

Abstract
The tendency for flying organisms to possess small genomes has been interpreted as evidence of natural selection acting on the physical size of the genome. Nonetheless, the flight–genome link and its mechanistic basis have yet to be well established by comparative studies within a volant clade. Is there a particular functional aspect of flight such as brisk metabolism, lift production or maneuverability that impinges on the physical genome? We measured genome sizes, wing dimensions and heart, flight muscle and body masses from a phylogenetically diverse set of bird species. In phylogenetically controlled analyses, we found that genome size was negatively correlated with relative flight muscle size and heart index (i.e. ratio of heart to body mass), but positively correlated with body mass and wing loading. The proportional masses of the flight muscles and heart were the most important parameters explaining variation in genome size in multivariate models. Hence, the metabolic intensity of powered flight appears to have driven genome size reduction in birds.
Keywords: C-value, genome size, flight, heart index, flight muscles
1. Introduction
(a). Genome size evolution
The causes and consequences of variability in nuclear genome size have been a subject of research and discussion for many decades [1,2], but much remains to be learned. Several intriguing comparative patterns have been identified that provide insights into the evolutionary forces that have shaped genome size diversity. As a notable example, a proposed link between powered flight and reduced genome sizes in bats, birds and pterosaurs has been interpreted as evidence that the metabolic demands of flight exerted selective pressures for small cells with reduced DNA content [3–5]. These three historical instances of flight origins and genome constriction are suggestive, but comprise meagre statistical evidence for a mechanistic link. Furthermore, ancestral state estimates for the archosaur phylogeny revealed that much of the genome size reduction in the ancestors of modern birds predated the origin of flight [6].
If flight ability imposes strong evolutionary constraints on genome size, then interspecific diversity in flight ability should explain at least some of the variability in genome size within volant clades. Indeed, evidence to this effect has begun to accumulate, with some caveats. Hughes & Hughes [3] found that flightless birds have larger genomes than volant relatives, but this pattern became non-significant after accounting for phylogenetic inertia [4]. Hovering hummingbirds exhibit the highest mass-specific metabolic rates among vertebrates [7], and a large sample of hummingbird species was found to have uniformly small genomes and to include the smallest amniote genome measured to date [8]. In 74 species of temperate, migratory passerine birds, genome size was positively associated with wing-loading index, indicating that species that have evolved larger wings for flight efficiency have also evolved smaller genomes [9]. Additional components of flight performance beyond wing size and shape have yet to be investigated for potential effects on genome size.
The metabolic rate hypothesis is the leading explanation for reduced genomes in volant organisms [3]. According to this hypothesis, the size of the genome imposes a minimum size constraint on nucleated cells, and the higher proportional surface areas of smaller cells make them conducive to higher metabolic flux [10]. The energy required to produce lift and thrust requires sustained high metabolic output [11], resulting in indirect selection for smaller genomes to reduce the size of nucleated cells [10]. In keeping with this hypothesis, genome size and red blood cell size are negatively correlated with resting metabolic rate across vertebrates and within vertebrate classes, including birds [12]. Aside from the putative metabolic rate effect, a reduction in cellular DNA content and a concomitant decrease in cell size might allow flying organisms to achieve a reduction in body mass [12] and may even enhance efficiency of neural function associated with maneuverability [12,13]. Under the latter mechanisms, wing loading and wing shape would be predicted to correlate strongly with genome size.
(b). The avian flight ‘engines’
The pectoral and cardiac muscles are the metabolic ‘engines’ of avian flight, and therefore might illuminate the causes of genome size evolution. The avian pectoral flight muscles, comprising the pectoralis major and supracoracoideus, provide the power for downstroke and upstroke, respectively. The pectoral flight muscles vary greatly in size across birds, ranging from approximately 5% of the body mass in rails and ground cuckoos to over 30% in some hummingbirds and pigeons [14,15]. The ratio of heart mass to body mass, the heart index [16], indicates the relative allocation of resources for oxidative metabolism. Both maximum cardiac output and aerobic power input scale with heart mass to a power of approximately 0.88 [17,18], and heart mass accurately predicts maximum metabolic rate and aerobic scope [19]. Although the flight muscles and heart generate and supply power for a substantial portion of locomotory and thermogenic activity [20–23], species variation in relative flight muscle size and heart index indicates the degree of importance of burst power [24] and endurance flight performance [17], respectively. Previous studies using non-phylogenetic methods have found that genome size was negatively correlated with heart index in birds (n = 53), mammals, non-avian reptiles and amphibians [16].
(c). This study
The link between flight and reduced genome size has received substantial attention [3–5,12,25], but there is not yet a broad comparative analysis of variation in the flight and metabolic structures as they relate to genome size within a volant clade. In this study, we developed the largest internally consistent dataset on bird genome sizes, comprising 18 orders, 76 families and 422 species. Species were selected to represent a diversity of environments, ecological niches and life histories. We sought to elucidate the effects of flight on genome size evolution during the diversification of birds using measurements of three flight-related structures: the heart, the flight muscles and the wings.
2. Material and methods
(a). Data collection
We captured wild birds and drew blood from the brachial vein to prepare blood smear slides. Birds were euthanized, prepared as museum specimens with frozen tissues and deposited at the Museum of Southwestern Biology (University of New Mexico, USA) and/or Centro de Ornithología y Biodiversidad (Lima, Peru). Slides of blood blotted from the liver were prepared for individuals from which we were unable to draw blood. One investigator (N.A.W.) estimated genome size by Feulgen image analysis densitometry using the protocol of Hardie et al. [26], with chicken, Gallus gallus (1.25 pg), as the standard, and more than 200 nuclei measured per individual. For species represented by more than one individual, mean within-species standard deviation of genome size estimates was 0.035, whereas the among-species standard deviation in genome size was 0.138. The low intraspecific variation, relative to interspecific variation, suggests that estimates of genome size using only one individual should be sufficiently representative of the species. Genome size estimates are available in the electronic supplementary material, table S1.
We weighed each bird and extracted and weighed whole hearts (after blotting to remove blood) and the pectoralis major and supracoracoideus flight muscles. Relative flight muscle size was calculated by dividing total flight muscle mass by body mass. Heart index was calculated by dividing the heart mass by the body mass [16]. We used proxies for aspect ratio and wing loading that could be measured from museum study skins. The hand-wing index, or Kipp's index [27,28], is equal to the distance between the tip of the longest primary and the tip of the first secondary divided by the wing chord [27]; larger values indicate more pointed wings. This can be calculated as (WL − SL)/WL, where WL is the wing chord and SL is the distance from the wrist joint to the tip of the first secondary [27]. This index is correlated with dispersal ability in interspecific comparisons [27,29,30]. WL comprises the lengths of the manus and the longest primary feather, both of which are strongly correlated with overall wing length and scale isometrically with other wing components such as the ulna [31–33]. Bird wings are generally shaped as one-quarter of an oval. Thus, wing area was estimated as the area of an oval divided by four (WL × SL × π/4). Wing area estimates were used to approximate wing loading (body mass/total wing area).
Species averages for the above morphological measurements were obtained from specimens at the Museum of Southwestern Biology and Florida Museum of Natural History. We were unable to obtain all morphological measurements for all species. Thus, with genome size estimates for 422 species, we analysed two overlapping datasets of species-average values: (i) relative flight muscle size, heart index and body mass for 289 species; and (ii) relative flight muscle size, heart index, body mass, hand-wing index and wing loading for 193 species.
(b). Data analysis
We used a time-calibrated phylogeny based on published DNA sequences for over 6000 species of birds [34] that we trimmed to include only the species in our genome size dataset. We placed the approximately 5% of the species in our dataset that were not represented in this tree following the methods of Sibly et al. [35]. We used this tree to calculate phylogenetic independent contrasts, test for phylogenetic signal in the data and perform phylogenetic generalized least-squares (PGLS) models under a Brownian motion model in R v. 2.15.1 [36] using packages ape [37], caper [38] and nlme [39]. We log-transformed body mass prior to analyses. We used Akaike's Information Criterion (AIC) to select the best model(s) for non-phylogenetic linear regression and PGLS. Multi-parameter models were discarded from consideration if a nested model, containing a subset of the same parameters, had a better AIC score [40,41].
We tested for constraints on maximum genome size that might be associated with high flight ability (i.e. high values of relative heart mass, relative flight muscle size and hand-wing index or low values of body mass and wing loading). For each measure of flight ability, we binned values into 15 equally spaced bins, following Derryberry et al. [42]. We selected the species with the maximum genome size value in each bin to include in the upper-bound set. We then tested whether these upper-bound values of genome size decline with increasing flight ability more dramatically than do all other values. To do this, we used ANCOVA to test for the significance of an interaction term between the measure of flight ability and whether a sample was included in the upper-bound set.
3. Results
There was significant phylogenetic inertia in genome size across the entire 422-species dataset. Pagel's lambda was 0.88 (95% CI: 0.788, 0.943), indicating that phylogenetic non-independence should be accounted for in comparative analyses. Blomberg's K, which estimates the amount of phylogenetic signal in the data relative to the amount expected for a trait evolving along the same tree by Brownian motion [43,44], was 0.451 (significantly greater than zero, p = 0.001).
Body mass was negatively correlated with heart index (PGLS: d.f. = 286, p < 0.001, R2 = 0.043), but not with relative flight muscle size (PGLS: d.f. = 286, p = 0.13, R2 < 0.01). Heart mass scaled with body mass to a power of 0.78 in our dataset (95% CI: 0.75, 0.81).
In single variable phylogenetic models, genome size was negatively correlated with heart index and relative flight muscle size and positively correlated with body mass and wing loading (figures 1 and 2; the electronic supplementary material, table S2). In the larger dataset (289 species), the top ranking PGLS models (i.e. those within 95% cumulative AIC weight after excluding models with uninformative parameters) included heart index and relative flight muscle size as significant predictors of genome size (table 1). In the reduced dataset (193 species), the top ranking PGLS models included relative flight muscle size, wing loading, heart index and body mass as important predictors of genome size; wing loading was unique among these variables in that it was not statistically significant (p < 0.05) in any of the top models (tables 2 and 3).
Figure 1.
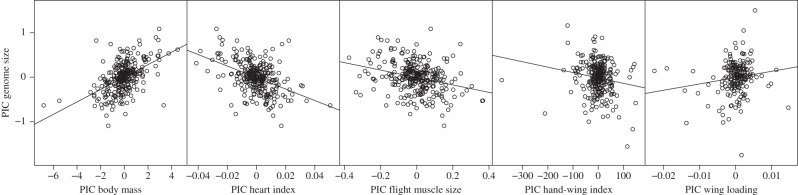
Phylogenetic independent contrasts (PICs) of species mean values for genome size plotted as a function of heart index, relative flight muscle size, body mass, hand-wing index and wing loading.
Figure 2.

Species mean values for genome size plotted as a function of heart index, relative flight muscle size, wing loading, hand-wing index and body mass, colour-coded by order. Solid lines are all-points regression lines; dashed lines are upper bound regression lines.
Table 1.
Models predicting genome size within 95% cumulative AIC weight after excluding models with uninformative parameters for the 289-species dataset (f, relative flight muscle size; h, heart index; m, body mass). PGLS, phylogenetic generalized linear model; regression, conventional (non-phylogenetically corrected) linear regression.
| model type | model | adj. R2 | d.f. | p-values | AIC | AIC weight |
|---|---|---|---|---|---|---|
| PGLS | f + h | 0.073 | 286 | f: 0.0038; h: 0.0043 | −557.31 | 0.91 |
| PGLS | h | 0.049 | 287 | h < 0.001 | −522.19 | 0.07 |
| regression | f + m | 0.38 | 286 | f < 0.001; m < 0.001 | −486.49 | 0.999 |
Table 2.
Models predicting genome size within 95% cumulative AIC weight after excluding models with uninformative parameters for the reduced dataset (N = 193 species; f, relative flight muscle size; h, heart index; wl, wing loading; hw, hand-wing index; m, mass). PGLS, phylogenetic generalized linear model; regression, conventional (non-phylogenetically corrected) linear regression.
| model type | model | adj. R2 | d.f. | p-values | AIC | AIC weight |
|---|---|---|---|---|---|---|
| PGLS | f + h + m + wl | 0.12 | 188 | f < 0.001; h: 0.49; m: 0.006; wl: 0.26 | −369.18 | 0.46 |
| PGLS | f + h + wl | 0.088 | 189 | f: 0.005; h: 0.057; wl: 0.40 | −368.03 | 0.26 |
| PGLS | f + m + wl | 0.12 | 189 | f < 0.001; m: 0.001; wl: 0.26 | −366.75 | 0.13 |
| PGLS | f + h + m | 0.119 | 189 | f < 0.001; h: 0.49; m: 0.007 | −364.52 | 0.04 |
| PGLS | f + h | 0.09 | 190 | f: 0.006; h: 0.019 | −364.41 | 0.04 |
| PGLS | h + wl | 0.054 | 190 | h: 0.001; wl: 0.66 | −363.06 | 0.02 |
| regression | f + m + hw + wl | 0.41 | 188 | f < 0.001; m < 0.001; hw: 0.041; wl: 0.063 | −346.44 | 0.52 |
| regression | f + m + hw | 0.40 | 189 | f < 0.001; m < 0.001; hw: 0.033 | −344.88 | 0.24 |
| regression | f + m + wl | 0.40 | 189 | f < 0.001; m < 0.001; wl: 0.051 | −344.14 | 0.17 |
| regression | f + m | 0.39 | 190 | f: 0.002; m < 0.001 | −342.24 | 0.06 |
Table 3.
Cumulative model weights for each predictor variable in the five-parameter (N = 193 species) and three-parameter (N = 289 species) datasets, calculated by summing Akaike model weights for all models that included the variable of interest, following Arnold [40]. PGLS, phylogenetic generalized linear model; regression, conventional (non-phylogenetically corrected) linear regression.
| five-parameter (N = 193) |
three-parameter (N = 289) |
|||
|---|---|---|---|---|
| PGLS | regression | PGLS | regression | |
| relative flight muscle size | 0.966 | 0.996 | 0.93 | 0.999 |
| wing loading | 0.894 | 0.691 | n.a. | n.a. |
| heart index | 0.834 | 0.302 | 0.99 | 0.27 |
| body mass | 0.657 | 0.999 | 0.06 | 0.999 |
| hand-wing index | 0.002 | 0.742 | n.a. | n.a. |
We found evidence of constraints on genome size with respect to some measures of flight ability. The slopes of regressions of genome size by heart index and hand-wing index, respectively, were more steeply negative for upper-bound points than for other points (table 4 and figure 2), indicating that large genome sizes were particularly unlikely to occur in species with large hearts or pointed wings.
Table 4.
Comparison of upper bound regressions to standard linear regressions for genome size by each flight ability predictor variable. We used an ANCOVA with an interaction term between the predictor variable and whether a sample was included in the upper-bound set.
| predictor variable | 95% CI for upper bound slope | 95% CI for all points slope | two-tailed p-value for upper bound interaction |
|---|---|---|---|
| heart index | (−32.3, −19.1) | (−18.1, −12.1) | p = 0.018 |
| relative flight muscle size | (−1.78, −0.07) | (−1.33, −0.70) | p = 0.78 |
| body mass | (0.04, 0.21) | (0.15, 0.20) | p = 0.24 |
| hand-wing index | (−0.0095, −0.0049) | (−0.0028, −0.00090) | p = 0.003 |
| wing loading | (−10.26, 19.96) | (20.42, 33.71) | p = 0.86 |
Visualization of the evolution of genome size across the avian tree revealed lability among families and orders (figure 3). Orders differed significantly from one another in average genome size (ANOVA: p < 0.001, d.f. = 404; figure 3; the electronic supplementary material, table S3), with Apodiformes exhibiting the smallest average genome sizes. Of the orders represented by at least five species, Piciformes had the largest and most variable genomes (figures 2 and 3; the electronic supplementary material, table S3). Passerine genomes were significantly smaller than those of non-passerine orders (t-test: p = 0.006; d.f. = 180; 95% CI of mean difference: (−0.08, −0.013)). Deep phylogenetic structure in genome size was evident among major clades of passerines. Nine-primaried oscines had larger genomes, on average, than other passerines (t-test: p < 0.001; d.f. = 128; 95% CI of mean difference: (0.019, 0.071)). However, extreme values were not necessarily consistent with these broader patterns. For example, out of all passerine families, the smallest average genome sizes were found in two oscine families, the vangas (Dicruridae; C-value 1.13 pg) and the indigobirds and whydahs (Viduidae; C-value 1.14 pg); and the largest passerine genome was not found in a nine-primaried oscine, but rather the phainopepla (Phainopepla nitens; C-value 1.83 pg).
Figure 3.

Family-level phylogeny with terminal branches colour-coded by the average genome size for each family. Internal branches are colour-coded by the estimated ancestral genome size, using maximum-likelihood ancestral character state estimation in the R package ape [37]. The tree is from Jetz et al. [45] and was downloaded from www.birdtree.org with data preference set to ‘Hackett all species’.
4. Discussion
(a). Which aspects of flight are related to genome size?
Flight muscle size, heart size, body size, wing aspect ratio and wing loading reveal different aspects of flight ability in birds. Each of these parameters, with the exception of wing aspect ratio, is individually correlated with genome size across a wide swath of the avian tree (figures 1 and 2; the electronic supplementary material, table S2), strongly supporting the idea that small genomes and overall flight ability are linked. In multivariate models, flight muscle size, heart size, body size and wing loading explain unique fractions of genome size variation, with flight muscle size and heart size being the most consistently important predictors of genome size. There was no indication that wing aspect ratio (measured by hand-wing index) explains any additional variation. These findings are consistent with the hypothesis that the cellular metabolic rate during flight, rather than constraints on aerial maneuverability or lift, was the main cause of genome size reduction in birds.
Relative flight muscle size was the single best predictor of genome size: it was significant in almost all top models in both the 289- and 193-species datasets (tables 1–3). Flight muscles provide the power for flapping flight, and as such are most important in strong take-offs and bursts of speed in flight, where power requirements are highest [24]. Thus, large flight muscles are expected to indicate an evolutionary response to selection for rapid take-off ability or powered acceleration during flapping flight. Additionally, the size of the flight muscles is a primary determinant of exercise-induced maximum metabolic rate or thermogenic capacity in intraspecific comparisons for at least five species of birds that have been tested, representing three orders and a wide range of body sizes [20–23,46].
Heart index was an important and statistically significant predictor of genome size in top PGLS models (tables 1–3). The size of the heart constrains its stroke volume and, as a result, limits maximum cardiac output, aerobic power, exercise-induced maximum metabolic rate and aerobic scope [17–19]. Routine powered flight incurs a 10- to 20-fold sustained increase in metabolic rate [11]. Therefore, heart index in birds should closely reflect aerobic power requirements for sustained flight.
Body mass generally shows strong phylogenetic signal [43] and correspondingly was present and significant in all top ranking non-phylogenetic models, but not all PGLS ones (tables 1–3). Either body mass or heart index was significant (or nearly so) in every top ranking model, but in no model were both parameters significant (tables 1 and 2). Heart mass scales with body mass to a power of 0.78 in our dataset, very close to the three-quarter power scaling of metabolic rate [47]. Heart size may be a more precise predictor of metabolic rate than body mass because it is more directly, mechanistically connected to metabolism than is body mass, in part because it scales isometrically with cardiac stroke volume [18,48]. Indeed, heart mass has been shown to explain variation in exercise-induced maximum metabolic rate after accounting for the effects of body mass [19]. Consistent with this, for our largest dataset, AIC decisively indicated that heart index was the best single predictor of genome size and that body mass did not explain residual variation after heart index was included in the model (table 1).
Hand-wing index did not appear in top ranking PGLS models (tables 2 and 3). The link between genome size and hand-wing index in conventional regression (figure 2 and table 3; the electronic supplementary material, table S2) appears to have been driven by phylogenetic inertia, specifically the confluence of extreme values in the hummingbirds and swifts (Apodiformes). Accordingly, the effect of hand-wing index disappears when covariance due to phylogeny is taken into account. Long, pointed wings improve high-speed flight performance by reducing drag, especially at higher speeds [49]. High aspect ratio is typical of bird species that are long-distance migrants or aerial foragers [50,51]. However, some birds with high aspect ratios engage in flight styles with low energetic demand (e.g. dynamic soarers) and accordingly would not achieve the metabolic intensity associated with flapping flight.
Wing loading was included as an important predictor variable in several top PGLS models, but was never a significant variable in these models (p > 0.25; tables 2 and 3). Birds with low wing loading are able to produce lift with low metabolic energy input [52]. The tendency for aerial specialists to have low wing loading is thought to be the basis for the previous finding that passerines with low wing loading tend to have small genomes [13]. However, wing loading reflects a balance of ecological and biomechanical pressures and many strong, energetic flyers have high wing loading (e.g. pigeons, doves and many ducks [52]). Thus, wing loading, like aspect ratio, is at least partly decoupled from flight metabolism and is unlikely to explain genome size variation if the maximum sustained rate of cellular metabolism is the key driver of genome size reduction.
(b). The importance of accounting for phylogenetic inertia
Genome size exhibited phylogenetic signal across the avian tree, as indicated by Pagel's lambda and Blomberg's K-values that were significantly greater than zero. Related taxa were less similar in genome size than expected under a model of evolution by Brownian motion, as indicated by the Blomberg's K-value being less than one [43,44]. This apparent lability is more typical of behavioural traits than constrained life-history traits such as body mass [43] and it suggests that divergent selection has acted on genome size, although measurement error is also expected to contribute to this effect. Nonetheless, as a result of the pervasive phylogenetic inertia in genome size, the PGLS analyses are expected to be more informative regarding potential mechanisms in cases where they differ from non-phylogenetically controlled analyses [53]. Hence, in the 289-species analysis, the inclusion of body mass and exclusion of heart index in the top ranking non-phylogenetic model are likely examples of type I and type II errors, respectively (table 1).
(c). Constraints on genome size associated with extreme flight ability
Genome size appears to be constrained with respect to heart index and hand-wing index, as indicated by the different slope of the regression for upper bound points (figure 2 and table 4). Species with small hearts or rounded wings may have either large or small genomes, but strong flyers with large hearts or pointed wings appear to be constrained to have small genomes. This pattern suggests that genome size evolves neutrally in poor flyers, but that large genomes impose a fitness cost on birds that experience high metabolic intensity during flight.
(d). Genome size variation across the tree of birds
The hummingbirds have exceptionally small genomes with low variation in genome size [8]. This study is the first to show that the nearest relatives of hummingbirds, the swifts (Apodidae), also have small genomes (figure 3 and the electronic supplementary material, table S3). Like many other traits of hummingbirds that are thought to be adaptive for metabolically intensive flight (e.g. high aspect ratio, reduced hindlimbs, large pectoral flight muscles), reduced genome size probably evolved in the common ancestor of hummingbirds and swifts (figure 3).
The Pici clade, comprising the woodpeckers, honeyguides, barbets and toucans, exhibits exceptionally large genomes, whereas members of its sister taxon, the Galbulae (Galbulidae and Bucconidae), have fairly small genomes (figure 3 and the electronic supplementary material, table S3). Members of the clade including Piciformes and Coraciiformes are anomalously variable, with sister lineages sometimes differing substantially in genome size (figure 3 and the electronic supplementary material, tables S1 and S2). For example, the two honeyguide species included in this study, both in the genus Indicator, exhibit strikingly different genome sizes (1.38 versus 2.05 pg; the electronic supplementary material, table S1). Han et al. [54] found large numbers of transposable elements in the Piciformes–Coraciiformes clade, which may contribute to the large average genome sizes and the high degree of variation observed in the group [4,55]. The macromutations that may have caused fluctuations in genome size among Piciformes deserve investigation by comparison of genome sequences. To this end, frozen tissues are archived for all of our voucher specimens (see electronic supplementary material, table S1).
5. Conclusion
The long-recognized link between flight and small genome size has been decisively confirmed using interspecific comparisons within the most diverse volant group. Four flight-related characteristics show that the evolution of enhanced flight ability tends to be accompanied by genome size reduction. In comparisons among phylogenetically corrected models, the best predictors of genome size proved to be the sizes of the flight muscles and heart, rather than the size or shape of the wings. The avian flight muscles generate cellular metabolic energy in proportion to their enormous bulk (5–30% of total body mass), providing the majority of power output for aerial locomotion and thermogenesis. Cardiac output constrains the power input for cellular metabolism and is in turn limited by heart size. Consistent with the metabolic rate hypothesis, these two components of the metabolic flight ‘engine’ implicate the rate of energy use as a key driver of repeated evolutionary reductions in avian genome size.
Acknowledgements
We thank N. Jeffery for assistance in the laboratory, C. E. Gunning for help with R code and J. Hammond for statistics advice. This study was made possible by data, specimens and curatorial staff from the Museum of Southwestern Biology, the Field Museum of Natural History, and the Florida Museum of Natural History. We thank the individuals who collected key samples: J. M. Bates, E. Bautista, E. J. Beckman, P. M. Benham, S. V. Brant, D. DeSwardt, R. W. Dickerman, S. G. DuBay, S. C. Galen, E. Gendron, S. J. Hackett, A. B. Johnson, M. R. Jones, B. D. Marks, R. Nuttall, C. J. Schmitt, D. C. Schmitt, C. G. Schmitt, A. Smiley, E. W. Valdez, T. Valquí, W. Vargas, J. D. Weckstein and D. E. Willard. B. O. Wolf, J. H. Brown and two anonymous reviewers provided valuable feedback on the manuscript. This work was conducted under UNM IACUC protocol number 11-100742-mcc. We thank the management authorities of Peru (135-2009-AG-DGFFS-DGEFFS, 0377-2010-AG-DGFFS-DGEFFS, 0199-2012-AG-DGFFS-DGEFFS) and Free State, South Africa (01/8233, 01/12248).
Data accessibility
Genome size estimates are available in Table S1 and on the Animal Genome Size Database (http://www.genomesize.com/). Table S1 includes museum accession numbers for each specimen used in this study. Specimen data are archived in museum databases (http://arctos.database.museum/SpecimenSearch.cfm).
Funding statement
We acknowledge NSF DEB-1146491, the Center for Evolutionary and Theoretical Immunology and R. W. Dickerman for funding support. N.A.W. is supported by the Program in Interdisciplinary Biomedical and Biological Sciences through the University of New Mexico award number T32EB009414 from the National Institute of Biomedical Imaging and Bioengineering. T.R.G. is supported by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada. The content is the sole responsibility of the authors and does not necessarily represent the official views of the National Institute of Biomedical Imaging and Bioengineering or the National Institutes of Health.
References
- 1.Mirsky AE, Ris H. 1951. The deoxyribonucleic acid content of animal cells and its evolutionary significance. J. Gen. Physiol. 34, 451–462 (doi:10.1085/jgp.34.4.451) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas CA. 1971. The genetic organization of chromosomes. Annu. Rev. Genet. 5, 237–256 (doi:10.1146/annurev.ge.05.120171.001321) [DOI] [PubMed] [Google Scholar]
- 3.Hughes AL, Hughes MK. 1995. Small genomes for better flyers. Nature 377, 391 (doi:10.1038/377391a0) [DOI] [PubMed] [Google Scholar]
- 4.Organ CL, Shedlock AM. 2009. Palaeogenomics of pterosaurs and the evolution of small genome size in flying vertebrates. Biol. Lett. 5, 47–50 (doi:10.1098/rsbl.2008.0491) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Q, Edwards SV. 2012. The evolution of intron size in amniotes: a role for powered flight? Genome Biol. Evol. 4, 1033–1043 (doi:10.1093/gbe/evs070) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Organ CL, Shedlock AM, Meade A, Pagel M, Edwards SV. 2007. Origin of avian genome size and structure in non-avian dinosaurs. Nature 446, 180–184 (doi:10.1038/nature05621) [DOI] [PubMed] [Google Scholar]
- 7.Suarez RK. 1992. Hummingbird flight: sustaining the highest mass-specific metabolic rates among vertebrates. Experientia 48, 565–570 (doi:10.1007/BF01920240) [DOI] [PubMed] [Google Scholar]
- 8.Gregory TR, Andrews CB, McGuire JA, Witt CC. 2009. The smallest avian genomes are found in hummingbirds. Proc. R. Soc. B 276, 3753–3757 (doi:10.1098/rspb.2009.1004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andrews CB, Mackenzie SA, Gregory TR. 2009. Genome size and wing parameters in passerine birds. Proc. R. Soc. B 276, 55–61 (doi:10.1098/rspb.2008.1012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gregory TR. 2001. The bigger the C-value, the larger the cell: genome size and red blood cell size in vertebrates. Blood Cells Mol. Dis. 27, 830–843 (doi:10.1006/bcmd.2001.0457) [DOI] [PubMed] [Google Scholar]
- 11.Ward S, Bishop CM, Woakes AJ, Butler PJ. 2002. Heart rate and the rate of oxygen consumption of flying and walking barnacle geese (Branta leucopsis) and bar-headed geese (Anser indicus). J. Exp. Biol. 205, 3347–3356 [DOI] [PubMed] [Google Scholar]
- 12.Gregory TR. 2002. A bird's-eye view of the C-value enigma: genome size, cell size, and metabolic rate in the class Aves. Evolution 56, 121–130 [DOI] [PubMed] [Google Scholar]
- 13.Andrews CB, Gregory TR. 2009. Genome size is inversely correlated with relative brain size in parrots and cockatoos. Genome 52, 261–267 (doi:10.1139/G09-003) [DOI] [PubMed] [Google Scholar]
- 14.Hartman FA. 1961. Locomotor mechanisms of birds. Smithsonian Misc. Coll. 143, 1–9 [Google Scholar]
- 15.2013. Museum of Southwestern Biology specimen data See http://arctos.database.museum/SpecimenSearch.cfm.
- 16.Vinogradov AE, Anatskaya OV. 2006. Genome size and metabolic intensity in tetrapods: a tale of two lines. Proc. R. Soc. B 273, 27–32 (doi:10.1098/rspb.2005.3266) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bishop CM. 1997. Heart mass and the maximum cardiac output of birds and mammals: implications for estimating the maximum aerobic power input of flying animals. Phil. Trans. R. Soc. Lond. B 352, 447–456 (doi:10.1098/rstb.1997.0032) [Google Scholar]
- 18.Bishop CM, Butler PJ. 1995. Physiological modeling of oxygen consumption in birds during flight. J. Exp. Biol. 198, 2153–2163 [DOI] [PubMed] [Google Scholar]
- 19.Bishop CM. 1999. The maximum oxygen consumption and aerobic scope of birds and mammals: getting to the heart of the matter. Proc. R. Soc. Lond. B 266, 2275–2281 (doi:10.1098/rspb.1999.0919) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chappell MA, Bech C, Buttemer WA. 1999. The relationship of central and peripheral organ masses to aerobic performance variation in house sparrows. J. Exp. Biol. 202, 2269–2279 [DOI] [PubMed] [Google Scholar]
- 21.Hohtola E. 1982. Thermal and electromyographic correlates of shivering thermogenesis in the pigeon. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 73, 159–166 (doi:10.1016/0300-9629(82)90049-4) [DOI] [PubMed] [Google Scholar]
- 22.Hohtola E, Stevens ED. 1986. The relationship of muscle electrical activity, tremor and heat production to shivering thermogenesis in the Japanese quail. J. Exp. Biol. 125, 119–135 [DOI] [PubMed] [Google Scholar]
- 23.Swanson DL, Zhang Y, King MO. 2013. Individual variation in thermogenic capacity is correlated with flight muscle size but not cellular metabolic capacity in American goldfinches (Spinus tristis). Physiol. Biochem. Zool. 86, 421–431 (doi:10.1086/671447) [DOI] [PubMed] [Google Scholar]
- 24.Tobalske BW, Hedrick TL, Dial KP, Biewener AA. 2003. Comparative power curves in bird flight. Nature 421, 363–366 (doi:10.1038/nature01284) [DOI] [PubMed] [Google Scholar]
- 25.Altshuler DL, Stiles FG, Dudley R. 2004. Of hummingbirds and helicopters: hovering costs, competitive ability, and foraging strategies. Am. Nat. 163, 16–25 (doi:10.1086/380511) [DOI] [PubMed] [Google Scholar]
- 26.Hardie DC, Gregory TR, Hebert PDN. 2002. From pixels to picograms: a beginners’ guide to genome quantification by Feulgen image analysis densitometry. J. Histochem. Cytochem. 50, 735–749 (doi:10.1177/002215540205000601) [DOI] [PubMed] [Google Scholar]
- 27.Claramunt S, Derryberry EP, Remsen JV, Jr, Brumfield RT. 2012. High dispersal ability inhibits speciation in a continental radiation of passerine birds. Proc. R. Soc. B 279, 1567–1574 (doi:10.1098/rspb.2011.1922) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kipp FA. 1959. Der Handflügel-Index als flugbiologisches MaB. Vogelwarte 20, 77–86 [Google Scholar]
- 29.Burney CW, Brumfield RT. 2009. Ecology predicts levels of genetic differentiation in neotropical birds. Am. Nat. 174, 358–368 (doi:10.1086/603613) [DOI] [PubMed] [Google Scholar]
- 30.Dawideit BA, Phillimore AB, Laube I, Leisler B, Böhning-Gaese K. 2009. Ecomorphological predictors of natal dispersal distances in birds. J. Anim. Ecol. 78, 388–395 (doi:10.1111/j.1365-2656.2008.01504.x) [DOI] [PubMed] [Google Scholar]
- 31.Nudds RL, Kaiser GW, Dyke GJ. 2011. Scaling of avian primary feather length. PLoS ONE 6, e15665 (doi:10.1371/journal.pone.0015665.g001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X, McGowan AJ, Dyke GJ. 2011. Avian wing proportions and flight styles: first step towards predicting the flight modes of Mesozoic birds. PLoS ONE 6, e28672 (doi:10.1371/journal.pone.0028672.g001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nudds RL. 2007. Wing–bone length allometry in birds. J. Avian Biol. 38, 515–519 (doi:10.1111/j.0908-8857.2007.03913.x) [Google Scholar]
- 34.Burleigh G, Braun EL, Kimball RT. In preparation. A new supermatrix tree for over 6,000 species of birds. [Google Scholar]
- 35.Sibly RM, Witt CC, Wright NA, Venditti C, Jetz W, Brown JH. 2012. Energetics, lifestyle, and reproduction in birds. Proc. Natl Acad. Sci. USA 109, 10 937–10 941 (doi:10.1073/pnas.1206512109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.R Core Team 2012. R: a language and environment for statistical computing See http://www.R-project.org/
- 37.Paradis E, Claude J, Strimmer K. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290 (doi:10.1093/bioinformatics/btg412) [DOI] [PubMed] [Google Scholar]
- 38.Orme D, Freckleton RP, Thomas GH, Petzoldt T, Fritz SA, Isaac NJ, Pearse W. 2012. caper: comparative analyses of phylogenetics and evolution in R. R package version 0.5 See http://CRANR-project.org/package=caper
- 39.Pinheiro J, Bates D, DebRoy S, Sarkar D, R Development Core Team 2012. nlme: linear and nonlineaer mixed effects models R package version 3.1-104
- 40.Arnold TW. 2010. Uninformative parameters and model selection using Akaike's Information Criterion. J. Wildl. Manage. 74, 1175–1178 (doi:10.2193/2009-367) [Google Scholar]
- 41.Burnham KP, Anderson DR. 2002. Model selection and multimodal inference: a practical information-theoretic approach, 2nd edn New York, NY: Springer [Google Scholar]
- 42.Derryberry EP, Seddon N, Claramunt S, Tobias JA, Baker A, Aleixo A, Brumfield RT. 2012. Correlated evolution of beak morphology and song in the neotropical woodcreeper radiation. Evolution 66, 2784–2797 (doi:10.1111/j.1558-5646.2012.01642.x) [DOI] [PubMed] [Google Scholar]
- 43.Blomberg SP, Garland T, Jr, Ives AR. 2003. Testing for phylogenetic signal in comparative data: behavioral traits are more labile. Evolution 57, 717–745 [DOI] [PubMed] [Google Scholar]
- 44.Garland T, Jr, Bennett AF, Rezende EL. 2005. Phylogenetic approaches in comparative physiology. J. Exp. Biol. 208, 3015–3035 (doi:10.1242/jeb.01745) [DOI] [PubMed] [Google Scholar]
- 45.Jetz W, Thomas GH, Joy JB, Hartmann K, Mooers AO. 2012. The global diversity of birds in space and time. Nature 491, 444–448 (doi:10.1038/nature11631) [DOI] [PubMed] [Google Scholar]
- 46.Hammond KA, Chappell MA, Cardullo RA, Lin R-S, Johnsen T. 2000. The mechanistic basis of aerobic performance variation in red junglefowl. J. Exp. Biol. 203, 2053–2064 [DOI] [PubMed] [Google Scholar]
- 47.West GB, Brown JH, Enquist BJ. 1997. A general model for the origin of allometric scaling laws in biology. Science 276, 122–126 (doi:10.1126/science.276.5309.122) [DOI] [PubMed] [Google Scholar]
- 48.Grubb BR. 1983. Allometric relations of cardiovascular function in birds. Am. J. Phys. 245, H567–H572 [DOI] [PubMed] [Google Scholar]
- 49.Videler JJ. 2005. Avian flight. Oxford, UK: Oxford University Press [Google Scholar]
- 50.Lockwood R, Swaddle JP, Rayner JMV. 1998. Avian wingtip shape reconsidered: wingtip shape indices and morphological adaptations to migration. J. Avian Biol. 29, 273–292 (doi:10.2307/3677110) [Google Scholar]
- 51.Saino N, Rubolini D, von Hardenberg J, Ambrosini R, Provenzale A, Romano M, Spina F. 2010. Spring migration decisions in relation to weather are predicted by wing morphology among trans-Mediterranean migratory birds. Funct. Ecol. 24, 658–669 (doi:10.1111/j.1365-2435.2009.01659.x) [Google Scholar]
- 52.Rayner JMV. 1988. Form and function in avian flight. Curr. Ornithol. 5, 1–66 [Google Scholar]
- 53.Rohlf FJ. 2006. A comment on phylogenetic correction. Evolution 60, 1509–1515 (doi:10.1554/05-550.1) [DOI] [PubMed] [Google Scholar]
- 54.Han KL, et al. 2011. Are transposable element insertions homoplasy free?: an examination using the avian tree of life. Syst. Biol. 60, 375–386 (doi:10.1093/sysbio/syq100) [DOI] [PubMed] [Google Scholar]
- 55.Kidwell MG. 2002. Transposable elements and the evolution of genome size in eukaryotes. Genetica 115, 49–63 (doi:10.1023/A:1016072014259) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Genome size estimates are available in Table S1 and on the Animal Genome Size Database (http://www.genomesize.com/). Table S1 includes museum accession numbers for each specimen used in this study. Specimen data are archived in museum databases (http://arctos.database.museum/SpecimenSearch.cfm).