) genetic variance, but also non-genetic inheritance processes such as epigenetic or social inheritance, whereas non-transmitted variance includes other environmental effects [20,21]. Because of the difficulty to tease apart genetic versus non-genetic inheritance processes in the wild, we will use this conservative view of inheritance (i.e. ‘inclusive inheritance’ [20]). Predicting the evolutionary potential of resistance and tolerance in wild populations hence requires assessment of quantitative parameters related to the inheritance of these traits from one generation to another (i.e. transmitted variance, VT; and inclusive heritability, H). Moreover, because resistance and tolerance are often costly [3], a negative genetic correlation is expected between these two traits [2,11]. Such negative genetic correlation may constrain the evolution of one of the two traits [22], and is hence an additional key parameter that has to be assessed to understand their evolution [3].
Here, we aimed at (i) quantifying transmitted versus non-transmitted variance in resistance and tolerance, and (ii) estimating the genetic correlation between these two traits in a freshwater fish (the dace Leuciscus burdigalensis) parasitized by an ectoparasite copepod (Tracheliastes polycolpus). Our analyses are based on long-term monitoring of a wild dace population from which genotypic and phenotypic data have been gathered. Tracheliastes polycolpus has been introduced relatively recently to western Europe (approx. 50–60 years ago, i.e. approx. 200 parasite generations ago; S.B. & G.L. 2013, unpublished data), and it is hence likely that the Leuciscus–Tracheliastes interaction has not yet reached its evolutionary equilibrium. However, interindividual variation in both resistance and tolerance to this parasite has been detected in dace, and this variation has been suggested to be—at least partially—under genetic control [14,23]. We first reconstructed a dace pedigree using genetic markers to estimate relatedness among sampled individuals [24]. We confronted our empirical pedigrees to simulated data to estimate the reliability of molecular-based pedigrees drawn from large populations with overlapping generations (two characteristics of our L. burdigalensis population). Second, we sought to estimate inclusive heritability of resistance and tolerance in dace using ‘animal models’ [24]. Animal models allow decomposition of the phenotypic variance (VP) in different variance components from multigenerational information [24]. Here, VP is decomposed into its transmitted part (i.e. VT; genetic and non-genetic processes were not disentangled) and its non-transmitted part (VNT) that here corresponded to environmental effects and unknown sources of error. Theory predicts that VT should be higher for resistance than for tolerance, with tolerance possibly lacking significant VT [3]; this is what we expect in our wild population if an evolutionary equilibrium has been reached in the Leuciscus–Tracheliastes interaction. Additionally, we hypothesized that we would find strong and significant VNT, notably for tolerance, because this trait has been shown to be under strong environmental effects [25]. Finally, bivariate animal models were used to test the working hypothesis of a significantly transmitted correlation between tolerance and resistance.
2. Methods
(a). Biological models
We focused on L. burdigalensis because it is the major host species of T. polycolpus in the study area [26]. Leuciscus burdigalensis is a freshwater cyprinid fish species endemic to western Europe, commonly distributed in streams and rivers of France. This is a highly mobile species living in shoals of two to five individuals in patches with relatively high water velocity. Tracheliastes polycolpus is a generalist parasite that is anchored to fish fins where it feeds on the epithelial cells and mucus [26,27]. Only adult females are parasitic, and they often cause the partial destruction of fins [27]. This lowers the dace fitness through a reduction in feeding success and decreased growth rate, and possibly incurs size-selective mortality towards younger hosts [23,28]. After approximately three to four months, females lay their eggs in the water column and die. The infective stage of the parasite rapidly develops and infects (within 3 days) other individual hosts through passive encounter. There is no evidence of increased likelihood of parasite transmission either within a host or from parents to offspring. Most likely, the parasite transmits at random in the host population (S.B. & G.L. 2013, unpublished data).
(b). Measurements of host resistance and tolerance
Resistance can be measured as the inverse of parasite burden [3]. Two main ways of measuring tolerance exist in the literature. First, ‘range tolerance’ is defined as the slope of the relationship between a trait capturing host fitness (e.g. growth rate or survivorship) and parasite burden [7,11]. The use of ‘range tolerance’ is particularly appropriate when the fitness proxy examined is affected by factors other than parasite burden [3]. Second, when no other factors than parasitism affect the fitness proxy examined, ‘point tolerance’ is simply the host's ability to preserve its fitness when infected by a given number of parasites [7,11,27]. Here, resistance was estimated as the inverse of parasite burden (i.e. the higher the number of ectoparasites on a host, the less it is resistant), and the extent of fin damage (while accounting for parasite number) was used as a surrogate of tolerance (i.e. ‘point tolerance’ method). Indeed, the presence of T. polycolpus strongly affects the fitness of its host by causing fin damage [29], and fin degradation is only caused by T. polycolpus and not by other factors that would concurrently affect host fitness (i.e. uninfected fish do not present degradation on their fins, at least not the kind of degradations caused by T. polycopus). We have previously demonstrated a significant and positive phenotypic correlation between parasite burden and fin degradation (i.e. the higher the parasite burden, the higher the fin degradation [28]). Therefore, in our study, we use point tolerance because it relates to a phenotypic trait measured at the individual level. This gives us greater statistical power to test for the existence of tolerance heritability and genetic correlations.
(c). Field sampling protocol
A wild population of dace was investigated on the Viaur River (southwestern France). Host sampling was conducted by electric-fishing according to French legislation and prefectoral decrees from the Direction Départementale des Territoires at eight sampling sites which cover the whole distribution of dace in the river (see the electronic supplementary material, figure S1). We consider individuals from these sampling sites to belong to the same population, because we previously showed that genetic spatial structure in this river was weak (with a mean pairwise FST = 3.06% [30]). These eight sampling sites have been monitored at the end of June for each year from 2005 to 2011. The database collected includes measures of resistance and tolerance, as well as other phenotypic and genotypic host characteristics. On each site and for each year, up to 35 daces were sampled, for a total of 670 individuals sampled (table 1). For each specimen, total body length was measured to the nearest mm (as an index of fish age; see below), all T. polycolpus were counted (i.e. parasite burden), and fin degradation was quantified as a score related to the proportion of degraded fin tissue. A score ranging from 0 (0% of the fin is degraded) to 4 (100% of the fin is degraded) was visually attributed to each fin, and the scores were summed over all fins so as to obtain a single total score per host (i.e. for a host, the total score varies from 0 to 7 × 4 = 28; see [14] and the electronic supplementary material, figure S2 for details). We used age–size relationships inferred from this population (using scale reading; see [29]) to assign individuals to the generation to which they belong. A small cut of one pelvic fin was also collected and stored in alcohol for the subsequent genetic analyses. On average, parasite prevalence was 89.6%, parasite load (the number of parasite per individual) was 11.2 ± 11.9 and fin degradation was 1.3 ± 2.2 (see the electronic supplementary material, table S1). We previously demonstrated that there is no association between parasite prevalence, parasite load, fin degradation and the position of sampling sites along the upstream–downstream gradient (e.g. sites with high prevalence during a sampling year do not necessarily harbour high prevalence in successive years [31]). All individuals were released alive in their original sampling site.
Table 1.
Number of sampled and successfully amplified daces (Leuciscus burdigalensis) for each sampling year (2005–2011) and each sampling site on the Viaur River.
| sampling site | 2005 | 2006 | 2007 | 2008 | 2009 | 2010 | 2011 |
|---|---|---|---|---|---|---|---|
| Albinet | 18 | 20 | 10 | 12 | 0 | 0 | 0 |
| Bannnes | 10 | 16 | 0 | 5 | 8 | 0 | 0 |
| Calquiere | 16 | 13 | 15 | 11 | 17 | 14 | 2 |
| Capelle | 22 | 19 | 20 | 32 | 29 | 17 | 13 |
| Fuel | 10 | 18 | 12 | 10 | 0 | 0 | 1 |
| Navech | 14 | 20 | 20 | 25 | 19 | 5 | 7 |
| St Just | 16 | 20 | 20 | 35 | 17 | 17 | 2 |
| Serres | 11 | 17 | 4 | 8 | 0 | 3 | 0 |
| total | 117 | 143 | 101 | 138 | 90 | 56 | 25 |
(d). Genetic analyses
We used neutral microsatellite genetic markers to infer relatedness and reconstruct pedigrees in the studied population. After DNA extraction, individual genotypes were obtained at 13 polymorphic microsatellite loci (see [23] for details on microsatellites). To avoid wrong family assignments and to improve the quality of our pedigrees [32], we paid particular attention to errors related to genotyping and profile reading (i.e. several genotypes were amplified twice and reading was done by three people independently) so as to reduce those errors below 0.1%. No linkage disequilibrium or null alleles were detected in this set of microsatellites, and they were all in Hardy–Weinberg equilibrium [23]. Because individuals from the studied population may have been repeatedly sampled over several years, a preliminary analysis was performed using Genecap [33] to identify potential recaptures. This software identifies perfect matches from microsatellites genotypes within a dataset, which represent recaptured individuals over years. We detected 24 individuals that were recaptured twice or more. To avoid pseudo-replication, the first capture event was kept in the final pedigree database, whereas subsequent recaptures were removed from the database. Nevertheless, these 24 individuals were used to estimate repeatability values [19] on both tolerance and resistance (see below).
(e). Pedigree reconstruction
Relatedness was inferred using Pedigree v. 2.2 [34], which uses a likelihood estimator to allocate or partition all individuals into groups of putative siblings in different family configurations. Initially, all individuals are assumed unrelated and are allocated to full-sib groups of size 1 (called singletons). According to Herbinger et al. [35], a full-sib partition contains a grouping of full-sib individuals defined using the rules of Mendelian inheritance from one cross of two parents. Thus, Pedigree v. 2.2 produces a full-sib partition by sampling the space of possible partitions through the Markov chain Monte Carlo (MCMC) procedure. We used 20–40 different starting points for the MCMC procedure. The chain was iterated 106 times and allocated a weight of 1 to avoid false full-sib assignments [35]. We then assessed the consistency of the reconstructed groups across the different starting points [35]. From these results, we retained one ‘best’ sibship configuration (with the maximized partition score) of full-sib families.
Dace is a long-lived species (up to 10 years old) with overlapping generations and high migration rate [30], and we probably sampled only a fraction of the population (effective population size was estimated as approximately 500 individuals; see the electronic supplementary material, figure S3). Although these characteristics are likely to pose difficulties for the inference of relatedness using microsatellite data, a recent study demonstrated this approach is appropriate and robust to these population parameters (high migration rate and large population size), even with a relatively low number of markers (less than 20 loci [32]). To ensure that our partitions mostly contain full-sibs (but not half-sibs, cousins or other relationships), we developed an approach aiming at maximizing the certainty of the reconstructed relationships by reducing the possible range of sibship relationships being inferred. To do so, individuals were first assigned to the generation they belong to (i.e. using age–size relationships; see above). We then built a pedigree for each generation separately (17 pedigrees from the 17 generations identified from 1994 to 2010), so as to ensure that each pedigree was a ‘first-generation’ pedigree. These pedigrees were finally assembled into a single pedigree for the estimation of quantitative transmitted parameters. This within-generation pedigree reconstruction allowed us to maximize the probability to reconstruct full-sib relationships and hence to detect some of the variance components constituting the measured phenotypes.
The reliability of the final pedigree was tested by confronting the optimal partitions against a null hypothesis of no relatedness. Each pedigree is tested for significance using 100 randomization trials of the original genotype dataset. The overall significance (p-value) of each pedigree retained was estimated by the proportion of the 100 randomized trials having a pedigree score at least as high as the true partition score observed.
(f). Simulation: testing the accuracy of pedigree reconstruction
We simulated a genetic dataset to test the reliability of the pedigree built from the approach described above. First, we used the program spip v. 1.0 [36] to simulate a population defined by life-history parameters approximating our dace population. This program uses an individual-based algorithm to simulate the transmission of genes from parents to offspring in a population under a demographic dynamic. The simulated population is based on life-history parameters that are detailed in table 2. Parameters were age- and sex-specific, and we assumed that the maximum lifespan was 10 years. In this population, males and females had equal initial population size, relative fecundity and absolute probability to reproduce and survive (table 2). The life-history parameters were gathered from unpublished literature (S.B. & G.L. 2013, unpublished data). The initial number of females and males were distributed among generations to represent the distribution in our natural population. We assumed that around 6000 individuals are produced at each generation. We then characterized individuals by 13 loci with allelic frequencies similar to those observed in the natural population. Simulations were run for 50 generations. Because our simulations were individual-based, relatedness information (i.e. parents' identity) was available for each individual of each generation. Upon simulations, we haphazardly sampled 650 individuals from the last 10 generations to mimic the fact that we sampled only a fraction of our dace population. From this subset, we identified ‘true’ family links from information on simulated parental identities to set a ‘true’ pedigree made of groups including only full-sibs. In parallel, the genetic data from the same simulated subdataset were used as input in Pedigree v. 2.2 to infer molecular-based pedigrees (using the within-generation approach described above). The final molecular-based pedigree was then compared with the ‘true’ pedigree and congruency between pedigrees was calculated using PedAgree v. 1.05 [37].
Table 2.
Life-history parameters used to simulate a population of a long-lived species with large effective population size and overlapping generations.
| age | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| initial number of females/males | 100 | 100 | 334 | 456 | 770 | 708 | 348 | 272 | 160 | 106 | n.a. |
| relative fecundity | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0.5 | 0.5 | 0.5 |
| absolute probability to reproduce | 0 | 0 | 0 | 0.1 | 0.1 | 0.2 | 0.2 | 0.1 | 0.1 | 0.1 | 0.1 |
| probability of survival | 0.3 | 0.3 | 0.4 | 0.5 | 0.5 | 0.5 | 0.5 | 0.2 | 0.2 | 0.1 | 0 |
2. Statistical analysis
Quantitative genetic analyses are based on the partitioning of the total phenotypic variance into different components. We considered the total phenotypic variance (hereafter VP) to be
| 2.1 |
where VT is the transmitted variance, VNT is the non-transmitted variance and VR is the residual variance [20].
In the statistical models presented below, the pedigree (or family membership) was used to estimate VT, and the sampling years, the sampling sites and the resulting two-term interaction accounted for VNT in equation (2.1). All these variables were included as random effects in subsequent models. It is noteworthy that VNT not only includes environmental factors affecting (directly or indirectly via resource availability) host defence strategies, but also potential differences in parasite virulence between sampling sites and/or sampling years (e.g. parasite virulence can vary across sites because of a spatial structure of the parasite, or because of differences in environmental conditions affecting virulence). Because body size is highly correlated to age in fishes, and because parasites can accumulate over time on a host, we added dace body size (a continuous trait) as a fixed covariate in the models. Parasite burden was also included as a fixed covariate in the tolerance models to measure fin degradation while statistically accounting for the number of parasites [13,14,29].
Afterwards, inclusive heritability (H) was calculated as the ratio of the estimated transmitted variance to total phenotypic variance (i.e. H = VT/VP). Similarly, the proportion of variance explained by each term composing VNT was quantified. Models are based on non-Gaussian error-term distributions (see below), meaning that ‘deviance’ (rather than variance) should be used. However, for the sake of clarity, we will hereafter use the term ‘variance’ to define deviance components.
For each trait, we ran a univariate model (see the electronic supplementary material, S5 for R scripts) so as to quantify the proportion of variance explained by each components of equation (2.1) using animal models as developed in the R-Gui package MCMCglmm [38]. A Poisson error-term distribution was assumed for resistance, whereas an over-dispersed Poisson error-term distribution was assumed for our measure of tolerance. Usually, 106 iterations of the MCMC resulted in model convergence, with no evident autocorrelation within chains (lower than 0.05). The posterior distribution of the animal models was sampled every 1000 iterations after a burn-in period of 50 000 iterations for a total of 950 samples. We used non-informative priors (V = 1, ν = 1.002) for all animal models [38] (models with informative priors led to very similar results and are hence not shown here). Variance components were estimated as the mode of the posterior distribution and 95% credible intervals (CIs) are given. Ratios (VT/VP and VNT/VP) were calculated for each MCMC sample, and we reported the mode and 95% CIs of these posterior distributions. Covariance components were considered meaningful when 95% CIs excluded zero.
This quantitative analysis was complemented by a measure of repeatability [19]. Repeatability was inferred for resistance and tolerance using the 24 recaptures detected previously. Repeatability can be inferred by partitioning the phenotypic variance into within-individual variance and between-individual variance [19]. We used the REML framework as implemented in the R package rptR [39] to estimate repeatability for both tolerance and resistance according to the following equation:
| 2.2 |
where 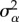 is the between-group variance and
is the between-group variance and 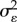 is the within-group variance. We used the adjusted repeatability measures that account simultaneously for the confounding effects of sampling year, sampling site and parasite burden (only for the repeatability related to our measure of tolerance).
is the within-group variance. We used the adjusted repeatability measures that account simultaneously for the confounding effects of sampling year, sampling site and parasite burden (only for the repeatability related to our measure of tolerance).
(a). Bivariate animal models: estimating genetic correlation
To disentangle the possible link between tolerance and resistance, we tested whether there was a genetic correlation between these two traits using bivariate animal models. The fixed and random effects used in the Bayesian univariate models described above were used in these analyses.
The genetic correlation (rg) is calculated as
| 2.3 |
where COVtrait1,trait2 is the genetic covariance between the traits, and VGtrait1 and VGtrait2 are the genetic variance, respectively, for trait 1 and trait 2.
3. Results
(a). Pedigree reconstruction
The pedigree included 661 individuals assigned in 396 presumed full-sib families (figure 1). There were 147 individuals assigned to a family of one individual (i.e. singletons; figure 1). These singletons were discarded from subsequent analyses. No family larger than three full-sibs was reconstructed, and most families were constituted of two full-sibs (figure 1).
Figure 1.
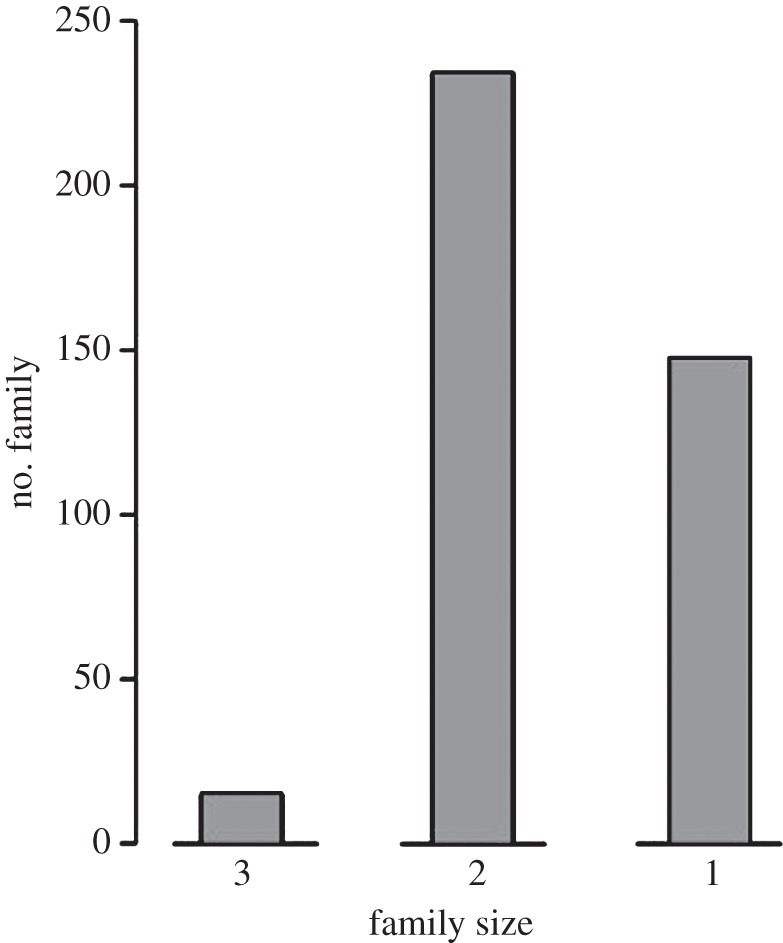
Distribution of family sizes for each of the pedigrees we reconstructed for dace (Leuciscus burdigalensis) in the Viaur River.
Importantly, our pedigree provides strong evidence that families were weakly spatially aggregated (i.e. members of a family were not all captured in a single sampling site). Indeed, for families of two full-sibs, only 22% of the families were spatially aggregated, whereas only 28% of the families were spatially aggregated for families of three full-sibs (not shown).
Our pedigree reached a score higher than any of the 100 randomized pedigrees, indicating that the full-sib partition could not be seen in a set of unrelated individuals (p < 0.02).
(b). Simulation and accuracy of pedigree reconstruction
According to our simulated dataset, the congruence between the ‘true’ pedigree (the one for which family links are gathered from parental identity information) and the reconstructed global pedigree was 53.60%, which indicated that our approach fails to match perfectly the ‘true’ pedigree.
(c). Univariate animal models
Animal models revealed significant inclusive heritability for resistance to parasites (posterior mean = 17.6%; 95% CI: 7.2–32.2%; figure 2a). All factors (sampling site, sampling year and the two-term interaction) related to non-transmitted variance explained a significant amount of variation in resistance (figure 2a). Overall non-transmitted variation accounted for 48.8% of the total variation observed in resistance. 33.6% of the phenotypic variance remains unexplained in our model (figure 2a). The influence of body size on the parasite burden was positive, although relatively weak (not shown).
Figure 2.

Proportion of phenotypic variance explained by transmitted (VT/VP) and non-transmitted factors (VNT/VP) on (a) resistance and (b) tolerance to the ectoparasite Tracheliastes polycolpus. Proportions of explained variance were obtained from univariate animal models. The proportions of unexplained variance (VR/VP) are also displayed. In each plot, we represented the posterior mode of each variable (dots) and the 95% CI for this effect (horizontal bars). See details in the electronic supplementary material, table S3.
There was also significant inclusive heritability for tolerance (posterior mean = 18.8%; 95% CI: 4.4–36.1%; figure 2b). Whole effects related to non-transmitted variance summarized 40.9% of the amount of phenotypic variation. Residual phenotypic variance was 40.3% (figure 2b). The effect of body size was not significant (95% CI overlap zero), but as expected there was a positive and significant relationship between parasite burden and fin degradation.
(d). Repeatability
We found strong and significant repeatability for both resistance (r = 0.613, 95% CI: 0.258–0.835, p < 0.001) and tolerance (r = 0.639, 95% CI: 0.033–0.854, p < 0.001).
(e). Bivariate animal model
Concerning genetic correlation between resistance and tolerance, we found no support for a genetic correlation, because the 95% CI overlapped zero (rT = 0.06; 95% CI: −0.28 to 0.65).
4. Discussion
To the best of our knowledge, our study is among the first to attempt quantifying the heritability of both resistance and tolerance to parasites in a wild animal host population. The finding that resistance to parasites is transmitted across generations (i.e. heritable) confirms theoretical predictions as well as empirical studies [15,22]. Indeed, the maintenance of heritable variation in resistance is expected because of (i) the antagonistic coevolution between host resistance and parasite virulence, and (ii) the energetic costs associated with resistance [2,11]. Although similar costs have been reported [40], few studies have yet reported heritability for this trait in wild animal populations (but see [41–43]).
The finding that tolerance to parasites was also heritable was, however, less expected. Indeed, theoretical models predict an erosion of the genetic variance associated with tolerance [1]. Despite these theoretical considerations, genetic variation in tolerance has previously been suggested in laboratory-based systems [11,44]. Our study is, however, the first to find significant heritable variation in tolerance in a wild host–parasite association. Two main hypotheses may explain the maintenance of heritable variation in tolerance. First, it is likely that the Leuciscus–Tracheliastes interaction has not yet reached its evolutionary equilibrium, because T. polycolpus has been introduced approximately 200 parasite generations ago (S.B. & G.L. 2013, unpublished data). Second, Miller et al. [2] suggested that polymorphisms in tolerance can be maintained if other factors (e.g. environmental factors) simultaneously influence patterns of selection. This second hypothesis is also highly likely since, in the wild, selective pressures are multiple and polymorphism can be maintained actively by heterogeneous natural selection [45].
Inclusive heritability values estimated for both traits (approx. 16–18% for both traits) were moderate and in the range of what is typically found for resistance (see [46] for a review on fish). Studies indicating higher heritability values of resistance (i.e. greater than 20%) have generally been conducted in experimental settings [47,48] whereby non-transmitted factors were controlled for. It is hence likely that our relatively low values of heritability are inherent to the natural settings in which we performed our investigation. Our relatively low heritability values may also be due to limitations of the analytical tools currently available to estimate pedigrees. Our reconstructed pedigree possibly included links other than full-sib relationships, and we think that for species with overlapping generations such as L. burdigalensis, reconstructing a pedigree from molecular data gathered from a portion of the population is a complex task (but see [32]). Indeed, according to our simulations, we found moderate congruencies (53.6%) between ‘true’ and reconstructed pedigrees, which hence indicate that half-sibs, cousins and even unrelated groups are probably incorporated in our molecular-based pedigree. Such types of bias (i.e. assigning individuals as full-sibs, while they are not) have been shown to reduce statistical power (type II errors, i.e. failing to detect a significant heritability value while it is actually significant) and to lead to underestimation of quantitative genetic parameters (approx. 10% for heritability [49]). This means that, in the worst case, our pedigree has weak statistical power (which we confirmed using simulations, not shown) and underestimates heritability estimates, which is hence conservative. Accordingly, repeatability values, which are expected to provide an upper limit of possible heritability values [19], were strikingly higher for both resistance and tolerance to parasites. This suggests that the actual heritability estimates for these two traits are probably between the estimates we reported using animal models and those gathered from repeatability analyses.
The weak statistical power of our pedigrees may explain why we failed to find a negative genetic correlation between tolerance and resistance to parasites. Although theoretical work predicts that tolerance and resistance should display significant genetic correlation [16,50], some studies in plants and animals have found no support for this hypothesis [15,51,52]. Despite the weakness of our pedigrees, our estimate of genetic correlation is far from significant. Our results may hence confirm that a genetic trade-off between resistance and tolerance is not necessarily the norm in wild populations. This result may be partly explained by environmental variation that may strongly modulate the shape and strength of genetic correlations [53]. Hence, as for the maintenance of variation in tolerance, environmental heterogeneity may affect genetic correlations. Furthermore, the lack of a genetic trade-off might be specific to our system, which involves an ectoparasite. First, while such parasites may be less affected by host immunological responses than endoparasites, they may be more vulnerable to other resistance mechanisms such as behavioural defences. Second, tolerance mechanisms are more likely to involve wound repairs than immune regulation in this system. Therefore, it is possible that, because tolerance and resistance do not rely on the same proximate mechanisms (the host immunological system), they are genetically decoupled in this system. Non-transmitted variance accounted for approximately 40–50% of variation in resistance and tolerance to parasites. It is noteworthy that families were weakly spatially aggregated, meaning that transmitted variation is statistically independent from non-transmitted sources of variation (and vice versa). Here, non-transmitted variation was expressed as the spatial and temporal variation observed in resistance and tolerance, which may reflect spatio-temporal variability in demographic processes (e.g. number of parasite propagules), population structure (e.g. age, size and density of the hosts) and/or the direct effects of specific—although unmeasured—environmental factors (e.g. water velocity, water temperature, etc.). Overall, this observation is in agreement with recent studies demonstrating a strong role of the environment in explaining variation in resistance and tolerance [14,25]. For instance, in the Leuciscus–Tracheliastes interactions, Cardon et al. [31] demonstrated that fin degradation tended to be higher under colder conditions, possibly owing to more virulent parasites or reduced tolerance under these thermal conditions. Our results confirm that the environment (sensu latto) may be an important modulator of the coevolution between host and parasite, both because the environment can influence genetic parameters [53], and because it can directly or indirectly influence resistance, tolerance and virulence [8,54].
In this study, we adopted the ‘inclusive inheritance’ framework [20] to distinguish between transmitted versus non-transmitted variation, rather than the genetic versus environmental variation approach of quantitative genetics [19]. This framework is particularly appropriate for the study of wild populations where genetic and non-genetic transmitted processes can be confounded [20,21]. For instance, Stopher et al. [55] demonstrated that accounting for social contacts between individuals strikingly reduces estimates of heritability in a wild ungulate population. Likewise, in wild populations, resistance and tolerance may be affected by many processes that are transmitted across generations but that are not related to additive (or non-additive) genetic variance. For instance, epigenetic inheritance may contribute to the inclusive heritability of resistance and tolerance (reviewed in [56]). Similarly, parental non-genetic effects have already been shown in resistance to parasites. For example, in the kittiwake (Rissa tridactyla), females transfer specific antibodies against a bacterial infection in their eggs' yolk [57]. Although distinguishing between those underlying processes is not possible given our sampling design, we believe that it is conservative to consider that not only genetic variance but also non-genetic variance may contribute to the patterns of heritability reported here. Identifying the contribution of each transmitted process—and notably the contribution related to additive genetic variance—to phenotypic variation may have drastic evolutionary implications, and will be a major challenge of future studies [20,21].
5. Evolutionary implications and conclusion
Our results provide strong support for a significant inclusive heritability for resistance and tolerance in a wild population of the host L. burdigalensis. However, we failed to find a significant genetic correlation between these traits even though genetic correlations based on a pedigree design such as ours are expected to be inflated by non-additive genetic effects [58]. In the meantime, non-transmitted variation explained a large amount of the total phenotypic variance, which suggests that environmental factors should be accounted for. These results have important implications at the local scale, but also more broadly for predicting coevolution of host–parasite interactions.
At the local scale, the finding of a heritable basis for traits related to parasite defence strategies, and the absence of an apparent trade-off, suggests that both resistance and tolerance have significant evolutionary potential in this dace population. This is an important conclusion because, as stated above, T. polycolpus is a non-native parasite that has only recently colonized western Europe. Hence, our results suggest that the dace population from the Viaur River may possess the transmitted variance required to adapt to this recent selective pressure. Future studies will confirm this local-scale result by assessing and comparing different dace populations. This should provide new insights into the role of environmental factors in modulating transmitted variance, and into the possibility for local adaptation in this host–parasite system.
More broadly, the heritable basis for resistance and tolerance is often assumed in theoretical work, although rarely quantified in the wild. Our results are therefore of prime importance as they confirm strong assumptions of theoretical models. Given uncoupled heritable variation for the two defence mechanisms, one may ask how the independent evolution of resistance and tolerance shapes coevolution with virulence [16]. According to Carval & Ferriere [16], two coevolutionary avenues are possible: (i) virulence is predicted to covary positively with resistance, and negatively with tolerance as the cost of resistance decreases; or (ii) virulence is predicted to covary negatively with resistance, and positively with tolerance as the cost of tolerance decreases. According to experimental studies focusing on pathogen virulence, considerable spatial variation is observed in this three-component interaction, as well as in the strength of local coevolutionary dynamics [59]. Hence, demonstrating significant transmission across generations for tolerance and resistance is an important prerequisite for understanding coevolution between virulence, resistance and tolerance. However, we call for more experimental work aiming at elucidating proximal mechanisms affecting these traits, as well as empirical work explicitly including the spatial component of host–parasite interactions [11,16].
To conclude, Wolinska & King [8] stressed that coevolutionary models should include further three-way interactions (host-genotype–parasite-genotype–environment interactions). We accompany this call by arguing that both proximal mechanisms and ultimate processes have to be studied in a broader framework to understand and predict the polymorphism in pathogens' attack and hosts' defence strategies.
Acknowledgements
We are grateful to students and colleagues who helped with fish and parasite sampling. We are grateful to Roselyne Etienne and Charlotte Veyssière, who helped with genotyping. We thank the anonymous reviewers for their constructive comments. We are also grateful to Dr Christophe Herbinger and Dr Jason Coombs for helpful comments and technical support with the pedigree v. 2.2 and spip software, respectively.
Data accessibility
Data available from the Dryad Digital Repository: http://doi.org/10.5061/dryad.s3k34.
Funding statement
This study is part of the project INCLIMPAR, which is financially supported by a grant from the Agence Nationale de la Recherche to G.L. (grant no. ANR-2011-JSV7-010-01). E.M.-G. is financially supported by a doctoral scholarship from the Ministère de l'enseignement supérieur et de la recherche. This work is part of the Laboratoire d'Excellence TULIP (ANR-10-LABX-41).
References
- 1.Roy BA, Kirchner JW. 2000. Evolutionary dynamics of pathogen resistance and tolerance. Evolution 54, 51–63 (doi:10.1111/j.0014-3820.2000.tb00007.x) [DOI] [PubMed] [Google Scholar]
- 2.Miller MR, Whiten A, Boots M. 2005. The evolution of host resistance: tolerance and control as distinct strategies. J. Theor. Biol. 236, 198–207 (doi:10.1016/j.jtbi.2005.03.005) [DOI] [PubMed] [Google Scholar]
- 3.Råberg L, Graham AL, Read AF. 2009. Decomposing health: tolerance and resistance to parasites in animals. Phil. Trans. R. Soc. B 364, 37–49 (doi:10.1098/rstb.2008.0184) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Medzhitov R, Schneider DS, Soares MP. 2012. Disease tolerance as a defense strategy. Science 335, 936–941 (doi:10.1126/science.1214935) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baucom RS, de Roode JC. 2011. Ecological immunology and tolerance in plants and animals. Funct. Ecol. 25, 18–28 (doi:10.1111/j.1365-2435.2010.01742.x) [Google Scholar]
- 6.Ayres JS, Schneider DS. 2012. Tolerance of Infections. Annu. Rev. Immunol. 30, 271–294 (doi:10.1146/annurev-immunol-020711-075030) [DOI] [PubMed] [Google Scholar]
- 7.Little TJ, Shuker DM, Colegrave N, Day T, Graham AL. 2010. The coevolution of virulence: tolerance in perspective. PLoS Pathog. 6, e1001006 (doi:10.1371/journal.ppat.1001006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolinska J, King KC. 2009. Environment can alter selection in host–parasite interactions. Trends Parasitol. 25, 236–244 (doi:10.1016/j.pt.2009.02.004) [DOI] [PubMed] [Google Scholar]
- 9.Boots M, Best A, Miller MR, White A. 2009. The role of ecological feedbacks in the evolution of host defence: what does theory tell us? Phil. Trans. R. Soc. B 364, 27–36 (doi:10.1098/rstb.2008.0160) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Best A, White A, Boots M. 2008. Maintenance of host variation in tolerance to pathogens and parasites. Proc. Natl Acad. Sci. USA 105, 20 786–20 791 (doi:10.1073/pnas.0809558105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Råberg L, Sim D, Read AF. 2007. Disentangling genetic variation for resistance and tolerance to infectious diseases in animals. Science 318, 812–814 (doi:10.1126/science.1148526) [DOI] [PubMed] [Google Scholar]
- 12.Ayres JS, Schneider DS. 2009. The role of anorexia in resistance and tolerance to infections in Drosophila. PLoS Biol. 7, e1000150 (doi:10.1371/journal.pbio.1000150) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ayres JS, Freitag N, Schneider DS. 2008. Identification of Drosophila mutants altering defense of and endurance to listeria monocytogenes infection. Genetics 178, 1807–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blanchet S, Rey F, Loot G. 2010. Evidence for host variation in parasite tolerance in a wild fish population. Evol. Ecol. 24, 1129–1139 (doi:10.1007/s10682-010-9353-x) [Google Scholar]
- 15.Lefevre T, Williams AJ, de Roode JC. 2011. Genetic variation in resistance, but not tolerance, to a protozoan parasite in the monarch butterfly. Proc. R. Soc. B 278, 751–759 (doi:10.1098/rspb.2010.1479) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carval D, Ferriere R. 2010. A unified model for the coevolution of resistance, tolerance, and virulence. Evolution 64, 2988–3009 (doi:10.1111/j.1558-5646.2010.01035.x) [DOI] [PubMed] [Google Scholar]
- 17.Fornoni J, Valverde PL, Nunez-Farfan J. 2003. Quantitative genetics of plant tolerance and resistance against natural enemies of two natural populations of Datura stramonium. Evolution 5, 1049–1065 [Google Scholar]
- 18.Juenger T, Bergelson J. 2000. The evolution of compensation to herbivory in scarlet gilia, Ipomopsis aggregata: herbivore-imposed natural selection and the quantitative genetics of tolerance. Evolution 54, 764–777 [DOI] [PubMed] [Google Scholar]
- 19.Falconer DS, MacKay TFC. 1996. Introduction to quantitative genetics, 4th edn. Harlow, UK: Prentice Hall [Google Scholar]
- 20.Danchin E, Charmantier A, Champagne FA, Mesoudi A, Pujol B, Blanchet S. 2011. Beyond DNA: integrating inclusive inheritance into an extended theory of evolution. Nat. Rev. Genet. 12, 475–486 (doi:10.1038/nrg3028) [DOI] [PubMed] [Google Scholar]
- 21.Bonduriansky R, Day T. 2009. Nongenetic inheritance and its evolutionary implications. Annu. Rev. Ecol. Evol. Syst. 40, 103–125 (doi:10.1146/annurev.ecolsys.39.110707.173441) [Google Scholar]
- 22.Fineblum WL, Rausher MD. 1995. Tradeoff between resistance and tolerance to herbivore damage in a morning glory. Nature 377, 517–520 (doi:10.1038/377517a0) [Google Scholar]
- 23.Blanchet S, Rey O, Berthier P, Lek S, Loot G. 2009. Evidence of parasite-mediated disruptive selection on genetic diversity in a wild fish population. Mol. Ecol. 18, 1112–1123 [DOI] [PubMed] [Google Scholar]
- 24.Wilson AJ, Reale D, Clements MN, Morrissey MM, Postma E, Walling CA, Kruuk LEB, Nussey DH. 2010. An ecologist's guide to the animal model. J. Anim. Ecol. 79, 13–26 (doi:10.1111/j.1365-2656.2009.01639.x) [DOI] [PubMed] [Google Scholar]
- 25.Sternberg ED, Lefèvre T, Li J, de Castillejo CLF, Li H, Hunter MD, de Roode JC. 2012. Food plant derived disease tolerance and resistance in a natural butterfly–plant–parasite interactions. Evolution 66, 3367–3376 (doi:10.1111/j.1558-5646.2012.01693.x) [DOI] [PubMed] [Google Scholar]
- 26.Lootvoet A, Blanchet S, Gevrey M, Buisson L, Tudesque L, Loot G. 2013. Patterns and processes of alternative host use in a generalist parasite: insights from a natural host–parasite interaction. Funct. Ecol. 27, 1403–1414 (doi:10.1111/1365-2435.12140) [Google Scholar]
- 27.Loot G, Poulet N, Reyjol Y, Blanchet S, Lek S. 2004. The effects of the ectoparasite Tracheliastes polycolpus (Copepoda: Lernaeopodidae) on the fins of rostrum dace (Leuciscus leuciscus burdigalensis). Parasitol. Res. 94, 16–23 (doi:10.1007/s00436-004-1166-9) [DOI] [PubMed] [Google Scholar]
- 28.Blanchet S, Mejean L, Bourque JF, Lek S, Thomas F, Marcogliese DJ, Dodson J, Loot G. 2009. Why parasitized hosts look different? Resolving the ‘chicken-egg’ dilemma. Oecologia 160, 37–47 [DOI] [PubMed] [Google Scholar]
- 29.Corby-Harris V, Habel KE, Ali FG, Promislow DEL. 2007. Alternative measures of response to Pseudomonas aeruginosa infection in Drosophila melanogaster. J. Evol. Biol. 20, 526–533 (doi:10.1111/j.1420-9101.2006.01267.x) [DOI] [PubMed] [Google Scholar]
- 30.Blanchet S, Rey O, Etienne RS, Lek S, Loot G. 2010. Species-specific responses to landscape fragmentation: implications for management strategies. Evol. Appl. 3, 291–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cardon M, Loot G, Grenouillet G, Blanchet S. 2011. Host characteristics and environmental factors differentially drive the burden and pathogenicity of an ectoparasite: a multilevel causal analysis. J. Anim. Ecol. 80, 657–667 (doi:10.1111/j.1365-2656.2011.01804.x) [DOI] [PubMed] [Google Scholar]
- 32.Harrison EC, Saenz-Agudelo P, Planes S, Jones GP, Berumen ML. 2013. Relative accuracy of three common methods of parentage analysis in natural populations. Mol. Ecol. 22, 1158–1170 (doi:10.1111/mec.12138) [DOI] [PubMed] [Google Scholar]
- 33.Wilberg MJ, Dreher BP. 2004. GENECAP: a program for analysis of multilocus genotype data for non-invasive sampling and capture-recapture population estimation. Mol. Ecol. Notes 4, 783–785 (doi:10.1111/j.1471-8286.2004.00797.x) [Google Scholar]
- 34.Herbinger C. 2005. Pedigree 2.2 help manual See http://herbinger.biology.dal.ca:5080/Pedigree [Google Scholar]
- 35.Herbinger CM, O'Reilly PT, Verspoor E. 2006. Unravelling first-generation pedigrees in wild endangered salmon populations using molecular genetic markers. Mol. Ecol. 15, 2261–2275 [DOI] [PubMed] [Google Scholar]
- 36.Andersson EC, Dunham KK. 2005. spip 1.0: a program for simulating pedigrees and genetic data in age-structured populations. Mol. Ecol. Notes 5, 459–461 (doi:10.1111/j.1471-8286.2005.00884.x) [Google Scholar]
- 37.Coombs JA, Letcher BH, Nislow KH. 2010. PedAgree: software to quantify error and assess accuracy and congruence for genetically reconstructed pedigree relationships. Conserv. Genet. Resour. 2, 147–150 (doi:10.1007/s12686-010-9202-9) [Google Scholar]
- 38.Hadfield JD. 2010. MCMC methods for multi-response generalised linear mixed models: the MCMCglmm R package. J. Stat. Soft. 33, 1–22 [Google Scholar]
- 39.Nakagawa S, Schielzeth H. 2010. Repeatability for Gaussian and non-Gaussian data: a practical guide for biologists. Biol. Rev. 85, 935–956 (doi:10.1111/j.1469-185X.2010.00141.x) [DOI] [PubMed] [Google Scholar]
- 40.Koskela T, Puustinen S, Salonen V, Mutikainen P. 2002. Resistance and tolerance in a host plant–holoparasitic plant interaction: genetic variation and costs. Evolution 56, 899–908 [DOI] [PubMed] [Google Scholar]
- 41.Boulinier T, Sorci G, Monnat J-Y, Danchin E. 1997. Parent–offspring regression suggests heritable susceptibility to ectoparasites in a natural population of kittiwake Rissa tridactyla. J. Evol. Biol. 10, 77–85 (doi:10.1046/j.1420-9101.1997.10010077.x) [Google Scholar]
- 42.Smith JA, Wilson K, Pilkington JG, Pemberton JM. 1999. Heritable variation in resistance to gastro-intestinal nematodes in an unmanaged mammal population. Proc. R. Soc. Lond. B 266, 1283–1290 (doi:10.1098/rspb.1999.0776) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uller T, Olsson M, Madsen T. 2003. Family and population effects on disease resistance in a reptile. Heredity 91, 112–116 (doi:10.1038/sj.hdy.6800288) [DOI] [PubMed] [Google Scholar]
- 44.Kover PX, Schaal BA. 2002. Genetic variation for disease resistance and tolerance among Arabidopsis thaliana accessions. Proc. Natl Acad. Sci. USA 99, 11 270–11 274 (doi:10.1073/pnas.102288999) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anderson JT, Lee C-R, Rushworth CA, Colautti RI, Mitchell-Olds T. 2013. Genetic trade-offs and conditional neutrality contribute to local adaptation: genetic basis of local adaptation. Mol. Ecol. 22, 699–708 (doi:10.1111/j.1365-294X.2012.05522.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carlson SM, Seamons TR. 2008. A review of quantitative genetic components of fitness in salmonids: implications for adaptation to future change. Evol. Appl. 1, 222–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kube PD, Taylor RS, Elliott NG. 2012. Genetic variation in parasite resistance of Atlantic salmon to amoebic gill disease over multiple infections. Aquaculture 364–365, 165–172 (doi:10.1016/j.aquaculture.2012.08.026) [Google Scholar]
- 48.Kause A, van Dalen S, Bovenhuis H. 2012. Genetics of Ascites resistance and tolerance in chicken: a random regression approach. Genes Genomes Genet. 2, 527–535 (doi:10.1534/g3.112.002311) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomas SC, Hill WG. 2000. Estimating quantitative genetic parameters using sibships reconstructed from marker data. Genetics 155, 1961–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Restif O, Koella JC. 2004. Concurrent evolution of resistance and tolerance to pathogens. Am. Nat. 164, E90–E102 (doi:10.1086/423713) [DOI] [PubMed] [Google Scholar]
- 51.Mauricio R, Rausher MD, Burdick DS. 1997. Variation in the defense strategies of plants: are resistance and tolerance mutually exclusive? Ecology 78, 1301–1311 (doi:10.1890/0012-9658(1997)078[1301:VITDSO]2.0.CO;2) [Google Scholar]
- 52.Simms EL, Triplett J. 1994. Costs and benefits of plant-responses to disease: resistance and tolerance. Evolution 48, 1973–1985 (doi:10.2307/2410521) [DOI] [PubMed] [Google Scholar]
- 53.Sgrò CM, Hoffmann AA. 2004. Genetic correlations, tradeoffs and environmental variation. Heredity 93, 241–248 (doi:10.1038/sj.hdy.6800532) [DOI] [PubMed] [Google Scholar]
- 54.Laine A-L. 2008. Temperature-mediated patterns of local adaptation in a natural plant–pathogen metapopulation. Ecol. Lett. 11, 327–337 (doi:10.1111/j.1461-0248.2007.01146.x) [DOI] [PubMed] [Google Scholar]
- 55.Stopher KV, Walling CA, Morris A, Guinness FE, Clutton-Brock TH, Pemberton JM, Nussey DH. 2012. Shared spatial effects on quantitative genetic parameters: accounting for spatial autocorrelation and home range overlap reduces estimates of heritability in wild red deer. Evolution 66, 2411–2426 (doi:10.1111/j.1558-5646.2012.01620.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gómez-Díaz E, Jordà M, Peinado MA, Rivero A. 2012. Epigenetics of host–pathogen interactions: the road ahead and the road behind. PLoS Pathog. 8, e1003007 (doi:10.1371/journal.ppat.1003007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gasparini J, McCoy KD, Haussy C, Tveraa T, Boulinier T. 2001. Induced maternal response to the Lyme disease spirochaete Borrelia burgdorferi sensu lato in a colonial seabird, the kittiwake Rissa tridactyla. Proc. R. Soc. Lond. B 268, 647–650 (doi:10.1098/rspb.2000.1411) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roff DA. 1996. The evolution of genetic correlations: an analysis of patterns. Evolution 50, 1392–1403 (doi:10.2307/2410877) [DOI] [PubMed] [Google Scholar]
- 59.Thrall PH, Laine A-L, Ravensdale M, Nemri A, Dodds PN, Barrett LG, Burdon JJ. 2012. Rapid genetic change underpins antagonistic coevolution in a natural host–pathogen metapopulation: coevolution in a wild host–pathogen system. Ecol. Lett. 15, 425–435 (doi:10.1111/j.1461-0248.2012.01749.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data available from the Dryad Digital Repository: http://doi.org/10.5061/dryad.s3k34.