

Abstract
Aims
Volume overload and venous congestion are typically viewed as a consequence of advanced and of acute heart failure (HF) and renal failure (RF) although it is possible that hypervolaemia itself might be a critical intermediate in the pathophysiology of these diseases. This study aimed at elucidating whether peripheral venous congestion is sufficient to promote changes in inflammatory, neurohormonal, and endothelial phenotype similar to those observed in HF and RF.
Methods
To experimentally model peripheral venous congestion, we developed a new method (so-called venous stress test) and applied the methodology on 24 healthy subjects (14 men, age 35 ± 2 years). Venous arm pressure was increased to ∼30 mmHg above the baseline level by inflating a tourniquet cuff around the dominant arm (test arm). Blood and endothelial cells (ECs) were sampled from test and control arm (lacking an inflated cuff) before and after 75 min of venous congestion, using angiocatheters and endovascular wires. Magnetic beads coated with EC-specific antibodies were used for EC separation; amplified mRNA was analysed by Affymetrix HG-U133 Plus 2.0 Microarray.
Results
Plasma interleukin-6 (IL-6), endothelin-1 (ET-1), angiotensin II (AII), vascular cell adhesion molecule-1 (VCAM-1), and chemokine (C-X-C motif) ligand 2 (CXCL2) were significantly increased in the congested arm. A total of 3437 mRNA probe sets were differentially expressed (P < 0.05) in venous ECs before vs. after testing, including ET-1, VCAM-1, and CXCL2.
Conclusion
Peripheral venous congestion causes release of inflammatory mediators, neurohormones, and activation of ECs. Overall, venous congestion mimicked, notable aspects of the phenotype typical of advanced and of acute HF and RF.
Keywords: Congestive heart failure, Endothelium, Endothelin, Inflammation
See page 413 for the editorial comment on this article (doi:10.1093/eurheartj/eht562)
Introduction
Heart failure (HF) and renal failure (RF) promote fluid retention and venous congestion thereby shifting human physiology from a healthy biosystem that operates at low pressures to a pathophysiological milieu where organs are forced to function with significantly elevated venous and interstitial pressures several times above normal.
Besides venous congestion, inflammation, neurohormonal activation, and endothelial cell (EC) activation are also notable aspects of the phenotype typical of advanced and of acute HF and RF. Plasma levels of cytokines, such as interleukin-6 (IL-6) and tumour necrosis factor-α (TNF-α),1–7 vasoactive peptides, such as endothelin-1 (ET-1),8,9 neurohormones, such as angiotensin II (AII),10,11 endothelial adhesion molecules, such as vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1),12–16 and endothelial coagulation factors, such as vonWillebrand factor (vWF),17–19 are elevated in advanced and in acute HF and RF. While prognostic and pathophysiologic relevance of these circulating biomarkers is now widely accepted, the site and source of their production remains the object of intense investigation.
In vitro evidence indicates that the endothelium may become activated and turn into a primary source of pro-inflammatory, vasoconstrictive, and pro-thrombotic mediators in response to biomechanical stress. Interleukin-6,20 TNF-α,21 ET-1,22 AII,23 VCAM-1,24 ICAM-1,25 and vWF26 can be secreted within hours of exposure of ECs to stretch. Whether mechanical stretch is sufficient to activate the vascular endothelium in humans, for example, in a setting of venous congestion, remains unclear.
We developed a new experimental model of acute, peripheral venous congestion (so-called venous stress test) to test the hypothesis that venous congestion is sufficient (i) to cause local release of cytokines, vasoactive peptides, neurohormones, endothelial adhesion molecules, and coagulation factors and (ii) to cause EC activation in otherwise healthy human subjects. For the second purpose, we coupled venous EC sampling with transcriptomic approaches using microarray technology.
Methods
Study population and protocol
We enrolled 24 subjects who were normotensive, non-smokers with no history of chronic illness, or chronic medication use.
Venous stress test
Blood and ECs27 were sampled from the antecubital or basilic vein of the non-dominant arm (control arm) at baseline (time 0) and from the dominant arm (test arm) after 75 min of peripheral venous congestion, using angiocatheters and endovascular wires. Local venous pressure was increased to ∼30 mmHg above the baseline level by inflating a tourniquet cuff around the arm proximally, just below the shoulder (Figure 1). As a control, blood was collected from the non-dominant (control) arm 75 min after baseline samplings and measurements. Vital signs (control arm) and arterial oxymetry (test arm) were also recorded at 75 min.
Figure 1.
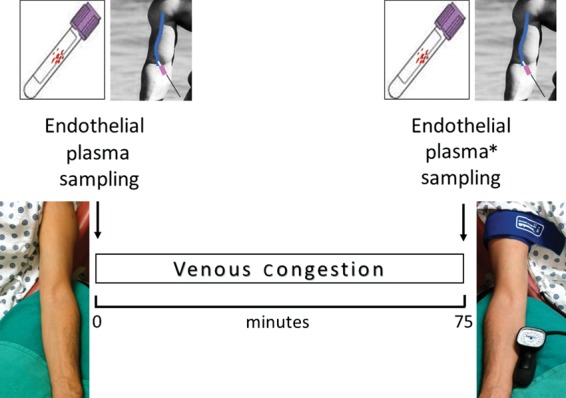
Venous stress test. Blood and endothelial cells were sampled from the antecubital or basilic vein of the non-dominant arm (control arm) at baseline and of the dominant arm (test arm) after 75 min of local venous congestion using angiocatheters and endovascular wires. Peripheral venous pressure was increased ∼30 mmHg above baseline levels by inflating a tourniquet cuff around the test arm, proximally, just below the shoulder. Blood was also obtained at 75 min from the control arm which was not exposed to venous congestion, thus serving as a control (*).
Study subjects were offered the option of undergoing EC and blood collection, or blood collection only.
Magnetic beads coated with EC-specific antibodies were used for EC separation. Amplified mRNA was analysed by Affymetrix HG-U133 2.0 Microarray.
A total of 24 normal subjects were enrolled and provided plasma samples. Of the 24, 16 also contributed EC samples 12 of which produced an mRNA product which was adequate for analysis in terms of quality and quantity. Therefore, our plasma protein results are based on a sample of 24 subjects while mRNA results are based on a subsample of 12 subjects.
Commercially available techniques were used to measure plasma IL-6, TNF-α, ET-1, AII, VCAM-1, ICAM-1, and vWF antigen (vWF:Ag).
An expanded Methods section is available in Supplementary material online.
Statistical analysis
All comparisons are based on within person biomarker differences (i.e. either plasma protein level or EC mRNA level) before vs. after induction of experimental venous congestion. mRNA fold-changes are defined by the ratio of post-experiment to pre-experiment absolute transcript levels.
Data are presented as means ± SEM in the text and tables. The Wilcoxon rank-sum test (continuous variables) or χ2 tests (categorical variables) were used to compare baseline clinical and laboratory characteristics between the 12 subjects with plasma data only, and the 12 subjects with both plasma and endothelial mRNA data. The Wilcoxon signed-rank test was used to test for significant differences in vital signs and plasma measurements before vs. after experimental venous congestion. For all analyses, a two-tailed P-value of 0.05 was used to infer statistical significance.
Endothelial cell mRNA (i.e. ‘gene expression’) data were first normalized and summarized using the log scale robust multi-array analysis28 with default settings. Differential expression in mRNA after vs. before experimental conditions was assessed using paired t-tests. Additional analyses were performed using random effects mixed models in which patients were conditioned as a random effect, gene expression was the dependent variable, and experimental state (pre- vs. post-experiment) was the independent variable. These models accounted for the within person correlation in gene expression and additionally allowed us to include measures of both leucocyte common antigen-1 and α-2-actin expression levels as covariates to minimize the possibility that gene expression from leucocyte or smooth muscle cell (SMC) populations influenced our findings (i.e. confounding by leucocyte or SMC contamination). Similar methods have been recently utilized to isolate effects from specific cell populations in studies of neurodegenerative disease.29 Statistical significance for gene expression analysis was based on α = 0.05 as well as false discovery rate (FDR)30 to minimize type 1 error. Gene set enrichment analysis was performed using DAVID to generate a Functional Annotation Chart identifying gene-term enrichment using default settings.31 Experimental details and results following the Minimum Information About a Microarray Experiment Standards are available at the Gene Expression Omnibus (accession number GSE38783).
Results
Subject characteristics
Table 1 summarizes the clinical characteristics of the 24 subjects enrolled: 12 subjects with plasma data available and 12 subjects with both plasma and endothelial mRNA data. Overall, study subjects had a normal body mass index, lipid profile, serum glucose, and renal function. Serum creatinine levels were lower in the plasma-only group owing to a higher percentage of women.
Table 1.
Clinical characteristics of the study population by sampling methodology (mean ± SEM)
| Variable | Plasma-only (n = 12) | Plasma and mRNA (n = 12) | P-value |
|---|---|---|---|
| Age (years) | 34 ± 3 | 36 ± 2 | 0.30 |
| Gender (no. male/female) | 4/8 | 10/2 | 0.02 |
| Body mass index (kg/m2) | 25 ± 1 | 24 ± 1 | 0.11 |
| Total cholesterol (mg/dL) | 168 ± 9 | 185 ± 9 | 0.31 |
| Low-density cholesterol (mg/dL) | 100 ± 8 | 111 ± 7 | 0.40 |
| High-density cholesterol (mg/dL) | 53 ± 4 | 60 ± 6 | 0.40 |
| Triglycerides (mg/dL) | 73 ± 7 | 72 ± 5 | 0.93 |
| Blood glucose (mg/dL) | 87 ± 2 | 90 ± 2 | 0.17 |
| Serum creatinine (mg/dL) | 0.71 ± 0.05 | 0.90 ± 0.04 | <0.01 |
Venous stress test
Cuff inflation raised peripheral venous pressure from 5 ± 0 mmHg at baseline to 36 ± 1 mmHg in the congested arm. Endothelial sampling was associated with minor discomfort; pain intensity was scored 0–3 in all cases using a 0–10 numeric scale. One subject developed a painful superficial phlebitis that was otherwise benign and resolved.
Systolic BP (119 ± 2 vs. 120 ± 2 mmHg, P = NS) and diastolic BP (78 ± 1 vs. 77 ± 1 mmHg, P = NS) did not change, while heart rate was higher at baseline than after 75 min of experimental venous congestion (70 ± 2 vs. 66 ± 2 b.p.m., P = 0.01).
Experimental venous congestion did not influence arterial O2 saturation (98 ± 0 vs. 99 ± 0%, P = NS) and venous lactic acid (7.5 ± 0.3 vs. 8.0 ± 0.2 mg/dL, P = NS) in the congested arm. These results suggest that experimental venous congestion did not induce local ischaemia in this model.
Acute venous congestion increases peripheral release of cytokines, vasoactive peptides, neurohormones, and endothelial adhesion molecules
The induction of venous congestion was related to a 60% increase in plasma levels of IL-6 and ET-1 (Table 2). Plasma levels of IL-6 also tended to be higher in the samples collected from the control arm compared with baseline values (P = 0.07). Angiotensin II increased by 20% in the congested arm. A marginal but significant increase of plasma VCAM-1 occurred in the congested (test) arm when compared with control arm. Plasma ICAM-1 levels also tended to be higher in the congested arm. Plasma TNF-α and vWF:Ag levels did not change. These results suggest that experimental venous congestion causes local release of cytokines, vasoactive peptides, neurohormones, and endothelial adhesion molecules. Of note, the significant increase in AII suggests volume-dependent activation of tissue angiotensin-converting enzyme and/or chymase in the periphery.
Table 2.
Plasma measurements (means ± SEM) before and after 75 min of venous congestion
| Variable | Baseline (B) (0 min) | Control arm (C) (75 min) | Test arm (T) (75 min) | P-value (T vs. B) | P-value (T vs. C) |
|---|---|---|---|---|---|
| IL-6 (pg/mL) | 1.37 ± 0.44 | 1.79 ± 0.53 | 2.26 ± 0.58 | <0.01 | <0.01 |
| TNF-α (pg/mL) | 1.35 ± 0.08 | 1.27 ± 0.08 | 1.35 ± 0.11 | 0.75 | 0.22 |
| ET-1 (pg/mL) | 1.46 ± 0.19 | 1.26 ± 0.13 | 2.43 ± 0.27 | <0.0001 | <0.0001 |
| AII (pg/mL) | 27 ± 3 | 25 ± 3 | 32 ± 4 | 0.01 | <0.01 |
| VCAM-1 (ng/mL) | 557 ± 26 | 544 ± 24 | 589 ± 25 | <0.01 | <0.01 |
| ICAM-1 (ng/mL) | 158 ± 9 | 158 ± 7 | 167 ± 9 | 0.09 | 0.06 |
| vWF:Ag (%) | 105 ± 9 | 100 ± 6 | 113 ± 9 | 0.37 | 0.15 |
IL-6, interleukin-6; TNF-α, tumor necrosis factor-α; ET-1, endothelin-1; AII, Angiotensin II; VCAM-1, vascular adhesion molecule-1; ICAM-1, intercellular adhesion molecule-1; vWF:Ag, vonWillebrand factor antigen.
Acute venous congestion promotes endothelial cell activation
Acute venous congestion was related to major changes in the EC transcriptome as assessed via Affymetrix genomic arrays. A total of 3437 probe sets were differentially expressed with P < 0.05 before vs. after venous congestion, 96 of which had an absolute fold-change >2.0 (Supplementary material online, Table S2). One thousand six hundred and twenty-eight probe sets were differentially expressed with FDR <0.05, among which 72 had an absolute fold-change value >2.0 (Figure 2). Results from mixed effects linear regression models that corrected for the presence of residual leucocytes and SMCs within the samples were even stronger than the aforementioned unadjusted results. Specifically, 5332 probe sets were differentially expressed before vs. after venous congestion (P < 0.05), among which 143 probe sets had a fold-change >2.0 (Supplementary material online, Table S3). Irrespective of the means of analysis, key signatures associated with venous congestion included genes relevant to inflammation, cell migration, and signal transduction, such as ET-1, VCAM-1, CD36, TNF receptor-associated factor 5 (TRAF5), and chemokine (C-X-C motif) ligand 2 (CXCL2). Gene ontology analysis is detailed in Supplementary material online, Table S4. On the basis of the above finding, we also assessed plasma CXCL2 (n = 15) and found that CXCL2 protein levels were higher in the congested arm compared with the control arm after 75 min of venous congestion (294 ± 110 vs. 254 ± 102 pg/mL, P = 0.04). These results suggest that acute venous congestion causes endothelial activation as evidence by significant up-regulation of several pro-inflammatory genes.
Figure 2.

Heat map of the 72 probe sets with an absolute fold-change >2.0 and a false discovery rate <0.05. The green header bar refers to endothelial cell gene expression in samples prior to venous congestion and the red header bar represents endothelial cell gene expression in samples after experimental venous congestion. Blue-coloured cells in the matrix represent relative under expression (i.e. lower expression values) and yellow-coloured cells represent relative over expression (i.e. higher expression values). FC, fold-change.
Discussion
The current study provides the first direct evidence that peripheral acute venous congestion, without ischaemia, causes inflammation and neurohormonal and EC activation in humans. Overall, this inflammatory, vasoconstrictive phenotype mimics, at least in part and on a local scale, notable aspects of the phenotype which is typical of patients with advanced and of those with acute HF and RF.
Several studies demonstrated elevations in circulating levels of cytokines, vasoactive peptides, neurohomones, endothelial adhesion molecules, and coagulation factors that correlate both with severity and acuity of HF and of RF.1–18 The source and site of their production is controversial,2,32–37 and no evidence has so far mechanistically linked peripheral venous congestion to this pro-inflammatory, vasoconstrictive, and activated vascular phenotype in humans. Herein, we introduce a new experimental model of peripheral venous congestion (so-called venous stress test) coupled with endothelial sampling27 and transcriptomic approaches using microarray technology. This approach enables, in a controlled setting, assessment of the acute inflammatory, neurohormonal, and endothelial response to biomechanical stress in humans. An increase in local peripheral venous pressure of ∼30 mmHg caused a >60% increase in plasma IL-6 and ET-1 as well as a 20% increase in plasma AII in the congested arm of healthy subjects. Plasma levels of IL-6 also tended to be higher in the sample collected from the control arm compared with baseline values. While we cannot provide a direct mechanistic justification for this latter result, it is possible that activation of circulating T cells and macrophages in response to a local inflammatory stimulus such as experimental venous congestion caused systemic in addition to local release of IL-6. Peripheral spillover from the congested arm into the systemic circulation may offer an alternatively explanation. However, it appears less likely as the concentration of the other plasma mediators did not increase in the control arm.
Venous congestion promoted deferential expression of >3000 probe sets in venous ECs, including key markers of EC activation such as ET-1 and VCAM-1. These results in ECs parallel and may, at least in part, explain the increase in plasma ET-1 and VCAM-1, which was observed in venous blood collected from the congested arm. Overall, these data expand and provide mechanistic insights to previous observational evidence of EC activation in HF patients who were hospitalized for acute HF.38
Whole-genome expression analysis in the EC samples also helped to identify other genes and biological pathways that might be involved in the mechanotransduction of high intravenous pressure. Among the observed probe sets, we recognized other key pro-inflammatory and vasoactive genes such as: (i) CD36, the endothelial receptor for thrombospondin, a potent antagonist of the angiogenic signalling downstream of nitric oxide;39 (ii) TRAF5 which is induced in atherosclerotic plaques and is one of the key mediators of the pro-inflammatory functions of CD40 ligand in ECs;40 and (iii) CXCL2, a small pro-inflammatory chemokine belonging to the CXC family. Of note, recent evidence suggests that TRAF5 prolongs the half-life of chemokines, including CXCL2.41 On the basis of the CXCL2 mRNA finding, we performed a data-driven a priori hypothesis test of CXCL2 plasma protein expression and found that CXCL2 was indeed increased in plasma collected from the congested arm compared with the control arm.
The present study has several limitations. First, the small sample size substantially limited our power to detect findings at the transcript level after adjustment for multiple comparisons. However, the primary aim of the experiment was to investigate the influence of venous congestion on plasma proteins, as they are more clinically relevant. Nevertheless, the mRNA data generated in our current study not only support our findings on plasma proteins, but also provide a rich database (see Supplementary material online, Tables S2–4) that will help us and others to develop focused mechanistic studies that can better explain how venous congestion leads to EC generation of pro-inflammatory mediators and, possibly, to neurohormonal activation. Secondly, this study was conducted in healthy subjects rather than in patients with HF and/or RF. However, the fact that we have studied healthy participants provides a strong proof-of-principle for the concept of hypervolaemia as a fundamental stimulus for the pathophysiology of HF and RF as it is not possible for other aspects of these disease phenotypes, for co-morbidities and/or for background therapy to confound our results. Finally, cuff inflation not only promoted venous congestion, but necessarily caused (i) hydrostatic pooling by reducing venous flow and (ii) reduction in arterial perfusion pressure by impinging on the brachial artery. These latter effects may have also influenced plasma concentration and/or peripheral release of the measured mediators. However, (i) the resulting change in local blood flow did not cause ischaemia in our model and (ii) reduction in perfusion pressure and hydrostatic pooling are typical clinical features of advanced HF where arterial blood pressure progressively declines42 and chronic venous insufficiency is frequent,43 thus making our model even more relevant, from a pathophysiological standpoint.
We developed a new human model of acute, local venous congestion (so-called venous stress test) where peripheral venous pressure was increased to ∼30 mmHg above baseline levels for 75 min by inflating a tourniquet cuff around the dominant arm. Transient venous congestion was sufficient to cause local release of inflammatory mediators, neurohormones, and endothelial activation. Additional studies are warranted to further test the hypothesis that venous and tissue congestion might be a causal contributor to (as opposed to a consequence of) the pathophysiology of advanced and of acute HF and RF via their influence on inflammation, neurohormonal, and EC activation. From a clinical perspective, if venous and tissue congestion prove to be fundamental stimuli for the pathophysiology of HF and RF, early and aggressive treatment of hypervolaemia may have a critical role not only in symptom relief, but also in disease progression. This early treatment strategy may include not only diuretics, but also, as one may infer from our results and test in future studies, adjuvant therapies such as short-term anti-inflammatory and vasoactive treatments that may prevent the development of overt decompensation. In the future, our experimental approach may also help identify ‘volume-sensitive’ patients, who have enhanced endothelial and neurohormonal responses to venous and tissue congestion. This group of patients may be more prone to decompensation episodes and, consequently, more likely to benefit from meticulous monitoring of their volume status, which may then be used to prompt early interventions.
Supplementary material
Supplementary material is available at European Heart Journal online.
Funding
This study was supported A. L. Mailman Family Foundation, by the NIH Grant Number HL092144, NIH Grant Number DE018739, and by the NIH Grant Number UL1 TR000040 (formerly the National Center for Research Resources Grant Number UL1 RR024156).
Conflict of interest: This study has no relationship with industry.
Supplementary Material
References
- 1.Munger MA, Johnson B, Amber IJ, Callahan KS, Gilbert EM. Circulating concentrations of proinflammatory cytokines in mild or moderate heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 1996;77:723–727. doi: 10.1016/s0002-9149(97)89206-5. [DOI] [PubMed] [Google Scholar]
- 2.Testa M, Yeh M, Lee P, Fanelli R, Loperfido F, Berman JW, LeJemtel TH. Circulating levels of cytokines and their endogenous modulators in patients with mild to severe congestive heart failure due to coronary artery disease or hypertension. J Am Coll Cardiol. 1996;28:964–971. doi: 10.1016/s0735-1097(96)00268-9. [DOI] [PubMed] [Google Scholar]
- 3.Packer M. Is tumor necrosis factor an important neurohormonal mechanism in chronic heart failure? Circulation. 1995;92:1379–1382. doi: 10.1161/01.cir.92.6.1379. [DOI] [PubMed] [Google Scholar]
- 4.Carlstedt F, Lind L, Lindahl B. Proinflammatory cytokines, measured in a mixed population on arrival in the emergency department, are related to mortality and severity of disease. J Intern Med. 1997;242:361–365. doi: 10.1046/j.1365-2796.1997.00209.x. [DOI] [PubMed] [Google Scholar]
- 5.Matsumoto M, Tsujino T, Lee-Kawabata M, Naito Y, Sakoda T, Ohyanagi M, Masuyama T. Serum interleukin-6 and c-reactive protein are markedly elevated in acute decompensated heart failure patients with left ventricular systolic dysfunction. Cytokine. 2010;49:264–268. doi: 10.1016/j.cyto.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 6.Barreto DV, Barreto FC, Liabeuf S, Temmar M, Lemke HD, Tribouilloy C, Choukroun G, Vanholder R, Massy ZA European Uremic Toxin Work G. Plasma interleukin-6 is independently associated with mortality in both hemodialysis and pre-dialysis patients with chronic kidney disease. Kidney Int. 2010;77:550–556. doi: 10.1038/ki.2009.503. [DOI] [PubMed] [Google Scholar]
- 7.Descamps-Latscha B, Herbelin A, Nguyen AT, Roux-Lombard P, Zingraff J, Moynot A, Verger C, Dahmane D, de Groote D, Jungers P, et al. Balance between il-1 beta, tnf-alpha, and their specific inhibitors in chronic renal failure and maintenance dialysis. relationships with activation markers of t cells, b cells, and monocytes. J Immunol. 1995;154:882–892. [PubMed] [Google Scholar]
- 8.Rodeheffer RJ, Lerman A, Heublein DM, Burnett JC., Jr Increased plasma concentrations of endothelin in congestive heart failure in humans. Mayo Clin Proc. 1992;67:719–724. doi: 10.1016/s0025-6196(12)60795-2. [DOI] [PubMed] [Google Scholar]
- 9.Cottone S, Mule G, Guarneri M, Palermo A, Lorito MC, Riccobene R, Arsena R, Vaccaro F, Vadala A, Nardi E, Cusimano P, Cerasola G. Endothelin-1 and f2-isoprostane relate to and predict renal dysfunction in hypertensive patients. Nephrol, Dial, Transplant. 2009;24:497–503. doi: 10.1093/ndt/gfn489. [DOI] [PubMed] [Google Scholar]
- 10.Dzau VJ, Colucci WS, Hollenberg NK, Williams GH. Relation of the renin-angiotensin-aldosterone system to clinical state in congestive heart failure. Circulation. 1981;63:645–651. doi: 10.1161/01.cir.63.3.645. [DOI] [PubMed] [Google Scholar]
- 11.Paton AM, Lever AF, Oliver NW, Medina A, Briggs JD, Morton JJ, Brown JJ, Robertson JI, Fraser R, Tree M, Gavras H. Plasma angiotensin ii, renin, renin-substrate and aldosterone concentrations in acute renal failure in man. Clin Nephrol. 1975;3:18–23. [PubMed] [Google Scholar]
- 12.Tsutamoto T, Hisanaga T, Fukai D, Wada A, Maeda Y, Maeda K, Kinoshita M. Prognostic value of plasma soluble intercellular adhesion molecule-1 and endothelin-1 concentration in patients with chronic congestive heart failure. Am J Cardiol. 1995;76:803–808. doi: 10.1016/s0002-9149(99)80231-8. [DOI] [PubMed] [Google Scholar]
- 13.Andreassen AK, Nordoy I, Simonsen S, Ueland T, Muller F, Froland SS, Gullestad L, Aukrust P. Levels of circulating adhesion molecules in congestive heart failure and after heart transplantation. Am J Cardiol. 1998;81:604–608. doi: 10.1016/s0002-9149(97)00972-7. [DOI] [PubMed] [Google Scholar]
- 14.Klein RM, Breuer R, Mundhenke M, Schwartzkopff B, Strauer BE. Circulating adhesion molecules (cicam-1, lcvcam-1) in patients with suspected inflammatory heart muscle disease. Z Kardiol. 1998;87:84–93. doi: 10.1007/s003920050158. [DOI] [PubMed] [Google Scholar]
- 15.Yin WH, Chen JW, Jen HL, Chiang MC, Huang WP, Feng AN, Lin SJ, Young MS. The prognostic value of circulating soluble cell adhesion molecules in patients with chronic congestive heart failure. Eur J Heart Fail. 2003;5:507–516. doi: 10.1016/s1388-9842(03)00009-6. [DOI] [PubMed] [Google Scholar]
- 16.Bonomini M, Reale M, Santarelli P, Stuard S, Settefrati N, Albertazzi A. Serum levels of soluble adhesion molecules in chronic renal failure and dialysis patients. Nephron. 1998;79:399–407. doi: 10.1159/000045084. [DOI] [PubMed] [Google Scholar]
- 17.Chin BS, Conway DS, Chung NA, Blann AD, Gibbs CR, Lip GY. Interleukin-6, tissue factor and von willebrand factor in acute decompensated heart failure: relationship to treatment and prognosis. Blood Coagul Fibrinolysis. 2003;14:515–521. doi: 10.1097/00001721-200309000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Galatius S, Wroblewski H, Sorensen VB, Bie P, Parving HH, Kastrup J. Endothelin and von willebrand factor as parameters of endothelial function in idiopathic dilated cardiomyopathy: different stimuli for release before and after heart transplantation? Am Heart J. 1999;137:549–554. doi: 10.1016/s0002-8703(99)70505-3. [DOI] [PubMed] [Google Scholar]
- 19.Shen L, Lu G, Dong N, Jiang L, Ma Z, Ruan C. Von willebrand factor, adamts13 activity, tnf-alpha and their relationships in patients with chronic kidney disease. Exp Ther Med. 2012;3:530–534. doi: 10.3892/etm.2011.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawai M, Naruse K, Komatsu S, Kobayashi S, Nagino M, Nimura Y, Sokabe M. Mechanical stress-dependent secretion of interleukin 6 by endothelial cells after portal vein embolization: clinical and experimental studies. J Hepatol. 2002;37:240–246. doi: 10.1016/s0168-8278(02)00171-x. [DOI] [PubMed] [Google Scholar]
- 21.Wang BW, Chang H, Lin S, Kuan P, Shyu KG. Induction of matrix metalloproteinases-14 and -2 by cyclical mechanical stretch is mediated by tumor necrosis factor-alpha in cultured human umbilical vein endothelial cells. Cardiovasc Res. 2003;59:460–469. doi: 10.1016/s0008-6363(03)00428-0. [DOI] [PubMed] [Google Scholar]
- 22.Hasdai D, Holmes DR, Jr, Garratt KN, Edwards WD, Lerman A. Mechanical pressure and stretch release endothelin-1 from human atherosclerotic coronary arteries in vivo. Circulation. 1997;95:357–362. doi: 10.1161/01.cir.95.2.357. [DOI] [PubMed] [Google Scholar]
- 23.Delli Gatti C, Osto E, Kouroedov A, Eto M, Shaw S, Volpe M, Luscher TF, Cosentino F. Pulsatile stretch induces release of angiotensin ii and oxidative stress in human endothelial cells: effects of ace inhibition and at1 receptor antagonism. Clin Exp Hypertens. 2008;30:616–627. doi: 10.1080/10641960802443183. [DOI] [PubMed] [Google Scholar]
- 24.Ali MH, Pearlstein DP, Mathieu CE, Schumacker PT. Mitochondrial requirement for endothelial responses to cyclic strain: implications for mechanotransduction. Am J Physiol Lung Cell Mol Physiol. 2004;287:L486–L496. doi: 10.1152/ajplung.00389.2003. [DOI] [PubMed] [Google Scholar]
- 25.Cheng JJ, Wung BS, Chao YJ, Wang DL. Cyclic strain enhances adhesion of monocytes to endothelial cells by increasing intercellular adhesion molecule-1 expression. Hypertension. 1996;28:386–391. doi: 10.1161/01.hyp.28.3.386. [DOI] [PubMed] [Google Scholar]
- 26.Galbusera M, Zoja C, Donadelli R, Paris S, Morigi M, Benigni A, Figliuzzi M, Remuzzi G, Remuzzi A. Fluid shear stress modulates von willebrand factor release from human vascular endothelium. Blood. 1997;90:1558–1564. [PubMed] [Google Scholar]
- 27.Colombo PC, Ashton AW, Celaj S, Talreja A, Banchs JE, Dubois NB, Marinaccio M, Malla S, Lachmann J, Ware JA, Le Jemtel TH. Biopsy coupled to quantitative immunofluorescence: a new method to study the human vascular endothelium. J Appl Physiol. 2002;92:1331–1338. doi: 10.1152/japplphysiol.00680.2001. [DOI] [PubMed] [Google Scholar]
- 28.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of affymetrix genechip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuhn A, Thu D, Waldvogel HJ, Faull RL, Luthi-Carter R. Population-specific expression analysis (psea) reveals molecular changes in diseased brain. Nat Methods. 2011;8:945–947. doi: 10.1038/nmeth.1710. [DOI] [PubMed] [Google Scholar]
- 30.Storey JD, Tibshirani R. Statistical methods for identifying differentially expressed genes in DNA microarrays. Methods Mol Biol. 2003;224:149–157. doi: 10.1385/1-59259-364-X:149. [DOI] [PubMed] [Google Scholar]
- 31.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 32.Kapadia S, Lee J, Torre-Amione G, Birdsall HH, Ma TS, Mann DL. Tumor necrosis factor-alpha gene and protein expression in adult feline myocardium after endotoxin administration. J Clin Invest. 1995;96:1042–1052. doi: 10.1172/JCI118090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X, Moody MR, Engel D, Walker S, Clubb FJ, Jr, Sivasubramanian N, Mann DL, Reid MB. Cardiac-specific overexpression of tumor necrosis factor-alpha causes oxidative stress and contractile dysfunction in mouse diaphragm. Circulation. 2000;102:1690–1696. doi: 10.1161/01.cir.102.14.1690. [DOI] [PubMed] [Google Scholar]
- 34.Mann DL. The effect of tumor necrosis factor-alpha on cardiac structure and function: A tale of two cytokines. J Card Fail. 1996;2:S165–S172. doi: 10.1016/s1071-9164(96)80073-x. [DOI] [PubMed] [Google Scholar]
- 35.Mann DL. Stress activated cytokines and the heart. Cytokine Growth Factor Rev. 1996;7:341–354. doi: 10.1016/s1359-6101(96)00043-3. [DOI] [PubMed] [Google Scholar]
- 36.Chang HJ, Chung J, Choi BJ, Choi TY, Choi SY, Yoon MH, Hwang GS, Shin JH, Tahk SJ, Choi BI. The origin of proinflammatory cytokines in patients with idiopathic dilated cardiomyopathy. J Korean Med Sci. 2003;18:791–796. doi: 10.3346/jkms.2003.18.6.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsutamoto T, Hisanaga T, Wada A, Maeda K, Ohnishi M, Fukai D, Mabuchi N, Sawaki M, Kinoshita M. Interleukin-6 spillover in the peripheral circulation increases with the severity of heart failure, and the high plasma level of interleukin-6 is an important prognostic predictor in patients with congestive heart failure. J Am Coll Cardiol. 1998;31:391–398. doi: 10.1016/s0735-1097(97)00494-4. [DOI] [PubMed] [Google Scholar]
- 38.Colombo PC, Banchs JE, Celaj S, Talreja A, Lachmann J, Malla S, DuBois NB, Ashton AW, Latif F, Jorde UP, Ware JA, LeJemtel TH. Endothelial cell activation in patients with decompensated heart failure. Circulation. 2005;111:58–62. doi: 10.1161/01.CIR.0000151611.89232.3B. [DOI] [PubMed] [Google Scholar]
- 39.Silverstein RL, Febbraio M. Cd36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal. 2009;2:re3. doi: 10.1126/scisignal.272re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zirlik A, Bavendiek U, Libby P, MacFarlane L, Gerdes N, Jagielska J, Ernst S, Aikawa M, Nakano H, Tsitsikov E, Schonbeck U. Traf-1, -2, -3, -5, and -6 are induced in atherosclerotic plaques and differentially mediate proinflammatory functions of cd40l in endothelial cells. Arterioscler Thromb Vasc Biol. 2007;27:1101–1107. doi: 10.1161/ATVBAHA.107.140566. [DOI] [PubMed] [Google Scholar]
- 41.Sun D, Novotny M, Bulek K, Liu C, Li X, Hamilton T. Treatment with il-17 prolongs the half-life of chemokine cxcl1 mRNA via the adaptor traf5 and the splicing-regulatory factor sf2 (asf) Nat Immunol. 2011;12:853–860. doi: 10.1038/ni.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barlera S, Tavazzi L, Franzosi MG, Marchioli R, Raimondi E, Masson S, Urso R, Lucci D, Nicolosi GL, Maggioni AP, Tognoni G. Predictors of mortality in 6975 patients with chronic heart failure in the gissi-hf trial: proposal for a nomogram. Circ Heart Fail. 2012;6:31–9. doi: 10.1161/CIRCHEARTFAILURE.112.967828. [DOI] [PubMed] [Google Scholar]
- 43.White JV, Ryjewski C. Chronic venous insufficiency. Perspect Vasc Surg Endovasc Ther. 2005;17:319–327. doi: 10.1177/153100350501700406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.