

Abstract
Aims
The aim of this study was to evaluate the haemodynamic effects of serelaxin (30 µg/kg/day 20-h infusion and 4-h post-infusion period) in patients with acute heart failure (AHF).
Methods and results
This double-blind, multicentre study randomized 71 AHF patients with pulmonary capillary wedge pressure (PCWP) ≥18 mmHg, systolic blood pressure (BP) ≥115 mmHg, and estimated glomerular filtration rate ≥30 mL/min/1.73 m2 to serelaxin (n = 34) or placebo (n = 37) within 48 h of hospitalization. Co-primary endpoints were peak change from baseline in PCWP and cardiac index (CI) during the first 8 h of infusion. Among 63 patients eligible for haemodynamic analysis (serelaxin, n = 32; placebo, n = 31), those treated with serelaxin had a significantly higher decrease in peak PCWP during the first 8 h of infusion (difference vs. placebo: −2.44 mmHg, P = 0.004). Serelaxin showed no significant effect on the peak change in CI vs. placebo. Among secondary haemodynamic endpoints, a highly significant reduction in pulmonary artery pressure (PAP) was observed throughout the serelaxin infusion (largest difference in mean PAP vs. placebo: −5.17 mmHg at 4 h, P < 0.0001). Right atrial pressure, systemic/pulmonary vascular resistance, and systolic/diastolic BP decreased from baseline with serelaxin vs. placebo and treatment differences reached statistical significance at some time points. Serelaxin administration improved renal function and decreased N-terminal pro-brain natriuretic peptide levels vs. placebo. Treatment with serelaxin was well tolerated with no apparent safety concerns.
Conclusion
The haemodynamic effects of serelaxin observed in the present study provide plausible mechanistic support for improvement in signs and symptoms of congestion observed with this agent in AHF patients.
ClinicalTrials.gov identifier NCT01543854.
Keywords: Serelaxin, Acute heart failure, Haemodynamics, Clinical trial
See page 410 for the editorial comment on this article (doi:10.1093/eurheartj/eht477)
Introduction
Acute heart failure (AHF) is a heterogeneous clinical syndrome with worldwide increasing prevalence, poor outcomes, and very high healthcare costs.1,2 Despite extensive research effort, there has been no significant pharmacological advance in the management of AHF patients with respect to mortality and dyspnoea relief for several decades.1 Indeed, current treatment aimed at early clinical stabilization and symptomatic improvement, namely loop diuretics with inotropes and vasodilators, does not have supporting data on long-term outcomes. This unmet need provides impetus to the on-going search for novel therapies that would be able to meet these therapeutic goals.
Serelaxin is a recombinant form of human relaxin-2, a naturally occurring peptide hormone that, in women, mediates maternal systemic haemodynamic and renal adaptations to an increase in intravascular volume during pregnancy.3 Relaxin-2 exerts numerous haemodynamic and renal effects in pregnant women, such as increase in arterial compliance with concomitant fall in systemic vascular resistance (SVR), and increase in renal blood flow, glomerular filtration rate, and cardiac output (CO).4–6 Since all these effects of relaxin-2 can be potentially beneficial for the treatment of AHF, clinical applicability of its analogue (serelaxin) has been evaluated in this setting.7,8 In the pilot Pre-RELAX-AHF phase II, dose-finding study, for example, treatment with serelaxin resulted in beneficial effects on both dyspnoea and post-discharge clinical outcomes in AHF patients admitted to the emergency room with evidence of congestion, normal-to-raised systolic blood pressure (BP), and mild-to-moderate renal dysfunction.7 These results were subsequently confirmed in RELAX-AHF, a larger phase III study, where early treatment with serelaxin was well tolerated and associated with significant dyspnoea relief and, among safety endpoints, a 37% reduction in 180-day mortality.8 An explanation of these beneficial clinical outcomes is desirable, and it has been postulated that prevention of end-organ damage and early relief from congestion resulting from serelaxin treatment may be associated with a reduction in all-cause mortality.9 It is tempting to speculate that the favourable haemodynamic effects, that serelaxin exerts in an early phase of AHF, may translate into relief of congestion, symptomatic improvement, and subsequent longer-term benefits. However, in the setting of heart failure, haemodynamic effects of serelaxin have only been characterized in a small group of patients with advanced, but stable clinical conditions.5 Thus, we have designed a study to evaluate the haemodynamic effects of serelaxin in patients with AHF at a dose rate of 30 µg/kg/day, as was administered in the RELAX-AHF study.8
Methods
Study participants
Male and female patients 18 years of age and above hospitalized for AHF who could be randomized within 48 h of presentation were eligible for participation. Acute heart failure was defined as new onset or worsening of signs and symptoms of heart failure requiring urgent therapy (e.g. dyspnoea at rest or on minimal exertion and pulmonary congestion at the time of presentation), and patients were non-electively admitted/required admission to hospital for AHF management. Patients were initially stabilized with furosemide, 40–120 mg/day i.v. (or other loop diuretics at equivalent dose) with no planned change in diuretic dose/use of i.v. vasodilator from 4 h prior to treatment initiation till 8 h thereafter. Central haemodynamic monitoring of the patients had to be initiated at least 1 h before randomization with a mean pulmonary capillary wedge pressure (PCWP) ≥18 mmHg.
The main exclusion criteria were systolic BP <115 mmHg or estimated GFR <30 mL/min/1.73 m2 (calculated using the simplified Modification of Diet in Renal Disease equation); history of acute coronary syndrome, major neurological event, or known significant valvular disease or AHF due to significant arrhythmias; acute myocarditis; or hypertrophic obstructive, restrictive, or constrictive cardiomyopathy; recent i.v. contrast radiography, acute contrast-induced nephropathy, or clinically significant hepatic impairment; planned treatment with i.v. therapies including inotropic agents, vasopressors, levosimendan, nesiritide, or analogues, or mechanical support (intra-aortic balloon pump, endotracheal intubation, mechanical ventilation, or any ventricular-assist device); current or planned organ transplant, ultrafiltration, haemofiltration, dialysis, or major surgery.
Study design
This was a randomized, double-blind, placebo-controlled, parallel-group, multicentre study. The study was conducted in compliance with Good Clinical Practice outlined in the principles of the Declaration of Helsinki, and was approved by an ethics committee or institutional review board of each participating centre and regulatory authorities. It was registered with ClinicalTrials.gov (NCT01543854). All participants provided written informed consent.
The study consisted of a 2-day screening period, a baseline period and a 20-h i.v. infusion treatment period with study drugs followed by a 4-h washout period. The study completion evaluation was done at 30 (±3) days after the start of the treatment. Patients meeting the selection criteria for enrolment during the screening period had a 7F Swan–Ganz Thermodilution catheter inserted to assess baseline PCWP on Day 1 of the study after stable haemodynamic measurements over at least 1 h. Baseline PCWP was calculated as the mean of the last three measurements taken at least 15 min apart immediately prior to randomization, which were required to be stable (within 15% of each other). Cardiac output measurements had to be stable (within 15% or no >0.5 L/min of each other) and baseline CO was calculated as the mean of up to three assessments in patients with sinus rhythm and five assessments in atrial fibrillation considering significant variability. Eligible patients were randomized in a 1:1 ratio to serelaxin or placebo, after baseline assessments were completed. Randomization numbers were generated (by the study sponsor) using an automated and validated system to assign treatment arms, which were concealed from patients and investigators. The placebo used was a buffer solution with an appearance identical to serelaxin to achieve blinding. Patients, investigator staff, and personnel performing the assessments were blinded to the identity of the treatment.
Serelaxin or matching placebo was administered as a 30 µg/kg/day infusion for 20 h (at a rate of 10 mL/h). If the patient's systolic BP decreased by >40 mmHg from baseline, but was >90 mmHg in two consecutive measurements 15 min apart, then the infusion rate was reduced to half for the rest of the administration. If systolic BP was reduced to <90 mmHg, infusion was terminated. Hypotension was reported as an adverse event (AE) by the investigators. Use of rescue medication, including i.v. vasodilators, to treat severe or serious conditions was allowed and recorded. Furosemide 40 mg (i.v. bolus) was administered ∼4 h before the start of the study drug infusion and repeated 8 h after the initiation of the infusion and haemodynamic assessment, if clinically indicated. On the next day furosemide, 40 mg i.v. was administered 4 h after the end of the study drug infusion and haemodynamic assessment, if required. For patients requiring furosemide 120 mg/day i.v., half the dose was advised to be given at 12-h intervals as mentioned earlier. Per study protocol, all oral medications were planned to be administered in the morning on Day 1 and after 4 h of completion of infusion and haemodynamic assessments on Day 2.
Haemodynamic assessments were carried out at baseline and at 0.5, 2, 4, 6, 8, 20, 21, 22 and 24 h after initiation of the study drug infusion, which covered the 20-h infusion period and the 4-h post-infusion (washout) period. Cardiac output was determined by the thermodilution technique. Brachial systolic and diastolic BP and pulse rate were measured using the A-PULSE CASPro® device (HealthSTATS International, Singapore) during haemodynamic assessments, and on Day 3 and Day 30 (follow-up). N-terminal pro-brain natriuretic peptide (NT-pro-BNP) levels were analysed using commercially available kits (Roche Diagnostics GmbH Mannheim, Germany) at baseline and at 8, 20, and 44 h after the study treatment initiation.
Efficacy assessments
Co-primary endpoints were peak changes from baseline in PCWP and CI during the first 8 h of infusion. Secondary haemodynamic endpoints included right atrial pressure (RAP), systolic, diastolic and mean pulmonary artery pressure (PAP), CO, SVR, pulmonary vascular resistance (PVR) and systolic/diastolic BP evaluated during the 24-h period after infusion initiation. Other endpoints included urine flow rate, urinary excretion of creatinine, and creatinine clearance measured from urine collection fractions over 24 h and cardio-renal biomarkers including NT-pro-BNP.
Safety monitoring
Safety assessments included monitoring of clinical signs and symptoms at screening and signs throughout the treatment phase, physical examination, electrocardiogram, standard clinical laboratory evaluations (haematology, blood chemistry, and urinalysis), and AE, and serious AE (SAE) monitoring. All the patients were followed up to Day 30 (±3) for safety assessments.
Statistical analysis
Assuming a standard deviation (SD) of 6 mmHg10,11 and the true drug effect of a reduction of ≥4.2 mmHg, this study had at least 80% power to detect a treatment difference from placebo in peak change from baseline PCWP (co-primary endpoint) that is considered to be both statistically (two-sided P < 0.05) and clinically significant (estimated treatment difference >3 mmHg) with a sample size of 64 patients (32 patients per group). In addition, with reported SD of 0.5 L/min/m2 for CI10,11 and 32 patients per group, assuming the true drug effect of an increase of ≥0.35 L/min/m2, the study also had at least 80% power to detect a statistically and clinically significant treatment difference (mean estimate >0.25 L/min/m2) in peak change from baseline in CI (second co-primary endpoint). Assuming a dropout rate of ∼10%, 35 patients per group needed to be randomized in order to obtain 32 analysable patients.
The safety population consisted of all patients who received the study drug infusion and had at least one post-baseline safety assessment. The efficacy population consisted of all patients who received at least 8 h of the study drug infusion, had at least one post-baseline assessment of a primary haemodynamic variable and no major protocol deviations. Peak change from baseline in PCWP and CI over 8 h of infusion was assessed by analysis of covariance (ANCOVA) using treatment as classification factor and corresponding baseline value as covariate through the SAS software (Version 9.3, Cary, NC, USA). Time-weighted average change from baseline and change from baseline for each scheduled time point were also analysed. Treatment difference in least squares means (LS means) and the associated two-sided 95% confidence intervals as well as P-values were calculated. Secondary endpoints were analysed similarly. For calculating time-weighted average change from baseline using area under the effect curve, missing values at the end time point of each interval were imputed using the last observation carried forward method before applying the trapezoidal rule. For urine flow rate, creatinine clearance, and NT-pro-BNP, data were log-transformed before analysis. The post hoc ANCOVA test was carried out to confirm results of the per-protocol analysis in the intention-to-treat data set (including all the patients who received drug infusion), using the same factors and covariates.
Results
Patient disposition and baseline characteristics
Patient enrolment was started in March 2012 and completed in January 2013. Out of a total of 120 patients screened, 71 patients were randomized (serelaxin, n = 34; placebo, n = 37) from 17 sites in 6 countries (Russia, Germany, Poland, Argentina, Italy, and The Netherlands). The remaining 49 patients did not meet eligibility criteria and were screening failures. Demographic and baseline characteristics were similar between serelaxin and placebo groups (Table 1). Patient disposition is summarized in Figure 1. Overall, 71 patients received study drug (serelaxin, n = 34; placebo, n = 37) and were included in the safety analysis; 63 patients (serelaxin, n = 32; placebo, n = 31) were eligible for haemodynamic analysis. One patient was excluded from the primary analysis in the serelaxin group because 8 h of drug infusion was not completed. Another serelaxin-treated patient and six placebo recipients were excluded due to protocol deviations that can affect efficacy assessments, the most common being use of furosemide during the initial 8 h of infusion (serelaxin, n = 1; placebo, n = 5).
Table 1.
Demographic data and baseline characteristics of randomized patients
| Serelaxin (n = 34) | Placebo (n = 37) | Total (n = 71) | |
|---|---|---|---|
| Age (years) | 66.6 (11.2) | 70.4 (12.4) | 68.6 (11.9) |
| Height (cm) | 169.9 (7.8) | 168.3 (9.4) | 169.1 (8.6) |
| Weight (kg) | 89.2 (20.2) | 86.0 (21.1) | 87.5 (20.6) |
| Sex: male; n (%) | 27 (79.4) | 26 (70.3) | 53 (74.6) |
| Body mass index (kg/m2) | 31.1 (7.6) | 30.6 (9.0) | 30.8 (8.3) |
| eGFR (mL/min/1.73 m2) | 71.7 (23.7) | 67.7 (24.1) | 69.7 (23.8) |
| Baseline SBP (mmHg) | 131.1 (14.7) | 131.6 (17.1) | 131.3 (15.9) |
| Baseline DBP (mmHg) | 84.3 (10.7) | 84.3 (13.0) | 84.3 (11.9) |
| Time from AHF hospitalization to start of infusion (h) | 27.9 (11.2) | 30.0 (11.2) | 29.0 (11.2) |
| Prior history of HF; n (%) | 34 (100) | 34 (91.9) | 68 (95.8) |
| NYHA classificationa; n (%) | |||
| Class III | 14 (41.2) | 20 (54.1) | 34 (47.9) |
| Class IV | 20 (58.8) | 17 (45.9) | 37 (52.1) |
| Prior HF hospitalization; n (%) | 25 (73.5) | 33 (89.2) | 58 (81.7) |
| Ejection fractionb (%) | 34.5 (15.3) | 32.5 (12.7) | 33.4 (13.7) |
| Primary aetiology; n (%) | |||
| Ischaemic | 16 (47.1) | 20 (54.1) | 36 (50.7) |
| Non-ischaemic | 10 (29.4) | 14 (37.8) | 24 (33.8) |
| Alcoholic | 1 (2.9) | 0 (0) | 1 (1.4) |
| Hypertensive | 4 (11.8) | 9 (24.3) | 13 (18.3) |
| Infectious/viral cardiomyopathy | 1 (2.9) | 2 (5.4) | 3 (4.2) |
| Other | 4 (11.7) | 9 (24.3) | 13 (18.2) |
| Not known | 2 (5.9) | 0 (0) | 2 (2.8) |
| Prior history of MI; n (%) | 17 (50.0) | 19 (51.4) | 36 (50.7) |
| Concomitant disorders; n (%) | |||
| Hypertension | 24 (70.6) | 20 (54.1) | 44 (62.0) |
| Atrial fibrillation | 12 (35.3) | 17 (45.9) | 29 (40.8) |
| Diabetes | 15 (44.1) | 14 (37.8) | 29 (40.8) |
| Chronic renal failure | 6 (17.6) | 6 (16.2) | 12 (16.9) |
| Chronic obstructive pulmonary disease | 7 (20.6) | 5 (13.5) | 12 (16.9) |
| Coronary artery disease | 7 (20.6) | 7 (18.9) | 14 (19.7) |
| Prior medications (on admission); n (%) | |||
| Diuretics | 34 (100) | 37 (100) | 71 (100) |
| ACE inhibitors | 24 (70.6) | 29 (78.4) | 53 (74.6) |
| Angiotensin II antagonists | 11 (32.4) | 6 (16.2) | 17 (23.9) |
| Beta-blocking agents | 15 (44.1) | 20 (54.1) | 35 (49.3) |
| Alpha- and beta-blocking agents | 20 (58.8) | 16 (43.2) | 36 (50.7) |
| Aldosterone antagonists | 18 (52.9) | 19 (51.4) | 37 (52.1) |
| Digitalis glycosides | 7 (20.6) | 12 (32.4) | 19 (26.8) |
| HMG CoA reductase inhibitors | 20 (58.8) | 21 (56.8) | 41 (57.7) |
| Concomitant medications (on randomization); n (%) | |||
| Loop diuretics | 34 (100.0) | 36 (97.3) | 70 (98.6) |
| Organic nitratesc | 5 (14.7) | 12 (32.4) | 17 (23.9) |
| Heparin | 20 (58.8) | 15 (40.5) | 35 (49.3) |
| Vitamin K antagonists | 14 (41.2) | 17 (45.9) | 31 (43.7) |
| Baseline haemodynamic parametersd | |||
| PCWP (mmHg) | 26.2 (5.9) | 26.5 (5.2) | — |
| CI (L/min/m2) | 2.4 (0.7) | 2.2 (0.6) | — |
| Systolic PAP (mmHg) | 56.1 (13.0) | 58.0 (13.8) | — |
| Diastolic PAP (mmHg) | 27.3 (6.2) | 28.8 (6.9) | — |
| Mean PAP (mmHg) | 36.9 (7.9) | 38.5 (8.1) | — |
| RAP (mmHg) | 12.7 (5.9) | 12.3 (5.5) | — |
| SVR (dynes × s/cm5) | 1530 (462) | 1720 (607) | — |
| PVR (dynes × s/cm5) | 210 (161) | 243 (166) | — |
Data presented as mean (SD), unless otherwise specified.
eGFR, estimated glomerular filtration rate; SBP, systolic blood pressure; DBP, diastolic blood pressure; AHF, acute heart failure; HF, heart failure; MI, myocardial infarction; NYHA, New York Heart Association; ACE, angiotensin-converting enzyme; HMG CoA, 5-hydroxyl-3-methylglutaryl-coenzyme A; PCWP, pulmonary capillary wedge pressure; CI, cardiac index; PAP, pulmonary arterial pressure; RAP, right atrial pressure; SVR, systemic vascular resistance; PVR, pulmonary vascular resistance.
aAt the time of screening, bserelaxin, n = 22; placebo, n = 30, coral or i.v., dserelaxin, n = 32; placebo, n = 31.
Figure 1.

Patient disposition. *Between Day 3 and Day 30 for all three patients.
Effects of serelaxin on haemodynamic parameters
Co-primary endpoints
Baseline mean (SD) PCWP was similar between treatment groups at 26.1 (5.9) mmHg in the serelaxin group and 26.5 (5.2) mmHg in the placebo group. Patients treated with serelaxin had significantly larger decrease in peak PCWP during the first 8 h of infusion when compared with those who received placebo (P = 0.004, Table 2).
Table 2.
Summary of changes from baseline in haemodynamic indices
| Haemodynamic parameter | Serelaxin (n = 32) | Placebo (n = 31) | Treatment difference (95% confidence interval) | P-value |
|---|---|---|---|---|
| PCWP (mmHg) | ||||
| Peak PCWP over 8 h | −6.69 (0.59) | −4.25 (0.60) | −2.44 (−4.10, −0.78) | 0.0040 |
| Time-weighted averagea | ||||
| 0–8 h | −3.79 (0.50) | −1.08 (0.51) | −2.70 (−4.10, −1.31) | 0.0001 |
| 8–20 h | −4.90 (0.73) | −2.67 (0.74) | −2.24 (−4.28, −0.19) | 0.0322 |
| 0–20 h | −4.46 (0.59) | −2.04 (0.60) | −2.42 (−4.08, −0.76) | 0.0042 |
| 20–24 h | −4.41 (0.83) | −3.11 (0.85) | −1.30 (−3.63, 1.03) | 0.27 |
| CI (L/min/m2) | ||||
| Peak CI over 8 h | 0.32 (0.05) | 0.30 (0.05) | 0.02 (−0.13, 0.16) | 0.79 |
| Time-weighted averagea | ||||
| 0–8 h | 0.12 (0.04) | 0.07 (0.04) | 0.04 (−0.07, 0.15) | 0.48 |
| 8–20 h | 0.05 (0.05) | 0.07 (0.05) | −0.02 (−0.15, 0.12) | 0.80 |
| 0–20 h | 0.08 (0.04) | 0.07 (0.04) | 0.01 (−0.11, 0.12) | 0.92 |
| 20–24 h | 0.08 (0.05) | 0.03 (0.05) | 0.05 (−0.09, 0.20) | 0.46 |
| Systolic PAP (mmHg) | ||||
| Peak systolic PAP over 8 h | −10.77 (1.03) | −4.59 (1.05) | −6.19 (−9.07, −3.30) | <0.0001 |
| Time-weighted averagea | ||||
| 0–8 h | −5.35 (0.93) | 0.64 (0.94) | −5.99 (−8.59, −3.39) | <0.0001 |
| 8–20 h | −5.87 (1.15) | −0.12 (1.17) | −5.74 (−8.97, −2.52) | 0.0005 |
| 0–20 h | −5.66 (0.98) | 0.18 (1.00) | −5.84 (−8.59, −3.09) | <0.0001 |
| 20–24 h | −5.05 (1.28) | −1.92 (1.30) | −3.13 (−6.71, 0.44) | 0.09 |
| Diastolic PAP (mmHg) | ||||
| Peak diastolic PAP over 8 h | −6.50 (0.70) | −3.89 (0.71) | −2.62 (−4.58, −0.66) | 0.0089 |
| Time-weighted averagea | ||||
| 0–8 h | −3.29 (0.59) | −0.22 (0.60) | −3.07 (−4.73, −1.42) | 0.0003 |
| 8–20 h | −3.95 (0.80) | −1.09 (0.81) | −2.86 (−5.10, −0.63) | 0.0119 |
| 0–20 h | −3.69 (0.64) | −0.74 (0.65) | −2.95 (−4.74, −1.16) | 0.0012 |
| 20–24 h | −3.93 (0.86) | −1.51 (0.88) | −2.42 (−4.84, 0.00) | 0.05 |
| Mean PAP (mmHg) | ||||
| Peak mean PAP over 8 h | −7.56 (0.72) | −3.63 (0.74) | −3.93 (−5.96, −1.90) | 0.0001 |
| Time-weighted averagea | ||||
| 0–8 h | −3.98 (0.65) | 0.06 (0.66) | −4.04 (−5.86, −2.22) | <0.0001 |
| 8–20 h | −4.56 (0.88) | −0.80 (0.89) | −3.76 (−6.22, −1.29) | 0.0028 |
| 0–20 h | −4.32 (0.72) | −0.45 (0.73) | −3.87 (−5.89, −1.85) | 0.0002 |
| 20–24 h | −4.29 (0.96) | −1.67 (0.98) | −2.62 (−5.31, 0.07) | 0.06 |
| RAP (mmHg) | ||||
| Peak mean RAP over 8 h | −3.24 (0.36) | −2.07 (0.36) | −1.16 (−2.16, −0.17) | 0.0216 |
| Time-weighted averagea | ||||
| 0–8 h | −1.12 (0.36) | −0.23 (0.36) | −0.89 (−1.89, 0.12) | 0.08 |
| 8–20 h | −1.12 (0.55) | −0.62 (0.55) | −0.49 (−2.02, 1.03) | 0.53 |
| 0–20 h | −1.12 (0.45) | −0.47 (0.45) | −0.65 (−1.89, 0.59) | 0.31 |
| 20–24 h | −1.26 (0.67) | −1.45 (0.67) | 0.19 (−1.68, 2.06) | 0.84 |
| SVR (dynes × s/cm5) | ||||
| Peak SVR over 8 h | −368.06 (45.92) | −284.62 (45.92) | −83.44 (−211.72, 44.85) | 0.20 |
| Time-weighted averagea | ||||
| 0–8 h | −166.15 (44.73) | −29.24 (44.73) | −136.91 (−261.89, −11.93) | 0.0318 |
| 8–20 h | −158.82 (57.04) | −27.20 (57.04) | −131.62 (−291.00, 27.76) | 0.11 |
| 0–20 h | −161.75 (48.44) | −28.02 (48.44) | −133.74 (−269.09, 1.62) | 0.05 |
| 20–24 h | −181.94 (67.36) | 70.01 (67.36) | −251.95 (−440.15, −63.75) | 0.0087 |
| PVR (dynes × s/cm5) | ||||
| Peak PVR over 8 h | −77.73 (10.01) | −52.69 (10.01) | −25.04 (−52.86, 2.78) | 0.08 |
| Time-weighted averagea | ||||
| 0–8 h | −20.30 (9.91) | 18.69 (9.91) | −38.99 (−66.53, −11.44) | 0.0055 |
| 8–20 h | −12.27 (12.28) | 30.45 (12.28) | −42.73 (−76.85, −8.60) | 0.0141 |
| 0–20 h | −15.48 (10.08) | 25.75 (10.08) | −41.23 (−69.25, −13.21) | 0.0039 |
| 20–24 h | −10.15 (16.20) | 30.14 (16.20) | −40.28 (−85.31, 4.74) | 0.08 |
Data represented as least squares mean (SE) change from baseline.
PCWP, pulmonary capillary wedge pressure; CI, cardiac index; PAP, pulmonary arterial pressure; RAP, right atrial pressure; SVR, systemic vascular resistance; PVR, pulmonary vascular resistance.
aBased on area under the effect curve for the corresponding time interval.
Baseline mean (SD) CI was 2.4 (0.7) L/min/m2 in the serelaxin group and 2.2 (0.6) L/min/m2 in the placebo group. There was no significant effect of serelaxin 8-h infusion on peak change in CI vs. placebo (P = 0.79, Table 2).
Secondary endpoints
Compared with placebo, treatment with serelaxin resulted in a statistically significant treatment difference in the time-weighted average PCWP change from baseline over 0–8 h (P = 0.0001) and over 8–20 h (P = 0.03, Table 2). Effects on PCWP in the serelaxin group were already apparent at 30 min, becoming statistically significant after 2 h (Figure 2). Effect of serelaxin on PCWP was sustained till completion of the drug infusion; however, there was no significant difference compared with placebo at time points between 20 and 24 h (Figure 2) and in the time-weighted average change from baseline over 20–24 h (P = 0.27, Table 2). Serelaxin leads to a small increase in CI that was also noted in the placebo group during the entire 20-h infusion period, and no significant treatment effect was observed (Table 2).
Figure 2.

Effects on haemodynamic indices during the course of study in both treatment groups. Data presented as means ± standard error.
Serelaxin significantly lowered PAP (systolic and diastolic) with the effect becoming evident and statistically significant already after the first 30 min of infusion [for mean PAP at 30 min, treatment effect as LS mean difference vs. placebo: −3.01 (95% confidence interval: −5.20, −0.81) mmHg, P = 0.0072; largest treatment effect −5.17 (−7.49, −2.86) mmHg occurred 4 h after the start of infusion, P < 0.0001] (Table 2), sustained throughout the whole infusion period and remained significant until the end of the 24-h haemodynamic evaluation (Table 2, Figure 2).
Changes in PAP and PCWP were mirrored by significant decrease in PVR in patients treated with serelaxin. For the time-weighted average PVR change from baseline, there were statistically significant decreases over 0–8 h (P = 0.0055) and 8–20 h (P = 0.014, Table 2). Peak mean RAP significantly decreased during 8 h of infusion in the serelaxin group compared with placebo (P = 0.0216). Systemic vascular resistance decreased from baseline in the serelaxin group vs. placebo, and treatment differences reached statistical significance at some time points (Table 2 and Figure 2).
Slight decreases in systolic/diastolic BP with serelaxin were observed already 30 min after the initiation of the infusion. Effects on peripheral BP were sustained for 24 h after the infusion was stopped in the serelaxin group compared with baseline. Indeed, treatment differences were statistically significant for systolic BP at 20, 21, and 27 h and for diastolic BP at 20, 21, 22, 24, 27, and 44 h (Figure 2). During the infusion with serelaxin, pulse rate increased to a maximum of 78 (16) b.p.m. at 2 h [placebo: 79 (18) b.p.m.; mean (SD)] and returned to baseline levels by the end of the 20-h infusion period. It was 74 (12) b.p.m. in the serelaxin group and 79 (18) b.p.m. in the placebo group at 44 h (Figure 2).
Results of haemodynamic parameters were similar in intention-to-treat patient population on post hoc ANCOVA see (Supplementary material online, Table S1).
Effects of serelaxin on parameters of renal function
The results on the effect of serelaxin on renal function are summarized in Figure 3. Prior to the start of infusion (−4 to 0 h), urine flow rates were lower in the serelaxin group than in the placebo group (geometric mean 168 vs. 215 mL/h). Urine flow rates decreased from baseline in both treatment groups during the 20-h infusion period. Over the entire 20-h infusion period, there was 39% decrease from baseline in urine flow rate in the serelaxin group compared with a 59% decrease in the placebo group [ratio of LS mean ratio to baseline 1.20 (0.98, 1.48), P = 0.07 on post hoc ANCOVA].
Figure 3.
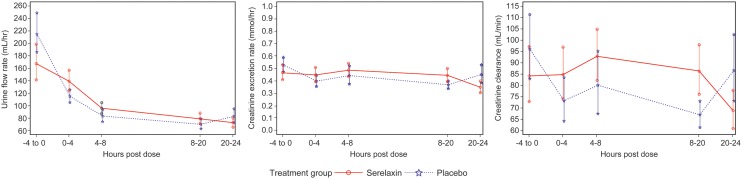
Effects on selected parameters of renal function in both treatment groups. Data presented as means ± standard error.
Baseline (−4 to 0 h) creatinine excretion rates were similar in the serelaxin and placebo groups (geometric mean 0.47 and 0.53 mmol/h, respectively). There was an 11% increase from baseline in urine creatinine excretion rate in the serelaxin group and a 21% decrease in the placebo group during the 20-h infusion period [ratio of LS mean ratio to baseline 1.25 (0.97, 1.61), P = 0.09 on post hoc ANCOVA].
Baseline (−4 to 0 h) creatinine clearance values were slightly lower in the serelaxin group than in the placebo group (geometric mean 84.2 vs. 96.1 mL/min). Over the entire 20-h infusion period with serelaxin, there was a 20% increase from baseline in creatinine clearance [geometric mean ratio 1.20 (0.90, 1.60)], whereas creatinine clearance decreased from baseline by 24% in the placebo group [geometric mean ratio 0.77 (0.58, 1.01)]. A post hoc ANCOVA confirmed a statistically significant treatment difference of 39% for 0–20 h in creatinine clearance change from baseline [ratio of LS mean ratio to baseline 1.39 (1.07, 1.81), P = 0.0143].
Effect of serelaxin on natriuretic peptides
Baseline values of NT-pro-BNP were lower in the serelaxin group compared with the placebo group (geometric mean 2702 and 3376 pg/mL, respectively). At the end of the 20-h infusion period, there was a 13% decrease from baseline in NT-pro-BNP in the serelaxin group and a 3% increase in the placebo group [ratio of LS mean ratio to baseline 0.83 (0.69, 0.99), P = 0.0366 on post hoc ANCOVA]. By 44 h, there was a 33% decrease from baseline in the serelaxin group and a 14% decrease in the placebo group [ratio of LS mean ratio to baseline 0.75 (0.60–0.93), P = 0.0101 on post hoc ANCOVA].
Adverse events
The drug infusions were well tolerated (Table 3). A total of 49 patients (69%) experienced at least one AE during the study, with a slightly lower proportion of patients in the serelaxin group (21 patients; 62%) than in the placebo group (28 patients; 76%). Adverse event of particular interest (hypotension/BP decreased/syncope) was reported by non-significantly more patients in the serelaxin group (five patients; 15%) compared with the placebo group (three patients; 9%); however, the majority of events was of mild-to-moderate severity. Serelaxin infusion was stopped prematurely in three patients as per the protocol-defined criteria of decrease in systolic BP. Eleven patients experienced 11 SAEs during the study: three (9%) patients in the serelaxin group and eight (22%) patients in the placebo group. None of these was suspected to be related to study drug by the investigators, while four events were fatal (serelaxin: one acute myocardial infarction on Day 8 and one acute pulmonary oedema 21 h post-dose; placebo: one spinal column injury on Day 22 and one pulmonary oedema on Day 23).
Table 3.
Adverse events reported in the study
| Adverse events, n (%) | Serelaxin (n = 34) | Placebo (n = 37) | Total (n = 71) |
|---|---|---|---|
| Hypokalaemia | 8 (23.5) | 6 (16.2) | 14 (19.7) |
| Haematuria | 4 (11.8) | 1 (2.7) | 5 (7.0) |
| Hypotension | 3 (8.8) | 1 (2.7) | 4 (5.6) |
| Constipation | 1 (2.9) | 3 (8.1) | 4 (5.6) |
| Headache | 0 (0.0) | 3 (8.1) | 3 (4.2) |
Adverse events reported by at least three patients in either of the group are mentioned.
Discussion
This is the first study specifically designed to evaluate the haemodynamic effects of serelaxin, given as a 20-h infusion, in patients hospitalized due to AHF. The major novel findings are that serelaxin exerted rapid haemodynamic effects, characterized by a reduction in PCWP and PAP with concomitant decrease in SVR and PVR. Most of these changes were apparent early (being detectable already after initial 30 min) and sustained throughout the entire infusion period. No significant changes in CI were detected. Consistent with earlier clinical studies, serelaxin therapy caused a slight decrease in systolic/diastolic BP, without a change in pulse rate and any drug-related serious tolerability concerns. Renal effects included significantly increased creatinine clearance; and NT-pro-BNP levels decreased with serelaxin infusion when compared with placebo. Again, these findings are consistent with earlier observations from the RELAX-AHF trial.8
Relaxin-2 is a naturally occurring peptide that possesses multiple systemic and renal vasodilatory properties, for example, mediating maternal adjustments to pregnancy.3 Importantly, numerous experimental and clinical studies using relaxin-2 have confirmed these effects, which form the background to believe that the physiological effects of this hormone may be pharmacologically useful in modulating cardiovascular and renal function in the setting of heart failure.4–6
Acute heart failure is a complex and heterogeneous clinical entity with numerous underlying pathophysiological mechanisms, among which vasoconstriction plays a central role.12 Constriction of the venous and arterial beds has deleterious effects on the end-organs and significantly contributes to the clinical progression of the disease and symptoms in the setting of AHF. Thus, an augmented vascular tone may become a target for treatment in selected AHF patients. In fact, all the epidemiological data confirm that the majority of patients admitted to the hospital due to AHF present with normal-to-elevated systolic BP and has a clinical profile labelled as ‘acute vascular failure’ that can be characterized by the evidence of vasoconstriction.13–15 Therefore, it may well be expected that in such a population, drugs with vasorelaxing properties to address key underlying pathophysiological abnormalities would be preferable. Importantly, although recently advocated, this ‘targeted approach’ has never been prospectively validated in clinical trials in AHF. Relaxin-2, with its potent biological vasorelaxing properties, seems to be an attractive therapeutic option.
Previous clinical experience with serelaxin in AHF is very encouraging. A phase II study, Pre-RELAX-AHF, demonstrated that in patients with AHF, normal-to-elevated systolic BP and mild-to-moderate renal dysfunction treatment with serelaxin significantly improved dyspnoea and had favourable effects on post-discharge clinical outcomes.7 These results were confirmed in the RELAX-AHF phase III trial, which demonstrated that a 48-h infusion of serelaxin in AHF patients with similar clinical characteristics resulted in improvement of dyspnoea by visual analogue scale and other clinical signs/symptoms of congestion; among secondary clinical and safety endpoints, a significant reduction in early worsening of heart failure and a 37% reduction in cardiovascular and all-cause mortality during the 180-day follow-up were observed.8 Several potential explanations of these favourable short- and long-term effects of serelaxin have been postulated, taking into account its numerous biological properties. It seems that early relief of congestion and possible prevention of end-organ damage may be associated with reduction of the risk of clinical deterioration during hospital stay and beneficial longer-term effects on mortality.9 One of the underlying mechanisms may be related to the favourable haemodynamic effects that serelaxin may exert in patients with AHF, as observed in the present study, along with improvements in biomarkers of end-organ damage.
Until now, haemodynamic effects of serelaxin have only been evaluated in a small, open-label study that recruited patients with advanced, stable heart failure.5 In the present placebo-controlled study, the vasorelaxant effects of serelaxin were shown with reductions in PCWP, mean PAP, and SVR without significant changes in CI compared with placebo and standard therapy. However, direct comparison between these two studies needs to be rather cautious, as the clinical responses in acute settings of heart failure may substantially differ to that in patients with stable disease.
In the present study, serelaxin was characterized by decreases in PCWP, PAP, RAP, SVR, and PVR that occurred early and were sustained throughout the whole infusion period. Serelaxin did not significantly affect CI, as measured by the thermodilution method. Very few patients required dose adjustment due to oligosymptomatic decreases in BP and despite the lower enrolment criterion for systolic BP (≥115 mmHg before randomization) compared with the previous trials in AHF patients, the decreases in systolic/diastolic BP were manageable and performed according to the same algorithm as in RELAX-AHF. We believe that such careful monitoring of changes in BP and appropriate adjustment of serelaxin therapy are of particular relevance in order to avoid potential adverse events related to hypotension. In fact, neither in this study nor in the RELAX-AHF trial was any trend observed towards an increase in clinically relevant adverse events related to hypotension.8 Importantly, changes in PAP did not seem to be a simple consequence of a passive drop in PCWP, as they were seen earlier and were more pronounced than changes in PCWP. It may be hypothesized that serelaxin also exerts a vasorelaxant effect on the pre-capillary pulmonary bed in patients with AHF and normal-elevated BP, and further mechanistic studies are warranted.
Renal changes during serelaxin infusion were characterized as a modest increase in creatinine clearance without any marked effects on serum levels of creatinine and urea (data not shown). Urine flow decreased in both treatment groups, particularly during the first 8 h of the infusion, but this seems to be related to our study design with planned i.v. loop diuretic (bolus) administration at 4 h before the initiation of infusion and after the first 8 h of the infusion. Interestingly, at the end of the infusion there was a statistically significant reduction in NT-pro-BNP in those receiving serelaxin. These data confirm similar observations derived from the larger RELAX-AHF trial.9
The interpretation of the favourable effects of any drug used in the AHF setting in the context of potential beneficial consequences on short- and long-term clinical outcomes remains controversial. Still, a direct relationship between changes in haemodynamics and improvement in clinical signs and symptoms in the early phase of AHF has not been confirmed. The results of studies with different drugs remain inconclusive, because in several reports favourable effects on haemodynamic indices were not directly related with better symptomatic outcomes. Only recently, Solomonica et al.16 documented in a retrospective analysis that among haemodynamic indices only acute changes in PCWP and PAP, but not SVR and CI, predicted improvement in dyspnoea severity in patients with AHF. Similarly, there are no haemodynamic indices for which changes have been able to predict favourable longer-term outcomes. However, the favourable haemodynamic ‘unloading’ profile of serelaxin demonstrated in this study may lead to more remarkable decongestion, and the effects on the systemic and pulmonary vasculature may translate into long-lasting improvement in end-organ perfusion with less damage and possibly longer-term benefits found in the RELAX-AHF study.8,9
Comparison of the present study results with reports of other agents that have demonstrated haemodynamic improvements, such as nesiritide, nitroglycerine (NTG), or ularitide would be of interest. In the VMAC study, for example, nesiritide resulted in a significant reduction in the mean PCWP of 5.8 vs. 2 mmHg with placebo at 3 h. Effects on CI were significantly different from placebo at 1 h with nesiritide but not at 3 h. The comparator group treated with NTG showed the mean PCWP reduction of 3.8 mmHg.10 In the SIRIUS II study, ularitide was reported to significantly reduce PCWP at 6 h (∼8.5 mmHg reduction with higher doses vs. ∼4 mmHg with placebo) along with increase in CI (∼0.5–0.6 L/min/m2 with higher doses vs. ∼0.3 L/min/m2 with placebo).11 Our results have shown smaller changes from baseline in PCWP (peak effect of −6.69 mmHg during 8 h vs. −4.25 mmHg with placebo; maximum effect of −5.03 mmHg vs. −1.5 mmHg with placebo at 8 h). However, due to noteworthy differences in patient populations studied, direct comparative clinical trials would be necessary to draw robust conclusions. It is of note that despite demonstration of haemodynamic improvements with these agents, there is no evidence to support any improvements in long-term outcomes (for example with nesiritide17) in contrast to what was observed with serelaxin in the RELAX-AHF trial.8
Potential limitations of the study include the small sample size due to the exploratory nature of the trial and use of per-protocol analysis, which does not necessarily reflect the real-world setting. Patients who received furosemide during the first 8 h of study drug infusion were excluded from the per-protocol analysis. However, per-protocol analysis was considered more appropriate for this study with a view to reducing data variability by excluding subjects with protocol deviations. To address potential limitations, an intention-to-treat analysis (post hoc) was performed which entirely confirmed the findings of the per-protocol analysis. Additionally, interpreting the haemodynamic results, it needs to be remembered that changes in PCWP were rather of modest magnitude, which indicates that they are not the only mechanism underlying favourable results with serelaxin in other studies.7,8 Further, interpretation of safety assessments is also limited by small sample size. These limitations aside, the study results are considered robust and provide a comprehensive assessment of the haemodynamic effects of serelaxin in patients with AHF.
Conclusions
Serelaxin was well tolerated with no apparent safety concerns and exerted favourable haemodynamic effects on PCWP and PAP, but did not significantly change CI, in patients with AHF and normal-to-elevated systolic BP. These effects are consistent with the changes in signs and symptoms of congestion observed with serelaxin in previous clinical studies.
Supplementary material
Supplementary material is available at European Heart Journal online.
Funding
This study was supported by Novartis Pharma AG, Switzerland. Funding to pay the Open Access publication charges for this article was provided by Novartis Pharma AG.
Conflict of interest: P.P. has received honoraria related to activities as principal investigator of this study; he has also received speaker fees and is a consultant to Novartis. V.M. has received honoraria, lecture fees and support for travel to meetings from Novartis. He is a board member, consultant, and has received lecture fees from Bayer. M.R. has no conflict of interest. A.F. has received fees for being national coordinator in Argentina from Sanatorio Modelo Quilmes. A.A.V. is a consultant to Novartis and has received grants, honoraria, fees for participation in review activities, and lecture fees from Novartis. A.V. has no conflict of interest. G.C. and O.M. are employees of Momentum Research, Inc. (MRI), which received research grants from Corthera and Novartis to provide services for this study, support for travel to meetings and fees for participation in review activities. MRI additionally has received research grants from Novacardia, Merck, Nile, Celadon, BioHeart, Cardio 3, Amgen, Trevena and NIH. R.Z. was a subinvestigator for the study and has no conflict of interest. M.M. is a board member with Novartis and has received lecture fees and honoraria (<1500 euros) to his institution. He is also a member of data monitoring committee and consultant to Bayer. He has received lecture fees from Abbott. U.L., Y.Z., and M.D. are employees of Novartis and have stock options.
Supplementary Material
Acknowledgements
The authors acknowledge Steve Winter, Novartis Pharma AG, Basel, Switzerland and Amit Kubavat, Novartis Healthcare Pvt. Ltd, Hyderabad, India, for medical writing assistance and subsequent revisions based on authors' feedback. The authors also acknowledge all the study coordinators at the participating centres and all the patients who participated in the study.
References
- 1.Gheorghiade M, Pang PS. Acute heart failure syndromes. J Am Coll Cardiol. 2009;53:557–573. doi: 10.1016/j.jacc.2008.10.041. [DOI] [PubMed] [Google Scholar]
- 2.Fang J, Mensah GA, Croft JB, Keenan NL. Heart failure-related hospitalization in the U.S., 1979 to 2004. J Am Coll Cardiol. 2008;52:428–434. doi: 10.1016/j.jacc.2008.03.061. [DOI] [PubMed] [Google Scholar]
- 3.Conrad KP. Maternal vasodilation in pregnancy: the emerging role of relaxin. Am J Physiol Regul Integr Comp Physiol. 2011;301:R267–R275. doi: 10.1152/ajpregu.00156.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teichman SL, Unemori E, Teerlink JR, Cotter G, Metra M. Relaxin: review of biology and potential role in treating heart failure. Curr Heart Fail Rep. 2010;7:75–82. doi: 10.1007/s11897-010-0010-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dschietzig T, Teichman S, Unemori E, Wood S, Boehmer J, Richter C, Baumann G, Stangl K. Intravenous recombinant human relaxin in compensated heart failure: a safety, tolerability, and pharmacodynamic trial. J Card Fail. 2009;15:182–190. doi: 10.1016/j.cardfail.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 6.Du XJ, Bathgate RA, Samuel CS, Dart AM, Summers RJ. Cardiovascular effects of relaxin: from basic science to clinical therapy. Nat Rev Cardiol. 2010;7:48–58. doi: 10.1038/nrcardio.2009.198. [DOI] [PubMed] [Google Scholar]
- 7.Teerlink JR, Metra M, Felker GM, Ponikowski P, Voors AA, Weatherley BD, Marmor A, Katz A, Grzybowski J, Unemori E, Teichman SL, Cotter G. Relaxin for the treatment of patients with acute heart failure (Pre-RELAX-AHF): a multicentre, randomised, placebo-controlled, parallel-group, dose-finding phase IIb study. Lancet. 2009;373:1429–1439. doi: 10.1016/S0140-6736(09)60622-X. [DOI] [PubMed] [Google Scholar]
- 8.Teerlink JR, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, Ponikowski P, Unemori E, Voors AA, Adams KF, Jr, Dorobantu MI, Grinfeld LR, Jondeau G, Marmor A, Masip J, Pang PS, Werdan K, Teichman SL, Trapani A, Bush CA, Saini R, Schumacher C, Severin TM, Metra M. Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet. 2013;381:29–39. doi: 10.1016/S0140-6736(12)61855-8. [DOI] [PubMed] [Google Scholar]
- 9.Metra M, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, Ponikowski P, Unemori E, Voors AA, Adams KF, Jr, Dorobantu MI, Grinfeld L, Jondeau G, Marmor A, Masip J, Pang PS, Werdan K, Prescott MF, Edwards C, Teichman SL, Trapani A, Bush CA, Saini R, Schumacher C, Severin T, Teerlink JR. Effect of serelaxin on cardiac, renal, and hepatic biomarkers in the Relaxin in Acute Heart Failure (RELAX-AHF) development program: correlation with outcomes. J Am Coll Cardiol. 2013;61:196–206. doi: 10.1016/j.jacc.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 10.Publication Committee for the VMAC Investigators (Vasodilatation in the Management of Acute CHF) Intravenous nesiritide vs. nitroglycerin for treatment of decompensated congestive heart failure: a randomized controlled trial. JAMA. 2002;287:1531–1540. doi: 10.1001/jama.287.12.1531. [DOI] [PubMed] [Google Scholar]
- 11.Mitrovic V, Seferovic PM, Simeunovic D, Ristic AD, Miric M, Moiseyev VS, Kobalava Z, Nitsche K, Forssmann WG, Luss H, Meyer M. Haemodynamic and clinical effects of ularitide in decompensated heart failure. Eur Heart J. 2006;27:2823–2832. doi: 10.1093/eurheartj/ehl337. [DOI] [PubMed] [Google Scholar]
- 12.Metra M, Teerlink JR, Voors AA, Felker GM, Milo-Cotter O, Weatherley B, Dittrich H, Cotter G. Vasodilators in the treatment of acute heart failure: what we know, what we don't. Heart Fail Rev. 2009;14:299–307. doi: 10.1007/s10741-008-9127-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gheorghiade M, Abraham WT, Albert NM, Greenberg BH, O'Connor CM, She L, Stough WG, Yancy CW, Young JB, Fonarow GC. Systolic blood pressure at admission, clinical characteristics, and outcomes in patients hospitalized with acute heart failure. JAMA. 2006;296:2217–2226. doi: 10.1001/jama.296.18.2217. [DOI] [PubMed] [Google Scholar]
- 14.Cotter G, Felker GM, Adams KF, Milo-Cotter O, O'Connor CM. The pathophysiology of acute heart failure—is it all about fluid accumulation? Am Heart J. 2008;155:9–18. doi: 10.1016/j.ahj.2006.02.038. [DOI] [PubMed] [Google Scholar]
- 15.Cotter G, Metra M, Milo-Cotter O, Dittrich HC, Gheorghiade M. Fluid overload in acute heart failure—re-distribution and other mechanisms beyond fluid accumulation. Eur J Heart Fail. 2008;10:165–169. doi: 10.1016/j.ejheart.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 16.Solomonica A, Burger AJ, Aronson D. Hemodynamic determinants of dyspnea improvement in acute decompensated heart failure. Circ Heart Fail. 2013;6:53–60. doi: 10.1161/CIRCHEARTFAILURE.112.970335. [DOI] [PubMed] [Google Scholar]
- 17.O'Connor CM, Starling RC, Hernandez AF, Armstrong PW, Dickstein K, Hasselblad V, Heizer GM, Komajda M, Massie BM, McMurray JJ, Nieminen MS, Reist CJ, Rouleau JL, Swedberg K, Adams KF, Jr, Anker SD, Atar D, Battler A, Botero R, Bohidar NR, Butler J, Clausell N, Corbalán R, Costanzo MR, Dahlstrom U, Deckelbaum LI, Diaz R, Dunlap ME, Ezekowitz JA, Feldman D, Felker GM, Fonarow GC, Gennevois D, Gottlieb SS, Hill JA, Hollander JE, Howlett JG, Hudson MP, Kociol RD, Krum H, Laucevicius A, Levy WC, Méndez GF, Metra M, Mittal S, Oh BH, Pereira NL, Ponikowski P, Tang WH, Tanomsup S, Teerlink JR, Triposkiadis F, Troughton RW, Voors AA, Whellan DJ, Zannad F, Califf RM. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med. 2011;365:32–43. doi: 10.1056/NEJMoa1100171. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.